
Two-step seed swelling polymerization to prepare poly(glycidyl methacrylate-divinylbenzene) microspheres and their sulfonation for chromatographic separation of rare earth elements†
Chen
Shen
ab,
Yuqing
Wei
ab,
Shuaishuai
Wang
ab,
Hongwei
Zhao
a,
Qifang
He
a and
Hongdeng
Qiu
 *abc
*abc
aKey Laboratory of Rare Earths, Ganjiang Innovation Academy, Chinese Academy of Sciences, Ganzhou 341119, China. E-mail: hdqiu@gia.cas.cn
bSchool of Rare Earths, University of Science and Technology of China, Hefei 230026, China
cResearch Center for Natural Medicine and Chemical Metrology, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, China
First published on 28th May 2025
Abstract
A two-step seed swelling polymerization method was used to successfully synthesize monodisperse porous poly(glycidyl methacrylate-divinylbenzene) (PGMA-DVB) microspheres, which can be sulfonated and then used as new cation-exchange stationary phases for the separation of rare earth elements (REEs), demonstrating high resolution, stability, and selective separation of the REEs.
Rare earth elements (REEs) comprise 17 metallic elements, including 15 lanthanides, along with scandium (Sc) and yttrium (Y) from group III B.1–3 Their exceptional optical, electrical, and magnetic properties make them indispensable in modern technology. REEs have widespread applications in high-tech fields such as catalysis,4–6 high-performance magnets,7 specialty ceramics,8–10 phosphors,11,12 and fiber-optic communication devices.13 Additionally, REEs are used in alloy manufacturing,14 precision glass polishing,15 laser technology,16 rechargeable batteries,17 high-efficiency motors,18etc. With advancements in electronics, clean energy, and laser fields, demand for REEs continues to rise, along with increased purity and performance requirements, solidifying their role as strategic resources. However, their similar properties and natural co-occurrence make separation challenging. Various technologies have been developed to extract REEs from secondary resources such as industrial residues, magnet scraps, and spent lighting equipment. Main purification methods include chemical precipitation,19 solvent extraction,20 fractional crystallization,21 and molten salt electrolysis.22,23 These processes often require multi-stage operations and large amounts of reagents, including strong acids and extractants, leading to high costs and environmental risks from toxic contamination.
High-performance liquid chromatography (HPLC) has become a promising and eco-friendly separation method, offering versatile applications in analysis and purification.24–26 HPLC enables rapid, high-resolution separation with superior sensitivity, column reusability, automation, and reproducibility.27–31 Campbell et al. first applied HPLC for REE separation using Dowex 50 resin.32 Fernández et al. achieved isocratic separation using anion-exchange resins by forming REE anionic complexes with ethylenediaminetetraacetic acid (EDTA).33 Inoue et al. successfully separated a series of REEs in 22 minutes using a nitrilotriacetate-type (NTA)-type chelating resin as the stationary phase and nitric acid as the eluent.34
The column is the core of chromatography, and its packing material is crucial for achieving efficient and selective separations. However, conventional materials such as silica gel and alumina are prone to hydrolysis under strongly acidic or basic conditions. To overcome these limitations, monodisperse porous polymeric microspheres have attracted extensive research interest. Compared to traditional materials, polymeric microspheres offer greater ligand flexibility, maximizing the interactions between the stationary phase and target analytes.
In this study, monodisperse porous poly(glycidyl methacrylate-divinylbenzene) (PGMA-DVB) microspheres were synthesized via a two-step seed swelling polymerization method, followed by sulfonation to introduce sulfonic acid functional groups. This study represents the first successful application of PGMA-DVB-based microspheres as stationary phases in liquid chromatography to achieve baseline separation of 15 REEs. Furthermore, the chromatographic column exhibited excellent long-term stability, confirming its reliability for extended use.
