
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative analysis of carbohydrate residues in dextran 40 from various sources: a comparative study using high-performance liquid chromatography coupled with a charged aerosol detector
Shenggu
Xie†
a,
Zhuyu
Jin†
 ab,
Yan
Huang
c and
Qiaoqiao
Huang
*a
ab,
Yan
Huang
c and
Qiaoqiao
Huang
*a
aZhejiang Institute for Food and Drug Control, National Medical Products Administration Key Laboratory for Core Technology of Generic Drug Evaluation, Zhejiang Key Laboratory of Biopharmaceutical Contact Materials, Hangzhou 310052, China
bSchool of Pharmacy, China Pharmaceutical University, Nanjing 211198, China
cZhejiang University of Technology, Hangzhou, 310014, China
First published on 21st January 2025
Abstract
Dextran 40, a typical high molecular weight carbohydrate drug refined through fermentation, is widely used in the clinical field in an injectable form. The final product obtained through fermentation may contain by-products such as fructose and residual sucrose, which carry a risk of adverse reactions. Current quality standards do not effectively control for the possible presence of carbohydrate residues, and methods for detecting such residues are lacking. This gap in quality control exists in the vast majority of existing carbohydrate drugs. This study established and compared liquid chromatography methods equipped with three different detectors (RID, MS, and CAD), selecting a convenient, rapid, and efficient HILIC-CAD method. This method combines the high sensitivity of the HILIC-MS method with the high throughput of the HILIC-RID method, using porous silica as the stationary phase and a high-precision charged aerosol detector in tandem, achieving rapid separation and quantification of fructose and sucrose. Additionally, pretreatment optimization was conducted to eliminate the impact of dextran 40 on the detection of fructose and sucrose. The method was validated, showing good repeatability, recovery, robustness, and linearity, capable of quantifying carbohydrate residues at approximately 3.3 ppm. This study compared the residual levels of fructose and sucrose in dextran 40 obtained from different purification processes, analyzing key purification operations that influence the extent of carbohydrate residues. These findings provide a reference for optimizing the production process of dextran 40, ensuring the quality of the drug and public drug safety. Furthermore, the approach used in this study for detecting carbohydrate residues is applicable to the quality control of other carbohydrate drugs produced via fermentation.
Introduction
Carbohydrate drugs such as hyaluronic acid,1 chondroitin sulfate,2 and dextran are often prepared using fermentation methods. Dextran 40 is a high molecular weight glucose polymer derived from sucrose through fermentation by Leuconostoc mesenteroides strain L.M-1226, followed by further processing and purification.3,4 The crude dextran powder obtained from sucrose fermentation is processed through acid hydrolysis, followed by separation, purification, and refinement to yield the active pharmaceutical ingredient (API). Dextran 40, a low molecular weight dextran, is extensively utilized in clinical medicine, primarily in the form of dextran 40 glucose injection, mainly for increasing plasma volume and anti-shock treatment.5,6 Currently, there are two mainstream purification processes: alcohol precipitation method and the membrane filter method.7,8In addition to dextran 40, the products obtained from fermentation may also contain the starting substrate sucrose, by-products such as fructose, and other residual carbohydrates from fermentation (Fig. 1 and 2).9 It has been reported that the presence of carbohydrate residues may cause abdominal pain and diarrhea in some patients,10 and may even lead to the Maillard reaction, exacerbating allergic reactions.11 Nonetheless, current quality standards for dextran 40, such as USP-NF 2024,12 EP11.0,13 and ChP 2020,14 do not have a control for these residual substances. Although many drugs, including dextran, are produced via fermentation,15 the potential residues in these drugs have not received adequate attention. Therefore, it is imperative to establish suitable analytical methods for assessing the main potential carbohydrate residues in dextran 40, to comprehensively evaluate the quality and associated risks of this class of drugs.
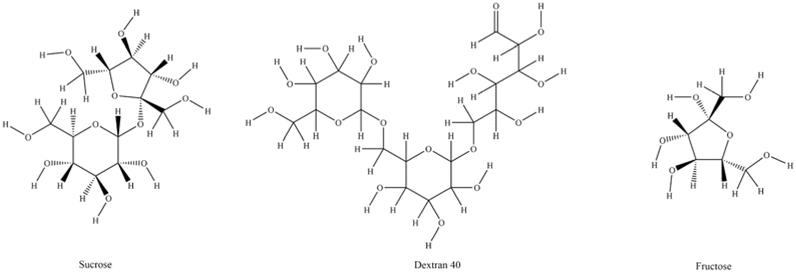 | ||
| Fig. 1 Chemical structure of carbohydrates (https://www.pubchem.ncbi.nlm.nih.gov/). | ||
 | ||
| Fig. 2 Reaction scheme of sucrose-to-dextran 40. | ||
Sucrose and fructose, both of which lack conjugated structures and are heavily hydroxylated, do not possess ultraviolet absorption and exhibit strong polarity. Thus, they are difficult to separate using conventional reversed-phase chromatography columns. Effective separation and detection of these carbohydrates in liquid chromatography systems present a significant challenge. Hydrophilic Interaction Liquid Chromatography (HILIC) utilizes bare silica or silica derivatized with various polar functional groups, such as amino or amide groups, in the packed columns, which can excellently retain polar compounds16–18. Therefore, it is possible to achieve the separation of these two carbohydrates by HILIC. Studies have employed techniques such as high-performance liquid chromato-graphy-mass spectrometer (HPLC-MS),19 HPLC-refractive index detector (RID), and HPLC-evaporative light-scattering detector (ELSD)20 to separate highly polar carbohydrates lacking chromophoric groups. However, all these methods have their limitations. For instance, MS requires expensive equipment; RID, while widely used for carbohydrate detection, demands a long equilibration time and has low sensitivity;21 ELSD suffers from poor repeatability, a narrow linear range, and suboptimal sensitivity.22 Consequently, it is necessary to find a detection method that is both reliable in sensitivity and precise in measurement for the determination of these carbohydrate residues.
