
Co-assembled biomimetic fibrils from collagen and chitosan for performance-enhancing hemostatic dressing†
Xingling
Zeng
ab,
Zhaohui
Sun
c,
Lidan
Chen
c,
Xiaoxia
Zhang
ab,
Xin
Guo
 ab and
Guoying
Li
*ab
ab and
Guoying
Li
*ab
aThe Key Laboratory of Leather Chemistry and Engineering (Ministry of Education), Sichuan University, Chengdu 610065, PR China. E-mail: liguoyings@163.com; Fax: +86-28-85405237; Tel: +86-28-85462568
bNational Engineering Laboratory for Clean Technology of Leather Manufacture, Sichuan University, Chengdu 610065, PR China
cDepartment of Laboratory Medicine General Hospital of Southern Theater Command, Guangzhou, Guangdong 510010, PR China
First published on 1st November 2024
Abstract
The development of safe and efficient hemostatic materials is medically important to prevent death due to trauma bleeding. Exploiting the synergistic effect between the D-periodic functional domain of collagen fibrils on platelet activation and cationic chitosan on erythrocyte aggregation is expected to develop performance-enhanced hemostatic materials. In this study, we prepared collagen fibrils and chitosan composite hemostatic materials by modulating the self-assembled bionic fibrillation of collagen with different degrees of deacetylation (DD, 50%, 70% and 85%) of chitosan. The findings indicated that chitosan promoted collagen self-assembly, with all the collagen fibrils demonstrating a typical D-periodical structure similar to that of the native collagen. Furthermore, the composite demonstrated enhanced structural integrity and procoagulant capacity along with good biocompatibility. Notably, the fibrillar composites with 70% DD of chitosan exhibited optimal mechanical properties, procoagulant activity, and adhesion of erythrocytes and platelets. Compared to pure collagen fibrils and the commercial hemostatic agent Celox™, the collagen/chitosan fibrillar composite treatment significantly accelerated hemostasis in the rat tail amputation model and liver injury model. This research offers new insights into the development of hemostatic materials and indicates that collagen-chitosan composites hold promising potential for clinical applications.
1. Introduction
Trauma hemorrhage is a common health problem in daily life and may cause various pathological changes, leading to organ failure and death.1–3 Physiological hemostatic response is only able to control minor bleeding and not heavy bleeding.2,4,5 Therefore, the additional use of hemostatic materials is required for the rapid control of hemorrhage. Various biomaterial-based hemostatic agents have been developed.6,7 Natural polymers, such as collagen, chitosan, cellulose and alginate, are widely used in the development of clinical hemostatic materials due to their excellent hemostatic properties, biodegradability and biocompatibility.8–10As one of the most commonly used natural polymers in biomedical material development, collagen is considered to be an effective hemostatic agent due to its excellent hemostatic properties and biocompatibility.11,12 It is an important structural protein in animals and a major component of the extracellular matrix (ECM), which usually exists in a fibrillar conformation with a typical D-periodicity (62–67 nm).13–16 Collagen is also known to participate in the physiological coagulation process of the body, inducing platelet adhesion and aggregation, initiating endogenous coagulation pathways, and promoting hemostasis and thrombosis.17–20 It is noteworthy that collagen fibrils with higher-order structure can provide more adhesion sites for platelets and induce more coagulation factor activation.14,19,21 Bernardo et al. found that collagen fibrils with bionic structure induced platelet aggregation 10-fold higher than that of collagen molecules.22 Moreover, in addition to their excellent hemostatic properties, collagen fibrils have superior bioactive and physicochemical properties. Therefore, the development of safe and efficient hemostatic materials is facilitated by replicating the structure of native collagen. It has been widely established that collagen molecules are capable of self-assembling into native ribbon fibrils under physiological conditions in vitro. The self-assembly process of collagen has been reported to be regulated by electrostatic interactions, hydrogen bonding and hydrophobic interactions.16,23,24 Polysaccharides, such as chitosan, chondroitin sulphate, hyaluronic acid and sodium alginate, can affect the reconstitution rate and structure of collagen fibrils to varying degrees through electrostatic interactions without disrupting their D-periodic cross-striations.25–28
Chitosan is a natural cationic polysaccharide with abundant sources and low cost, which has excellent antimicrobial, biocompatible and hemostatic properties, and is a recognized safe substance approved by the FDA.29,30 Notably, the hemostatic effect of cationic chitosan is not directly related to traditional intrinsic or extrinsic pathways. Also, it can directly stimulate platelet activation and promote the adhesion and aggregation of platelets and red blood cells as well as induce clot formation in the absence of coagulation factors.4,31 Therefore, chitosan is a promising and excellent hemostatic material. It was found that the hemostatic properties of chitosan mainly depend on its degree of deacetylation (DD).32,33 The deacetylation of chitosan leads to the formation of free amino groups, giving chitosan a positive charge.34–36 Thus, the DD affects the interaction of chitosan with negatively charged erythrocytes and platelets.33,37,38 Hu et al. reported that chitosan with moderate DD containing appropriate amino groups was more effective in initiating hemostasis.30
The coagulation mechanisms of collagen and chitosan are synergistic, and their combination can lead to composites with enhanced hemostatic properties.39,40 Moreover, numerous studies have shown that their combination might result in composites with enhanced mechanical properties, positive effects on cell proliferation, and controlled degradation rates.41–44 Therefore, combining collagen and chitosan through in vitro self-assembly may contribute to the development of safer and more efficient composite hemostatic materials. Although studies have been conducted to investigate the application of composites prepared by collagen and chitosan co-assembly in tissue engineering,45–47 the field of hemostasis is still relatively under-researched. Some progress has been made in studies on the impact of the ratio of collagen to chitosan on the kinetics of cofibrillogenesis and performances of the composites,28 but the specific effect of the DD of chitosan on these properties has not been thoroughly investigated.