The preparation of PGMA-DVB includes two stages: seed swelling and absorption polymerization. Initially, monodisperse polystyrene (PS) seed microspheres are synthesized via dispersion polymerization and activated using a swelling agent. Subsequently, an organic phase containing monomers, crosslinkers, and porogens is introduced, allowing the activated PS seed microspheres to fully absorb the organic phase and reach dynamic equilibrium, which is crucial for forming crosslinked polymer microspheres. After polymerization, the porogen-filled solvent phase occupies the internal pores of the microspheres, and upon removal, a well-developed porous structure is obtained. PGMA-DVB microspheres were further sulfonated using 1,3-propane sultone and sodium sulfite, which were designated as PGMA-DVB-S1 and PGMA-DVB-S2, respectively (Fig. 1). The detailed experimental steps can be found in the ESI.†
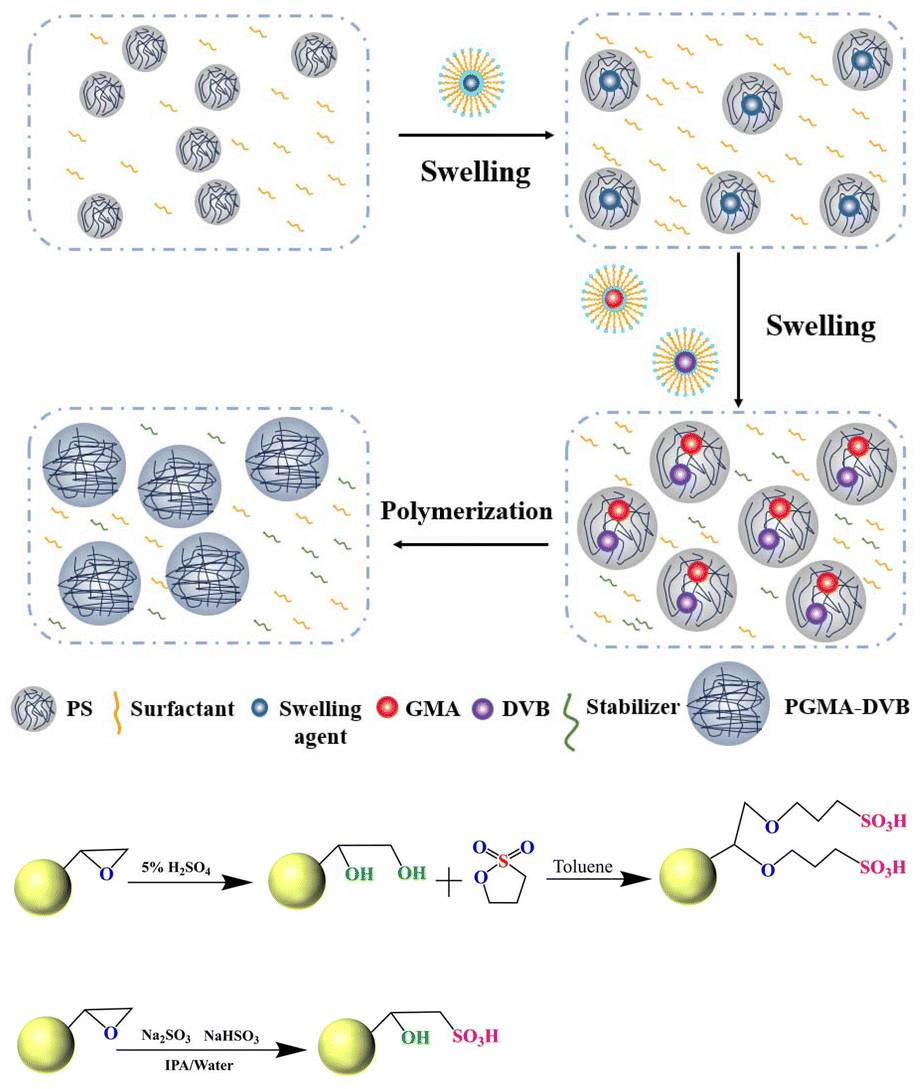 | ||
| Fig. 1 Synthesis of PGMA-DVB microspheres via two-step seed swelling polymerization and the synthesis of PGMA-DVB-S1 and PGMA-DVB-S2. | ||
The morphology of PS microspheres was characterized using scanning electron microscopy (SEM) (Fig. S1, ESI†). The polystyrene seed microspheres synthesized via dispersion polymerization exhibited smooth surfaces and excellent monodispersity, with an average diameter of approximately 1.2 μm. SEM was also used to characterize the morphology of PGMA-DVB, PGMA-DVB-S1, and PGMA-DVB-S2 microspheres (Fig. 2a–c). The PGMA-DVB microspheres synthesized via two-step seed swelling polymerization exhibited uniform particle size and a well-defined porous structure while maintaining good monodispersity, with an average diameter of approximately 5 μm. The introduction of sulfonic acid groups did not significantly alter the spherical morphology of the microspheres. Additionally, energy-dispersive X-ray spectroscopy (EDX) analysis (Fig. S2, ESI†) confirmed the uniform distribution of sulfur throughout the polymer microspheres, indicating the successful incorporation of sulfonic acid groups into the PGMA-DVB microspheres.