As a universal detector, the charged aerosol detector (CAD) demonstrates significant potential for detecting compounds that lack chromophoric groups. It is capable of detecting all non-volatile analytes, thus allowing for the detection of these two carbohydrate residues with great sensitivity and a broader linear range.23–25 Therefore, this study, for the first time, employs HILIC-CAD to detect carbohydrate residues in dextran 40, while also establishing and comparing the widely used HPLC-RID method and the HPLC-MS method, which have highly recognized sensitivity of impurities detection in the carbohydrate detection field. The goal is to assess the differences in detection capabilities among the three analytical methods, aiming to identify a cost-effective and highly sensitive detection method that ensures both the accuracy of results and exceptional sensitivity.
This study optimized the pretreatment process and chromatographic conditions and compared the determination results of fructose and sucrose in dextran 40 using HILIC methods equipped with three different detectors. The residual results were then analyzed in conjunction with the various purification processes of dextran 40, providing a reference for the process development, quality evaluation, and standard setting or revision of this drug. To our knowledge, this study is the first to investigate carbohydrate residues in drugs produced through fermentation methods, establishing the HILIC-CAD method to analyze these specific fermentation residues.
Materials and methods
Chemicals and reagents
Sucrose (batch number: 111507-202105, 99.8% purity) and fructose (batch number: 100231-202008, 99.9% purity) were obtained from the National Institute for Food and Drug Control (Beijing, China).Acetonitrile (CAS 75-05-8, HPLC grade) was purchased from Meck (Darmstadt, Germany). Ammonium acetate (CAS 631-61-8, LC-MS grade) and ammonia (CAS 1336-21-6, HPLC grade, 25% purity) were purchased from Sigma-Aldrich (St. Louis, MO, USA). De-ionized water was produced by a Millipore Milli-Q Gradient purification system (Burlington, MA, USA).
Dextran 40 samples were obtained from four companies: Company A, Shanghai Company (batch number: HM0503, HM0506 and HM0617), Company B, Shandong Company (batch number: 103170701. 103170708 and 103170923), Company C, Sichuan Company (batch number: A001180528, A001180529, A001180530) and Company D, Weifang Company (batch number: ST0514, ST0918 and ST0927).
HILIC-CAD conditions
The analysis was performed on the Alliance liquid chromatography system from Waters (Milford, MA, USA). The HPLC-CAD system includes a pump, auto-sampler, column oven, and Corona Veo CAD detector. Data processing was performed on Waters Empower 3.The separation of carbohydrate residues in dextran 40 was performed on an Agilent InfinityLab Poroshell 120 HILIC column (4.6 mm × 150 mm, 2.7 μm) (Agilent, USA). The column temperature was set at 40 °C. The mobile phase was acetonitrile–water (90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) solution. The flow rate was 0.5 mL min−1, and the injection volume was 5 μL.
:10) solution. The flow rate was 0.5 mL min−1, and the injection volume was 5 μL.
The following parameters of CAD were set: nebulization temperature of 50 °C, collection rate of 10 Hz, filter value of 5 and power function value of 1.0.
HILIC-RID conditions
The analysis was performed on the 1260 Infinity liquid chromatography system from Agilent (Santa Clara, CA, USA). Coupling the HPLC system with a differential refractive index detector. Data processing was performed on Agilent OpenLab CDS.Separation of carbohydrate residues in dextran 40 was performed on an Agilent ZORBAX-NH2 column (4.6 mm × 250 mm, 5 μm) (Agilent, USA). The column temperature was set at 40 °C same as the temperature of RID. The mobile phase was acetonitrile–water (75:25) solution. The flow rate was 1.0 mL min−1, and the injection volume was 50 μL.
HILIC-MS condition
1290 Infinity II Ultra-high-performance liquid chromatography coupled with 6470 Triple quaternary mass spectrometry was used. Using Waters XBridge BEH Amide Column (4.6 mm × 150 mm, 3.5 μm) (Waters, USA) to separate sucrose and fructose. The column temperature was set at 35 °C. The mobile phase A was 10 mM ammonium acetate acetonitrile–water (90:10) (containing 0.2% ammonia). The mobile phase B was 10 mM ammonium acetate acetonitrile–water (30:70) (containing 0.2% ammonia). The separation was achieved using gradient elution: 0–30 min, 8–40% B; 30–31 min, 40–8% B; 31–65 min, 8% B. The flow rate was 0.8 mL min−1, and the injection volume was 20 μL.