In this study, in order to develop performance-enhanced hemostatic dressings, biomimetic collagen fibrils/chitosan composites were prepared by the co-assembly of collagen and chitosan. The focus was to investigate the effect of the DD of chitosan on collagen fibrillogenesis and the overall properties of the composites. The mechanism of the fibrillogenesis process of collagen after incorporating chitosan with different DD was investigated via turbidity measurements, zeta potential measurements, hydroxyproline (Hyp) content, atomic force microscopy (AFM) and scanning electron microscopy (SEM). In addition, the mechanical properties and thermal stability of collagen/chitosan fibrillar composites were characterized. In vitro, the hemocompatibility, cytotoxicity and coagulation properties of the composites were also evaluated. The best formulated collagen/chitosan fibrillar composites were selected for animal experiments to assess their hemostatic effect in vivo. The results of the study help to provide useful information about the development and biomedical applications of collagen/chitosan composite hemostatic materials.
2. Materials and methods
2.1. Materials
Type I collagen was self-prepared from calfskin based on our previous report.48 Chitosan (Mw = 1000 kDa, deacetylation degree of 50%, 70% and 85%) was purchased from Shanghai Macklin Biochemical Co., Ltd. Unless otherwise noted, all other reagents were provided by Kelon (Chengdu, China) and were analytical grade.2.2. Preparation of collagen/chitosan co-assembled fibrils
A pre-experimentation phase was initially conducted to identify the optimal ratio of collagen and chitosan for co-assembly. Lyophilized collagen and chitosan powders were completely dissolved in a 0.1 M acetic acid solution to achieve a masterbatch concentration of 4 mg mL−1 for each component. The blend solutions were prepared by mixing the collagen solution with three types of chitosan solutions at varying ratios of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 8:2, 7:3, 6:4, and 5:5 (w/w), respectively. The final collagen concentration was adjusted to 1 mg mL−1 using a phosphate-buffered saline (PBS) solution and distilled water. The resultant blends contained 10 mM phosphate and 110 mM sodium chloride. Subsequently, 1 mol mL−1 NaOH solution was employed to adjust the pH to 7.2. The self-assembly process of all the samples was monitored at 37 °C using a UV-visible spectrophotometer (PerkinElmer Ltd, MA). The pre-experimental results demonstrated that the self-assembly of collagen exhibited a characteristic ‘S’ turbidity curve following the introduction of chitosan (Fig. S1†). Analysis of the turbidity curve revealed that the effects of chitosan with varying DD on collagen self-assembly converged, with an optimal degree of assembly observed for the 7:3 ratio. Consequently, this ratio was selected for further investigation into the co-assembly of collagen and chitosan with different DD for the preparation of hemostatic dressings with enhanced properties. The preparation method adhered to the aforementioned formulation, and the samples were designated as CC-50DD, CC-70DD, and CC-85DD based on the DD of chitosan. Pure collagen was prepared as a control and labeled as Col. All samples were incubated at 37 °C for 12 h to obtain the assembled products, which were subsequently washed, desalted, and freeze-dried, as required for testing.
:1, 8:2, 7:3, 6:4, and 5:5 (w/w), respectively. The final collagen concentration was adjusted to 1 mg mL−1 using a phosphate-buffered saline (PBS) solution and distilled water. The resultant blends contained 10 mM phosphate and 110 mM sodium chloride. Subsequently, 1 mol mL−1 NaOH solution was employed to adjust the pH to 7.2. The self-assembly process of all the samples was monitored at 37 °C using a UV-visible spectrophotometer (PerkinElmer Ltd, MA). The pre-experimental results demonstrated that the self-assembly of collagen exhibited a characteristic ‘S’ turbidity curve following the introduction of chitosan (Fig. S1†). Analysis of the turbidity curve revealed that the effects of chitosan with varying DD on collagen self-assembly converged, with an optimal degree of assembly observed for the 7:3 ratio. Consequently, this ratio was selected for further investigation into the co-assembly of collagen and chitosan with different DD for the preparation of hemostatic dressings with enhanced properties. The preparation method adhered to the aforementioned formulation, and the samples were designated as CC-50DD, CC-70DD, and CC-85DD based on the DD of chitosan. Pure collagen was prepared as a control and labeled as Col. All samples were incubated at 37 °C for 12 h to obtain the assembled products, which were subsequently washed, desalted, and freeze-dried, as required for testing.