 | ||
| Fig. 2 Characterization of PGMA-DVB, PGMA-DVB-S1, and PGMA-DVB-S2: (a–c) SEM, (d) FTIR spectra, (e) N2 adsorption–desorption isotherms, and (f and g) XPS survey spectra. | ||
The PGMA-DVB microspheres before and after sulfonation were characterized using Fourier transform infrared spectroscopy (FT-IR), X-ray photoelectron spectroscopy (XPS), N2 adsorption–desorption isotherms, and water contact angle measurements. The infrared absorption peaks of sulfonic acid groups are primarily attributed to the vibrations of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds (Fig. 2d). These vibrations split into two peaks, corresponding to the symmetric and asymmetric stretching vibrations of SO bonds. Specifically, 1340.3 cm−1 represents the characteristic peak of the SO asymmetric stretching vibration,35 while 1170.1 cm−1 and 1043.4 cm−1 correspond to the characteristic peaks of SO symmetric stretching vibrations.36 In the low-frequency region, 669.1 cm−1 is assigned to the C–S stretching vibration peak. The presence of the C–S stretching vibration peak confirms the successful grafting of sulfonic acid groups onto the polymer microspheres.
O bonds (Fig. 2d). These vibrations split into two peaks, corresponding to the symmetric and asymmetric stretching vibrations of SO bonds. Specifically, 1340.3 cm−1 represents the characteristic peak of the SO asymmetric stretching vibration,35 while 1170.1 cm−1 and 1043.4 cm−1 correspond to the characteristic peaks of SO symmetric stretching vibrations.36 In the low-frequency region, 669.1 cm−1 is assigned to the C–S stretching vibration peak. The presence of the C–S stretching vibration peak confirms the successful grafting of sulfonic acid groups onto the polymer microspheres.
The formation of sulfonic acid groups on the polymer surface was further confirmed by S 2p XPS spectral analysis (Fig. 2f and g). Due to spin–orbit splitting, the S 2p peak appears as a doublet. The deconvolution of the XPS peak for sulfonic acid groups reveals two spin–orbit components, with binding energies of 168.7 eV and 169.6 eV, corresponding to S 2p3/2 and S 2p1/2, respectively.37 The specific surface areas of PGMA-DVB, PGMA-DVB-S1, and PGMA-S2 were determined using the Brunauer–Emmett–Teller (BET) model by analyzing the adsorption branch within the P/P0 range of 0.05 to 0.35 (Fig. 2e). The calculated values were 353.06, 379.86, and 365.04 m2 g−1, respectively. These results indicate that the incorporation of sulfonic acid groups increased the specific surface area through chemical bonding. Fig. S4a–c† present the average pore diameters before and after sulfonation, calculated using the Barrett–Joyner–Halenda (BJH) model, which were 3.51, 4.85, and 3.76 nm, respectively. The increase in pore size after sulfonation is likely due to the introduction of sulfonic acid groups, which may have expanded the original void structure.
The static water contact angles of the polymer microspheres before and after sulfonation were measured. As shown in Fig. S5a,† the unmodified PGMA-DVB microspheres exhibited hydrophobicity, with a water contact angle of 123°. The static contact angles of PGMA-DVB-S1 and PGMA-DVB-S2 with water are shown in Fig. S5b and c†, respectively. After sulfonation, the contact angles of the polymer microspheres decreased, which can be attributed to the high polarity of sulfonic acid groups, enhancing the adsorption capacity for water molecules and thereby reducing the water contact angle.