The mass spectrometry was operated in a negative ion SIM detection mode, utilizing the ion m/z 179.1 for the quantitative analysis of fructose and the ion m/z 341.1 for the quantitative assessment of sucrose. Each ion channel was assigned a dwell time of 200 ms. The parameters for the mass spectrometry ion source were as follows: gas temperature at 300 °C, gas flow rate at 5 L min−1, nebulizer pressure set to 35 psi, sheath gas temperature at 250 °C, sheath gas flow rate at 11 L min−1, and the capillary voltage was maintained at 3500 V.
Preparation of solutions
:50).
000 rpm for 10 minutes, and the supernatant obtained was the sample solution with a concentration of 100 mg mL−1.
Method validation procedures
According to the ICH guidelines, the HILIC-CAD method for the determination of the carbohydrate residues in dextran 40 was validated for specificity, linearity, limit of detection (LOD) and limit of quantitation (LOQ), accuracy, and precision.26Results and discussion
Optimization of pretreatment methods
The carbohydrate residues in dextran 40 were present in trace amounts. To enhance the sensitivity during sample detection, the optimization was achieved by increasing the sample concentration. However, due to the high viscosity of dextran 40 solution, direct injection could easily lead to column blockage and contamination. The focus of this study was to measure the carbohydrate residues in dextran 40 without the need for quantification of dextran 40, thus, dextran 40 can be removed through purification steps to reduce risks and optimize the experiment. This study employed a counter-solvent method to remove dextran 40, taking advantage of its solubility characteristics: it dissolves easily in hot water but poorly in acetonitrile. A high-concentration dextran 40 sample solution was prepared in hot water, and after complete dissolution, an equal volume of acetonitrile was added to precipitate the dextran 40 completely. The mixture was then centrifuged at 10000 rpm for 15 minutes, and the supernatant was used as the sample solution. This method effectively retained sucrose and fructose in the supernatant, ensuring its clarity and transparency for direct injection analysis. The study also explored the proportion of acetonitrile added (Table 1), maintaining the same amount of dextran 40 dissolved in 5.0 mL of hot water and adding 2.5 mL, 5.0 mL, and 7.5 mL of acetonitrile, separately, followed by the same centrifugation procedure. The results showed that adding 2.5 mL of acetonitrile led to a turbid supernatant after centrifugation, indicating that the high-viscosity dextran 40 was not fully processed and was not suitable for injection analysis. In contrast, adding 7.5 mL of acetonitrile resulted in a clear supernatant, but the final concentration of the sample solution was lower compared to that with the 5 mL acetonitrile treatment, leading to reduced sensitivity. Therefore, it was determined that adding an equal volume of 5 mL acetonitrile for counter-solvent pretreatment was the optimal choice.
| Ratio 1 | Ratio 2 | Ratio 3 | |
|---|---|---|---|
| Volume of water(mL) | 5 | 5 | 5 |
| Volume of acetonitrile(mL) | 2.5 | 5 | 7.5 |
| Situation of supernatant | Turbid | Clear | Clear |
Comparison of detection capabilities of three analytical methods
Quantification of fructose and sucrose in dextran 40 prepared by the fermentation method was conducted using HILIC-RID, HILIC-MS, and HILIC-CAD methods. The detection capabilities of the three analytical methods are shown in Table 2. All three methods demonstrated good linearity in this study, but they exhibited different sensitivities for the detection of fructose and sucrose.| Carbohydrate | HILIC-RID | HILIC-MS | HILIC-CAD | |
|---|---|---|---|---|
| Linear equation | Fructose | Y = 4.774 × 105X + 2728 | Y = 0.8666X + 5.026 | Y = 5.911 × 106X + 4.308 × 106 |
| Sucrose | Y = 4.914 × 105X + 1471 | Y = 0.8900X + 2.220 | Y = 6.110 × 106X + 4.480 × 106 | |
| Correlation coefficient (r) | Fructose | 0.9999 | 0.9996 | 0.9991 |
| Sucrose | 0.9999 | 0.9991 | 0.9990 | |
| Range (μg mL−1) | Fructose | 100–1500 | 1.25–100 | 0.33–50 |
| Sucrose | 100–1500 | 0.12–2.54 | 0.33–50 | |
| Injection volume (μL) | Fructose | 50 | 20 | 5 |
| Sucrose | 50 | 20 | 5 | |
| Concentration of sample (mg mL−1) | Dextran 40 | 100 | 6 | 100 |
| LOQ (S/N = 10) (mean ± SD, n = 3) | Fructose | 9.91 ± 0.40 μg mL−1 (99.12 ± 4.02 ppm) | 1.27 ± 0.02 μg mL−1 (211.49 ± 2.89 ppm) | 0.33 ± 0.01 μg mL−1 (3.27 ± 0.08 ppm) |
| Sucrose | 10.06 ± 0.26 μg mL−1 (100.57 ± 2.63 ppm) | 0.36 ± 0.01 μg mL−1 (59.52 ± 1.50 ppm) | 0.34 ± 0.01 μg mL−1 (3.39 ± 0.08 ppm) | |
| LOD (S/N = 3) (n = 3) | Fructose | 4.00 μg mL−1 (40.00 ppm) | 0.42 μg mL−1 (70.42 ppm) | 0.10 μg mL−1 (1.01 ppm) |
| Sucrose | 4.25 μg mL−1 (42.46 ppm) | 0.12 μg mL−1 (20.13 ppm) | 0.11 μg mL−1 (1.08 ppm) |
Mass spectrometry, considered the “gold standard” for trace impurity detection, exhibits ultra-high sensitivity due to its detection principle. The HILIC-MS method established in this study, as anticipated, showed good sensitivity (Table 2). For sucrose detection, the LOD in the sample reached 20 ppm, which was superior to the HILIC-RID method (with an LOD of 40 ppm). However, when detecting fructose, an opposite situation was observed; LOD for fructose with the HILIC-MS method was 70 ppm, while the HILIC-RID method was 40 ppm, indicating that the sensitivity of the HILIC-RID method was superior when detecting the fructose residues in the sample.
When analyzing based on actual sample concentrations, the minimum detectable concentration of fructose in the solution for HILIC-MS was 0.42 μg mL−1, while for HILIC-RID, it was 4 μg mL−1. Therefore, from the perspective of the solution concentration, the MS method's detection capability remains undoubtedly sensitive. The reason the HILIC-MS method performed worse than the HILIC-RID method in detecting fructose in the sample is that the HILIC-RID method allowed for a larger injection volume and the concentration of the test sample could reach 100 mg mL−1, much higher than the 6 mg mL−1 achievable with the MS method. Thus, although the minimum detectable concentration for the RID method in solution is higher than that of the MS method, the LOD for fructose in samples using the RID method can be lower with a higher injection volume.
The limitation of introducing high-concentration samples into the MS system in this study was identified during the early method development phase. Even with the optimized counter-solvent method for sample preparation, the resulting supernatant exhibited a certain viscosity, which was directly proportional to the concentration. Increasing the injection volume or the concentration of the sample would lead to stronger signals in the mass spectrometer. However, after multiple injections, significant baseline fluctuations occurred, and the magnitude of the baseline fluctuation increased with the sample concentration. This indicated that high concentrations or large injection volumes of dextran 40 cannot be used to enhance the detection sensitivity of the HILIC-MS method.
The HILIC-CAD method combines the advantages of both HILIC-RID and HILIC-MS methods. The linear correlation coefficients obtained for sucrose and fructose through linear fitting were 0.9991 and 0.9990, respectively, demonstrating a good linear relationship within the concentration range of 0.33∼50 μg mL−1. This method accommodates high-concentration test samples, and under a 5 μL injection condition, the LOQ and LOD for fructose and sucrose reached 3.3 ppm and 1 ppm, respectively, which are significantly better than those of HILIC-RID and HILIC-MS methods. The advantages of this method are evident, thus further optimization and method validation are warranted.
Optimization of HILIC-CAD
:25) for equilibration, we found that the CAD chromatogram had a high baseline noise (Fig. 3A). After switching to another amino column, the baseline still had significant noise (Fig. 3B). We then used the column with amide-bonded silica, which resulted in reduced noise fluctuations, but the high noise levels were not improved (Fig. 3C). The results indicated that these three columns used for carbohydrate separation exhibited poor baseline stability when analyzed with CAD. The CAD detector is based on the nebulization-aerosol principle, where the HPLC eluent is nebulized by colliding with nitrogen in the CAD nebulizer, forming droplets that spray and contain analyte particles. These particles are dried in the drying tube to form solute particles, which collide with charged nitrogen, causing the analyte particles to acquire a positive charge and generate an electrical signal. Based on this, we chose the Poroshell HILIC column, which has porous silica without bonded polar groups. With an acetonitrile–water (75:25) system, the baseline was stable and met the sensitivity requirements (Fig. 3D). Thus, this core–shell column was selected to continue with subsequent separation method development.