The average sulfur loading of PGMA-DVB-S1 and PGMA-DVB-S2 was calculated to be 1.83 and 1.35 μmol m−2, respectively, based on the following formula:38
| α(μmol m−2) = S% × 106/(MS × NS × SBET) |
α-Hydroxyisobutyric acid (α-HIBA) is a weak organic acid with a pKa value of 3.72 (at 25 °C, ionic strength μ = 0.1). The molecule contains both hydroxyl (–OH) and carboxyl (–COOH) groups, which exhibit different degrees of dissociation at varying pH levels. When pH < pKa, α-HIBA predominantly exists in its protonated form ((CH3)2C(OH)COOH). In contrast, when pH > pKa, the carboxyl group undergoes deprotonation, forming the anionic species ((CH3)2C(OH)COO−), which can coordinate with REE3+ ions to form complexes. Under isocratic elution conditions with 400 mM α-HIBA, the effect of mobile phase pH (3.0–5.0) on the retention of REE3+ was systematically investigated using PGMA-DVB-S1 and PGMA-DVB-S2 columns. The effect of mobile phase pH on the retention of REE3+ is shown in Fig. 3a and b. The retention time of REEs is generally correlated with their ionic radii and complexation affinity with the eluting ligand, rather than with their atomic radii. In cation-exchange systems using α-HIBA as the complexing agent, REEs with smaller ionic radii typically exhibit stronger complexation and are retained longer on the column. As the pH of the mobile phase increases, the concentration of deprotonated IBA− ions also increases, leading to a gradual decrease in the retention time of REE3+. This phenomenon occurs because lanthanide ions form complexes with the ionized carboxyl groups (IBA−) present in the mobile phase. At higher pH values, the number of ionized carboxyl groups increases, resulting in stronger elution. α-HIBA forms stable complexes with REEs, and its concentration directly affects the abundance of free HIBA− ions in the mobile phase. Therefore, under isocratic elution conditions, the effect of the α-HIBA concentration (0.3–0.8 M) on the retention behavior of REE3+ was investigated. The pH of the mobile phase was maintained at 3.26 throughout the experiment. The influence of the mobile phase concentration on the retention factors of REEs is shown in Fig. 3c and d. An increase in the α-HIBA concentration leads to a decrease in REE3+ retention time without altering the elution order, which is expected. This is because a higher α-HIBA concentration (at a given pH) increases the amount of IBA− and enhances the elution efficiency of the mobile phase. As the α-HIBA concentration increases, the complexation ability is strengthened, making rare earth elements more likely to exist as complexes in the mobile phase, thereby reducing their adsorption onto the stationary phase, which ultimately results in a shorter retention time.
 | ||
| Fig. 3 Effect of mobile phase pH (a and b) and the concentration of α-HIBA (c and d) on the retention behaviour of REEs. | ||
By appropriately adjusting the pH and concentration of the α-HIBA mobile phase and designing a gradient elution program, continuous separation of REEs can be achieved through a strong cation-exchange mechanism. The effect of different sulfonation reagents on the retention capacity and separation efficiency of the resulting strong cation-exchange stationary phases was investigated.
For the PGMA-DVB strong cation-exchange stationary phases modified with two different sulfonation reagents, the elution order of REEs was consistent with the conclusions drawn in previous sections. As shown in Fig. 4a, PGMA-DVB-S1 exhibited distinguishable peaks for each REE, except for Dy and Y. However, significant peak tailing was observed at both the front and back ends of the peaks, as reflected by the asymmetry factor values listed in Table S3.† This peak tailing phenomenon is hypothesized to result from the hydrolysis of epoxy groups on PGMA-DVB, leading to the formation of two adjacent hydroxyl groups. These hydroxyl groups subsequently react with 1,3-propane sultone, generating closely spaced sulfonic acid groups, which increase local charge density. During the elution process, the strong electrostatic attraction between REEs and the continuous sulfonic acid groups may contribute to peak tailing. N2 adsorption–desorption characterization further revealed that the pore volume of 1,3-propane sultone-modified polymer microspheres was smaller than that of PGMA-DVB and PGMA-DVB-S2. The reduction in pore volume may be attributed to partial pore blockage caused by the introduction of 1,3-propane sultone, which could lead to reduced column efficiency and broader peaks. Additionally, nonspecific interactions between the propane segment of 1,3-propane sultone and REEs might also contribute to peak tailing.