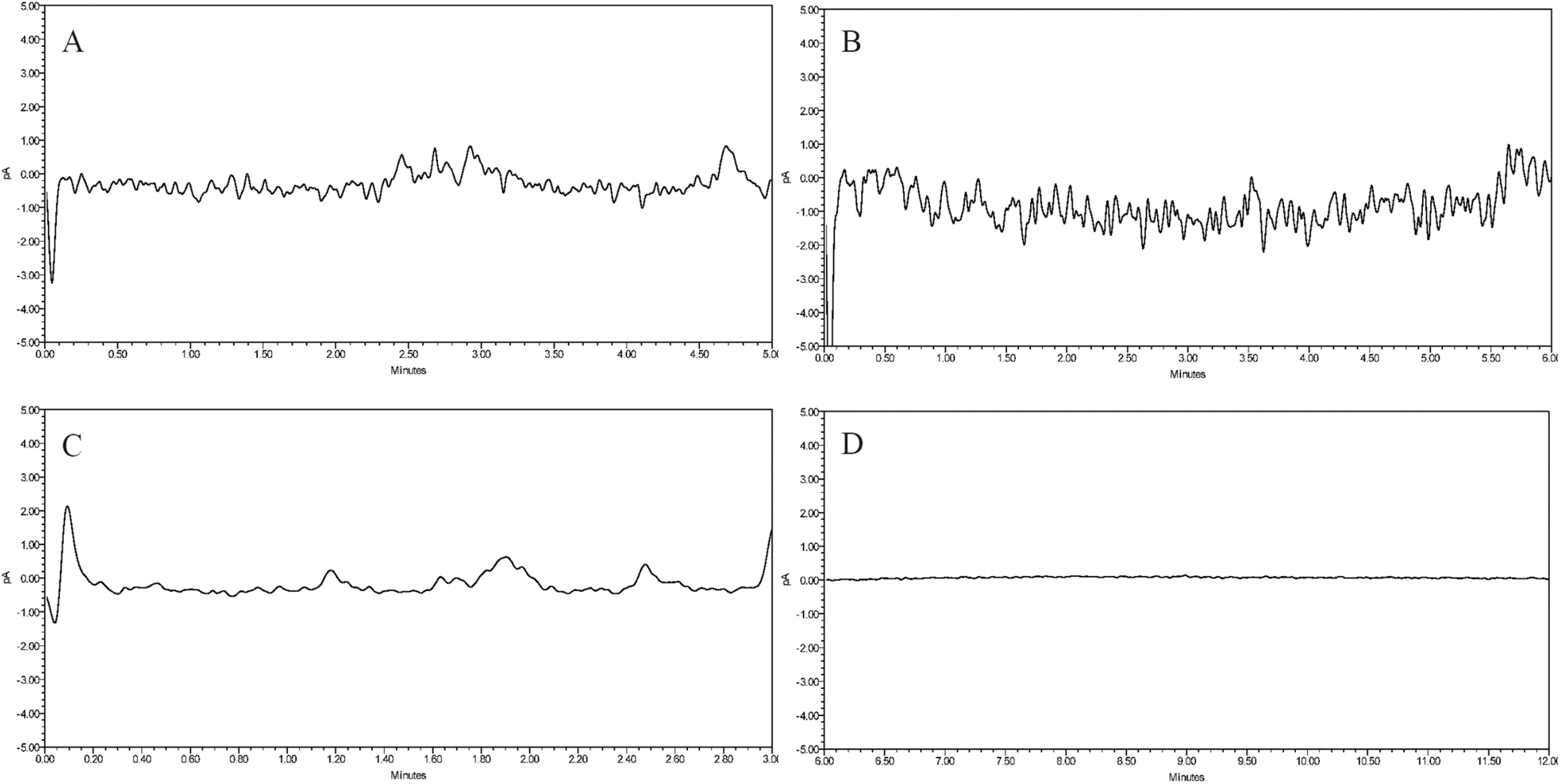 | ||
| Fig. 3 Comparison of the baseline results of different HILIC chromatographic columns. (A) Agilent ZORBAX-NH2 (4.6 mm × 250 mm, 5 μm), (B) Welch Ultimate XB-NH2 (4.6 mm × 250 mm, 5 μm), (C) waters XBridge BEH amide (4.6 mm × 150 mm, 3.5 μm), (D) Agilent InfinityLab Poroshell 120 HILIC (4.6 mm × 150 mm, 2.7 μm). | ||
:25) elution, sucrose and fructose could not be effectively separated. When the ratio was adjusted to 85:15, sucrose and fructose could be separated successfully, but the separation was poor, with a separation factor of only 1.73. Therefore, we continued to reduce the proportion of water, and when the ratio was adjusted to acetonitrile–water (90:10), the separation of sucrose and fructose was better, with a separation factor of 2.11. Further reducing the aqueous phase ratio to acetonitrile–water (95:5), although the separation of sucrose and fructose was further optimized at this point, the time required for a single injection was extended. Therefore, to balance the need for efficient experiments and separation efficiency, we determined to use an acetonitrile–water (90:10) mobile phase for elution.
Validation results of HILIC-CAD
 | ||
| Fig. 4 Purification processes diagram of dextran 40: (A) alcohol precipitation method and (B) membrane filter method (By Figdraw). | ||
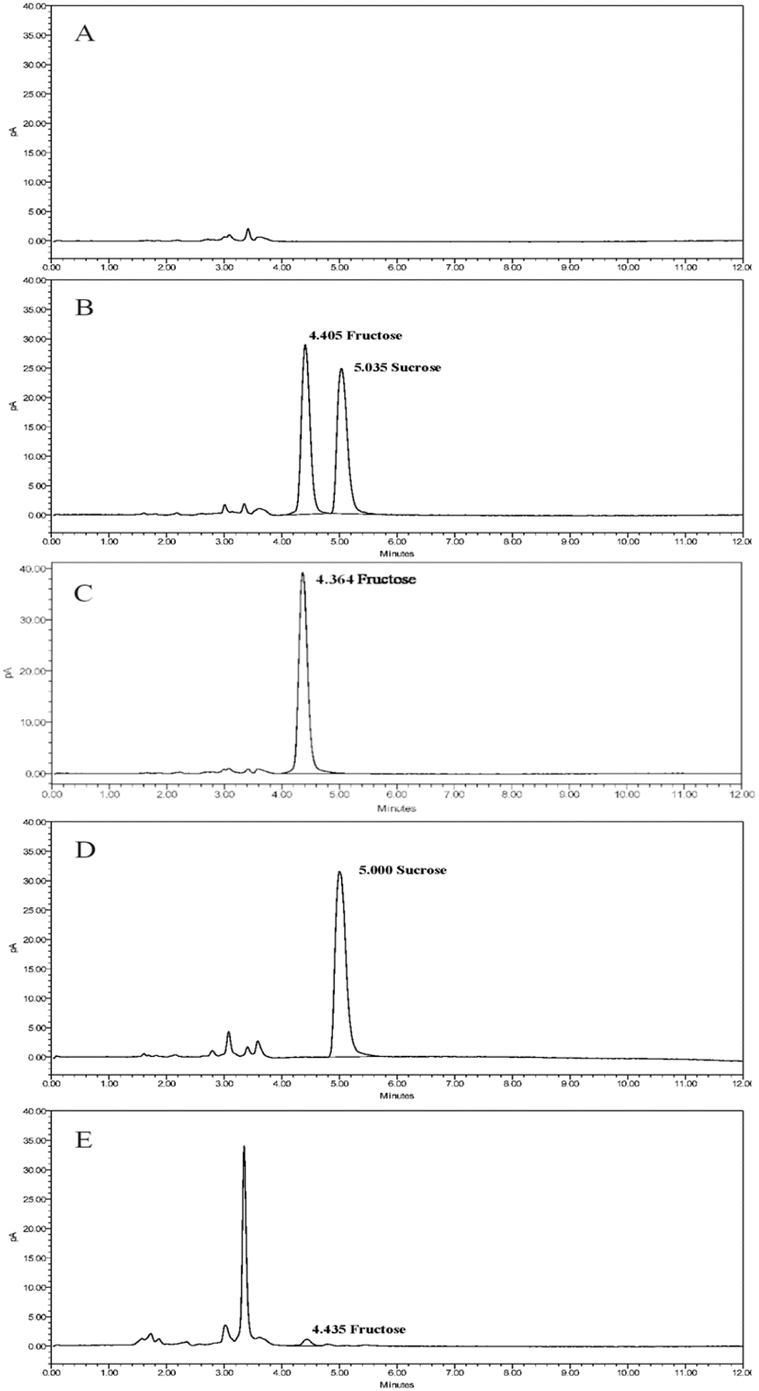 | ||
| Fig. 5 Chromatograms of the main carbohydrate residues in dextran 40 by HPLC-CAD method (A). blank solution, (B). standard solution, (C). fructose solution, (D). sucrose solution, and (E). sample solution of the company A HM0617. | ||
| Compound | Recovery (%) (n = 3 × 3) | Average | RSD (%) | ||
|---|---|---|---|---|---|
| Low concentration | Mid concentration | High concentration | |||
| Fructose | 109.7 | 104.6 | 100.0 | 104.8 | 4.61 |
| Sucrose | 108.6 | 104.6 | 100.0 | 104.4 | 4.13 |
| Parameter | Condition 1 | Condition 2 | Condition 3 | Condition 4 | Condition 5 | Condition 6 | Standard condition |
|---|---|---|---|---|---|---|---|
| Flow rate (mL min−1) | 0.45 | 0.55 | 0.50 | 0.50 | 0.50 | 0.50 | 0.50 |
| Column temperature (°C) | 40 | 40 | 35 | 45 | 40 | 40 | 40 |
| Nebulization temperature (°C) | 50 | 50 | 50 | 50 | 35 | 45 | 50 |
| Residual fructose in sample (%) | 0.301 | 0.284 | 0.291 | 0.289 | 0.290 | 0.289 | 0.296 |
| RSD (%) | 1.89 | ||||||
Comparison of residual carbohydrate levels in dextran 40 from different purification processes
There are two purification processes for the preparation of dextran 40 by fermentation methods: alcohol precipitation method and membrane filter method. The specific process steps are shown in Fig. 4. The dextran 40 collected from companies A and D in this study was purified by the membrane filter method, while the dextran 40 from companies B and C was purified by the other method. The residual results of fructose and sucrose measured in dextran 40 from companies A, B, C, and D are shown in Table 5. Sucrose, as the starting material for dextran 40 was thoroughly purified after sufficient reaction and multiple purification steps, and no sucrose residues were detected in the final products from the four sources of dextran 40. Fructose, an impurity produced in equal amounts during the production of dextran 40, is more likely to be residual in API, and the results confirmed this hypothesis. The average residues of fructose in dextran 40 from companies B and C were 0.297% and 0.025%, respectively, while the average residue of fructose in dextran 40 from company A was only 0.008%. No fructose was detected in the sample from company D. This indicates that the membrane filter method achieves a better purification effect than the alcohol precipitation method, as the latter results in higher fructose residue and poorer process stability.| Manufacturer | Production process | Lot number | Fructose (%) | Sucrose (%) |
|---|---|---|---|---|
| A | Membrane filter method | HM0503 | 0.009 | <LOD |
| HM0506 | 0.007 | <LOD | ||
| HM0617 | 0.008 | <LOD | ||
| B | Alcohol precipitation method | 103170701 | 0.296 | <LOD |
| 103170708 | 0.294 | <LOD | ||
| 103170923 | 0.300 | <LOD | ||
| C | Alcohol precipitation method | A001180528 | 0.025 | <LOD |
| A001180529 | 0.025 | <LOD | ||
| A001180530 | 0.024 | <LOD | ||
| D | Membrane filter method | ST0514 | <LOD | <LOD |
| ST0918 | <LOD | <LOD | ||
| ST0927 | <LOD | <LOD |
Company D improved the purification process by increasing the number of membrane filtration cycles and changing the type of filter membrane. The final product obtained showed no detectable sucrose or fructose residues, demonstrating excellent purification efficiency. From our previous research, it is evident that variations in purification processes can significantly impact the molecular weight and molecular weight distribution of dextran 40.5 Coupled with the findings of this study, it suggests a correlation between product quality and purification methodology. In products obtained from different purification processes, the starting material sucrose is nearly absent in the final product, while there are notable differences in the fructose residue levels. Therefore, the extent of the fructose residue can be utilized as a standard for evaluating the purification efficiency of different processes. Manufacturers should optimize production processes based on the residue results to ensure the safety and quality of the pharmaceutical product. For instance, products obtained via the membrane filter method have significantly lower fructose residue compared to those derived from the alcohol precipitation method. Even when employing the same purification principle, variations in the production equipment and process parameters among different manufacturers can lead to discrepancies in residue levels.