 | ||
| Fig. 4 Chromatograms of the separation of 16 rare earth elements using PGMA-DVB-S1 (a) and PGMA-DVB-S2 (b) and the chromatogram of the separation of Fe, U, and 16 rare earth elements using PGMA-DVB-S2 (c). | ||
In contrast, PGMA-DVB-S2 as another strong cation-exchange stationary phase achieved baseline separation for all REEs except Dy and Y (Fig. 4b). The retention factors, separation factors, resolution, asymmetry factors, and theoretical plate numbers are summarized in Table S3.† The chromatographic separation results indicate that the separation performance of sulfonated strong cation-exchange stationary phases depends not only on the bonded phase density, but also on proximal substituents such as alkyl groups attached to the sulfonic acid moiety.
The effect of injection volume on the separation of rare earth elements (REEs) using PGMA-DVB-S2 was investigated (Fig. S6a and b†). The experimental results showed that the chromatographic peak area of REEs increased linearly with the injection volume, while the retention time remained nearly unchanged. However, when the injection volume exceeded 20 μL, column efficiency began to decline, which may be attributed to the analyte load surpassing the column capacity. Therefore, to achieve optimal separation performance, the injection volume should not exceed the column capacity. The effect of column temperature on the retention and separation of REE3+ in HPLC was also examined (Fig. S7a and b†). With all other parameters held constant, the column temperature was varied from 25 to 50 °C to evaluate its impact on REE3+ retention and separation. As the temperature increased, the retention time of REEs exhibited a slight increase. The van't Hoff plot showed a distinct curvature, indicating that the enthalpy change (ΔH) of the separation process is not constant but rather a function of temperature. This suggests that the separation of REEs is an endothermic process. Notably, the retention factor (K) of heavy rare earth elements (HREEs) and Sc exhibited significant variation with increasing temperature, whereas light rare earth elements (LREEs) showed a much weaker dependence on temperature. Column reproducibility refers to the consistency of chromatographic system responses when multiple consecutive injections are performed under identical conditions. As shown in Fig. S8a,† the relative standard deviation (RSD) of retention time was calculated by comparing the retention time variations of rare earth elements (REEs) after 30 consecutive injections. The RSD values for REEs were 0.12%–3.56%. These results indicate that PGMA-DVB-S2, as a liquid chromatography stationary phase, exhibits excellent stability in the separation of rare earth elements. Additionally, chromatograms of the first, fiftieth, and hundredth injections were compared, as shown in Fig. S6b.† Even after 100 consecutive injections, the retention time and peak shape remained highly stable, further confirming the robustness and durability of the PGMA-DVB-S2 stationary phase.
Subsequently, the feasibility of using PGMA-DVB-S2 as a liquid chromatography stationary phase for the separation of rare earth elements (REEs) from Fe and U was investigated. The chromatographic analysis, as shown in Fig. 4c, demonstrated that the incorporation of Fe and U did not alter the elution order of REEs. The retention characteristics of Fe and U were significantly different from those of the REEs. Specifically, Fe eluted earlier, while U exhibited a pronounced delay, which can be attributed to differences in the binding constants between the stationary phase and the mobile phase. The retention factors, separation factors, resolution, asymmetry factors, and theoretical plate numbers for REEs, Fe, and U are summarized in Table S6.† The results indicate that REEs were completely baseline-separated from Fe and U, with sharp and symmetrical peaks and high theoretical plate numbers, confirming the effectiveness and practicality of this stationary phase in separating REEs in the presence of Fe and U as impurities.