Conclusions
Dextran 40 prepared by fermentation is widely used in industries such as food and pharmaceuticals,28 and as an injectable raw material, its quality requirements are more stringent compared to other fields. The trace carbohydrate residues in the final product should be effectively controlled to reduce the risk of adverse reactions. In ICH Q7, impurities are defined as any unwanted components present in intermediates or active pharmaceutical ingredients.29 According to the detection results, carbohydrate residues in some batches of samples were found to be as high as 0.3%, significantly exceeding the established threshold limit of 0.15% specified in Q3A.30 However, the existing standards do not effectively control such specific impurities, presenting potential safety risks. Therefore, this study established and optimized a user-friendly, highly efficient HILIC-CAD method for detecting carbohydrate residues in dextran 40 produced by fermentation. The method enables rapid separation and quantification of fructose and sucrose within 10 minutes. Compared to HILIC-MS and HILIC-RID methods, the established method demonstrates greater sensitivity, capable of quantitatively detecting carbohydrate impurities at concentrations as low as 3.3 ppm. Furthermore, the method has been validated to show good repeatability, recovery, durability, and linearity. Additionally, using the established method, this study compared the carbohydrate residue levels in dextran 40 obtained from different purification processes. It was found that the membrane filter method achieved superior purification compared to the alcohol precipitation method, although differences in filtration efficiency were observed among different membrane filtration processes. It has been observed that the residual carbohydrates in dextran 40 produced by some manufacturers have exceeded the threshold values. This situation has not yet received sufficient attention. Therefore, it is recommended that manufacturers using fermentation methods to produce dextran 40 employ the method established by this research to measure the levels of residual carbohydrates and establish reasonable release standards to ensure the quality. In cases where the threshold is exceeded, immediate improvements to the production process should be made. For example, the alcohol precipitation method could be replaced with the relatively safer membrane filter method, or further optimization of both the alcohol precipitation method and membrane filter processes could be conducted to ensure drug purity.Furthermore, regulatory agencies should increase their focus on such specific residual substances. The limits for these residues should be incorporated into the shelf-life standards. By using appropriate methods to determine their content, the quality control of drugs in circulation can be ensured, safeguarding public drug safety. The method established in this study can be utilized to evaluate the quality of samples from different sources, providing a reference for the optimization of various production processes. It effectively enables the quality control of dextran 40 and offers valuable insights into the quality control of carbohydrate drugs obtained through fermentation methods.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article. All data generated or analyzed during this study are included in the main text of the article. No additional datasets were generated or used in this study.Author contributions
Shenggu Xie: conceptualization, resources, writing – review & editing. Zhuyu Jin: data curation, formal analysis, methodology, validation, writing – original draft. Yan Huang: investigation. Qiaoqiao Huang: funding acquisition, project administration, supervision.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- G. Huang and J. Chen, Int. J. Biol. Macromol., 2019, 125, 478–484 CrossRef CAS PubMed.