In conclusion, this study developed monodisperse porous polymeric microspheres as an efficient stationary phase for REE separation. Two sulfonation strategies were used to functionalize the microspheres with sulfonic acid groups. Compared to PGMA-DVB-S1 sulfonated using 1,3-propane sultone, PGMA-DVB-S2 sulfonated using sodium sulfite exhibited superior selectivity and separation efficiency. Key chromatographic parameters—including mobile phase pH, α-HIBA concentration, column temperature, and injection volume—were investigated to understand REE retention. The van't Hoff analysis confirmed an endothermic separation process, with heavy REEs showing stronger temperature dependence than light REEs. PGMA-DVB-S2 demonstrated excellent reproducibility, maintaining high peak resolution and stable retention times over 100 injections. It also enabled baseline separation of REEs from Fe and U impurities, confirming its high selectivity.
Overall, this study proposes a novel strategy for REE separation using polymeric chromatographic materials, bridging the gap between polymer synthesis and chromatographic applications. The insights gained pave the way for advancements in sustainable REE extraction, magnet recycling, and nuclear waste processing, reinforcing the significance of polymeric microspheres as next-generation chromatographic materials.
Data availability
All the relevant data of this study are available within the manuscript and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDB1220000) and the National Natural Science Foundation of China (No. 22474141).References
- J. M. Klinger, Extr. Ind. Soc., 2015, 2, 572–580 Search PubMed.
- N. Haque, A. Hughes, S. Lim and C. Vernon, Resources, 2014, 3, 614–635 CrossRef.
- M. A. Müller, D. Schweizer and V. Seiler, J. Bus. Ethics., 2016, 138, 627–648 CrossRef.
- C. Li, P. Wang, M. He, X. Yuan, Z. Fang and Z. Li, Coord. Chem. Rev., 2023, 489, 215204 CrossRef.
- Y. Li, X. Yuan, P. Wang, L. Tang, M. He, P. Li, J. Li and Z. Li, J. Energy Chem., 2023, 83, 574–594 CrossRef CAS.
- S. Meng, G. Li, P. Wang, M. He, X. Sun and Z. Li, Mater. Chem. Front., 2023, 7, 806–827 RSC.
- J. M. D. Coey, Engineering, 2020, 6, 119–131 CrossRef CAS.
- K. Bodišová, R. Klement, D. Galusek, V. Pouchlý, D. Drdlík and K. Maca, J. Eur. Ceram. Soc., 2016, 36, 2975–2980 CrossRef.
- J. Cao, M. Zhang, X. Ma, S. Zhao, T. Jiang and W. Wen, Ceram. Int., 2024, 50, 42299–42308 CrossRef CAS.
- J. Hostaša, V. Nečina, T. Uhlířová and V. Biasini, J. Eur. Ceram. Soc., 2019, 39, 53–58 CrossRef.
- H. Wang, Z. Wei, Y. Zhao, S. Wang, L. Cao, F. Wang, K. Liu and Y. Sun, RSC Adv., 2023, 13, 27512–27519 RSC.
- L.-P. Yu, X. Zhang, D.-X. Wei, Q. Wu, X.-R. Jiang and G.-Q. Chen, Biomacromolecules, 2019, 20, 3233–3241 CrossRef CAS PubMed.
- M. Guo, S. Liu, W. Sun, M. Ren, F. Wang and M. Zhong, Front. Phys., 2023, 18, 21303 CrossRef.
- C. R. Borra, T. J. Vlugt, Y. Yang, J. Spooren, P. Nielsen, M. Amirthalingam and S. E. Offerman, Resour., Conserv. Recycl., 2021, 174, 105766 CrossRef CAS.
- B. V. Kheswa, Open Chem., 2023, 21, 20220345 CrossRef CAS.
- K. Miarabbas Kiani, H. C. Frankis, C. M. Naraine, D. B. Bonneville, A. P. Knights and J. D. B. Bradley, Laser Photonics Rev., 2022, 16, 2100348 CrossRef CAS.
- A. Sivakumaran, A. J. Samson, A. A. Bristi, V. Surendran, S. Butler, S. Reid and V. Thangadurai, J. Mater. Chem. A, 2023, 11, 15792–15801 RSC.
- S. S. R. Bonthu, A. K. M. Arafat and S. Choi, IEEE Trans. Ind. Electron., 2017, 64, 9729–9738 Search PubMed.
- M. Alemrajabi, Å. C. Rasmuson, K. Korkmaz and K. Forsberg, Hydrometallurgy, 2017, 169, 253–262 CrossRef CAS.
- S. Maes, W.-Q. Zhuang, K. Rabaey, L. Alvarez-Cohen and T. Hennebel, Environ. Sci. Technol., 2017, 51, 1654–1661 CrossRef CAS PubMed.
- X. Yin, Y. Wang, X. Bai, Y. Wang, L. Chen, C. Xiao, J. Diwu, S. Du, Z. Chai and T. E. Albrecht-Schmitt, Nat. Commun., 2017, 8, 14438 CrossRef CAS PubMed.
- Y. Kamimoto, T. Itoh, K. Kuroda and R. Ichino, J. Mater. Cycles Waste Manage., 2017, 19, 1017–1021 CrossRef CAS.
- T.-Q. Yin, Y. Xue, Y.-D. Yan, Z.-C. Ma, F.-Q. Ma, M.-L. Zhang, G.-L. Wang and M. Qiu, Int. J. Miner., Metall. Mater., 2021, 28, 899–914 CrossRef CAS.
- X. Sheng, J. Chen, J. Shao, X. Zhang, B. Wang, C.-F. Ding and Y. Yan, Analyst, 2025, 150, 395–404 Search PubMed.
- G. Sun, Y. Luo, Z. Yan, H. Qiu and W. Tang, Chin. Chem. Lett., 2024, 109787 CrossRef.
- Y. Yang, J. Chen, X. Liang, B. Liu, K. Quan, X. Liu and H. Qiu, J. Chromatogr. A, 2024, 1722, 464889 CrossRef PubMed.
- H. Li, X. Wang, C. Shi, L. Zhao, Z. Li and H. Qiu, Chin. Chem. Lett., 2023, 34, 107606 CrossRef.
- C. Liu, K. Quan, H. Li, X. Shi, J. Chen and H. Qiu, Chem. Commun., 2022, 58, 13111–13114 RSC.
- Y. Yang, H. Zhang, J. Chen, Z. Li, L. Zhao and H. Qiu, Analyst, 2020, 145, 1056–1061 RSC.
- C. Shi, H. Li, X. Shi, L. Zhao and H. Qiu, Chin. Chem. Lett., 2022, 33, 3613–3622 CrossRef.
- M. Xie, K. Quan, H. Li, B. Liu, J. Chen, Y. Yu, J. Wang and H. Qiu, Chem. Commun., 2023, 59, 314–317 RSC.
- D. O. Campbell and S. R. Buxton, Ind. Eng. Chem. Process Des. Dev., 1970, 9, 89–94 Search PubMed.
- R. G. Fernández and J. I. G. Alonso, J. Chromatogr. A, 2008, 1180, 59–65 CrossRef PubMed.
- Y. Inoue, H. Kumagai, Y. Shimomura, T. Yokoyama and T. M. Suzuki, Anal. Chem., 1996, 68, 1517–1520 Search PubMed.
- N. Iman, A. R. Razak and N. Nurhaeni, Kovalen, 2016, 2, 144455 Search PubMed.
- X.-P. Li, Z. Deng, Y. Li, H.-B. Zhang, S. Zhao, Y. Zhang, X.-Y. Wu and Z.-Z. Yu, Carbon, 2019, 147, 172–181 CrossRef.
- M. Matsuda, K. Funabashi, H. Yusa and M. Kikuchi, J. Nucl. Sci. Technol., 1987, 24, 124–128 CrossRef.
- Y. Yang, J. Wang, R. Liu, K. Quan, J. Chen, X. Liu and H. Qiu, Langmuir, 2022, 38, 14400–14408 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and additional data for the synthesised compounds. See DOI: https://doi.org/10.1039/d5an00388a |
| This journal is © The Royal Society of Chemistry 2025 |