- Q. Shen, Y. Guo, K. Wang, C. Zhang and Y. Ma, Molecules, 2023, 28, 7093 CrossRef CAS PubMed.
- T. Wang, Z. Jiang, Y. Wang, H. Wu, Y. Fang, W. Dong, B. Wu, J. Ma and M. Jiang, Front. Bioeng. Biotechnol., 2021, 9, 747602 CrossRef PubMed.
- A. J. Kareem and J. A. S. Salman, Rep. Biochem. Mol. Biol., 2019, 8, 287 Search PubMed.
- X. Sun, S. Xie, Y. Huang, N. Sun, S. Liu, Y. Wang, L. Lin and W. Ni, Chin. Pharm. J, 2022, 57, 301–305 Search PubMed.
- B. Kang, W. Shuai and D. Cheng, J. Pharm. Anal., 2021, 41, 1954–1959 Search PubMed.
- Q. Wang, T. Liu, X. Xu, H. Chen and S. Chen, Int. J. Biol. Macromol., 2021, 169, 60–66 CrossRef CAS PubMed.
- H. Chen, Y. Pu, Q. Zou, D. Hou and S. Chen, Polym. Bull., 2021, 78, 4863–4876 CrossRef CAS.
- Y. Miyagawa, H. Seto, K. Ohto and H. Kawakita, Biochem. Eng. J., 2012, 64, 17–21 CrossRef CAS.
- K. Ebert and H. Witt, Mol. Cell. Pediatr., 2016, 3, 10 CrossRef PubMed.
- R. K. Gupta, K. Gupta, A. Sharma, M. Das, I. A. Ansari and P. D. Dwivedi, Crit. Rev. Food Sci. Nutr., 2018, 58, 208–226 CrossRef CAS PubMed.
- The United States Pharmacopeial Convention, U.S. Pharmacopeia-National Formulary, 2024 Search PubMed.
- European Directorate for the Quality of Medicines and Healthcare, European Pharmacopoeia, Strasbourg, 11th edn, 2024 Search PubMed.
- National Pharmacopoeia Commission. Chinese Pharmacopoeia, 2020 Edition Part IV. China Medical Science and Technology Press, 2020 Search PubMed.
- Y. Wu, Z. Jin, S. Xie, Q. Shen and X. Liu, Medical Information, 2024, 43, 1315–1320 Search PubMed.
- F. Ianni, L. Pucciarini, A. Carotti, G. Saluti, S. Moretti, V. Ferrone, R. Sardella, R. Galarini and B. Natalini, Anal. Chim. Acta, 2018, 1044, 174–180 CrossRef CAS PubMed.
- K. Molnarova and P. Kozlík, Molecules, 2020, 25, 4655 CrossRef CAS PubMed.
- M. Abla, C. Ladavière and S. Trombotto, Chromatographia, 2022, 85, 55–63 CrossRef CAS.
- S. Sun, H. Wang, J. Xie and Y. Su, Chem. Cent. J., 2016, 10, 25 CrossRef PubMed.
- X. Luo, Y. Liu, J. Xing, X. Bi, J. Shen, S. Zhang, X. Xu, L. Mao and Y. Lou, Microchem. J., 2024, 201, 110666 CrossRef CAS.
- X. Q. Huang, D. W. Ji, Y. J. Qiu and F. T. Sun, China Pharm., 2024, 33, 71–74 Search PubMed.
- J. Gao, Q. Wang, X. Wei, B. Zhu, J. Wang and F. Wang, J. Pharm. Biomed. Anal., 2024, 249, 116384 CrossRef CAS PubMed.
- P. Lezana, M. F. García-Mayoral, B. Lamothe and M. Pena-Abaurrea, J. Chromatogr. A, 2021, 1639, 461927 CrossRef CAS PubMed.
- Y. Huang, H. Lu, Z. Li, Y. Zeng, Q. Xu and Y. Wu, J. Pharm. Biomed. Anal., 2023, 229, 115357 CrossRef CAS PubMed.
- Y. Lei, B. Wang, G. Zhu, G. Feng, W. Li, C. Han, Q. Qin, X. Yu, X. Song, Z. He, J. Zhang, H. Su and W. Wang, Nat. Prod. Commun., 2024, 19, 1934578X241232269 CrossRef CAS.
- ICH Guideline, Q2 (R2): Validation of Analytical Procedures, 2023 Search PubMed.
- M. Tiwari, S. Mhatre, T. Vyas, A. Bapna and G. Raghavan, Separations, 2023, 10, 199 CrossRef CAS.
- M. Naessens, A. Cerdobbel, W. Soetaert and E. J. Vandamme, J. Chem. Technol. Biotechnol., 2005, 80, 845–860 CrossRef CAS.
- ICH Guideline, Q7: Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients, 2000 Search PubMed.
- ICH Guideline, Q3A: Impurities in New Drug Substances, 2006 Search PubMed.
Footnote |
| † Shenggu Xie and Zhuyu Jin contributed equally to this article. |
| This journal is © The Royal Society of Chemistry 2025 |