2.3. Monitoring the co-assembly process of collagen and chitosan with different DD
The zeta potential was tested using a zeta potential analyzer (NANO ZS90, Malvern, UK) after diluting the samples 2-fold with phosphate buffer solution (10 mM PBS, 110 mM NaCl, pH = 7.2). The data were taken in triplicates.
000 rpm for 10 min, and 3 mL of the supernatant was hydrolyzed by mixing it with 3 mL of 12 M HCl at 110 °C for 24 h. The hydrolysate was neutralized with NaOH and the volume of deionized water was fixed at 25 mL. After chloramine-T oxidation and color development with Ehrlich's reagent for 25 min, the absorbance of the respective solutions was recorded by UV spectroscopy at 560 nm. The standard curve was determined according to the above method using high purity L-hydroxyproline as the standard, as detailed in the ESI (Fig. S2†).
2.4. SEM measurements
All assemblies were immobilized with 2% (v/v) glutaraldehyde in PBS for 12 h at room temperature and rinsed with deionized water several times. Stepwise dehydration was carried out in a series of ethanol concentrations (30, 50, 70, 90, 95 and 100%, v/v) and finally freeze-dried after replacing ethanol with tert-butanol. The morphology of the lyophilized samples was observed using scanning electron microscopy (SEM, Hitachi MC1000, Tokyo, Japan) at an accelerating voltage of 15 kV. The average diameter of the fibrils was measured with ImageJ software (V 1.8.0).2.5. Physicochemical characterization
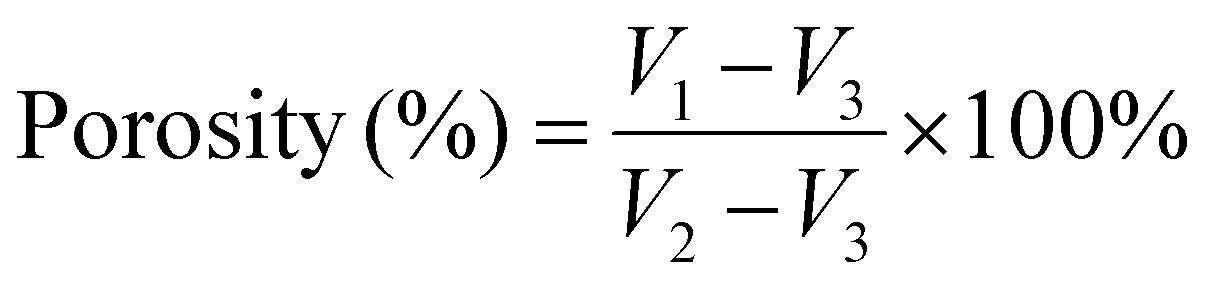 | (1) |
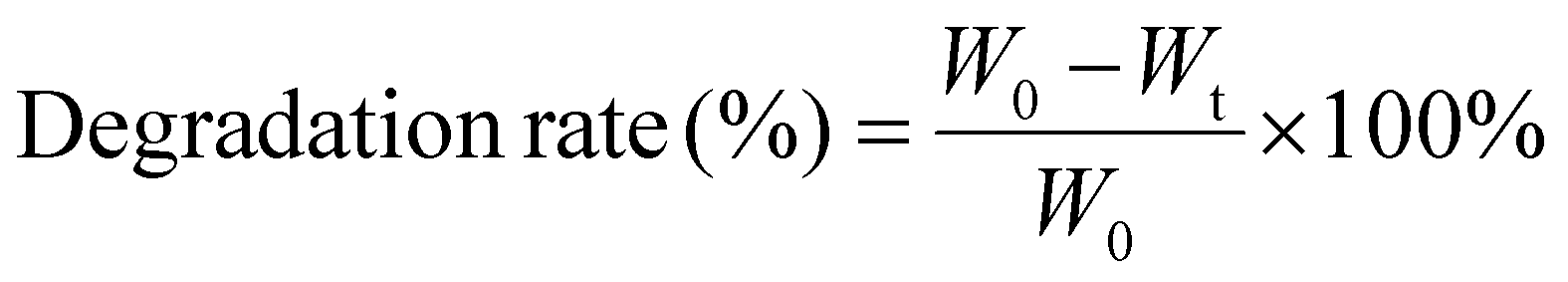 | (2) |
The thermal denaturation temperature of all the assemblies was measured with differential scanning calorimetry (DSC 200 PC, Netzsch, Germany). Approximately 2–3 mg of lyophilized assembled products were accurately weighed into aluminum crucibles and sealed. The temperature increased from 25 °C to 120 °C with a heating rate of 5 °C min−1 under a nitrogen atmosphere. The thermal degradation of the lyophilized fibrillar composites was determined with a thermogravimetric analyzer (TG209F1, Netzsch, Germany). Each sample (∼4 mg) was heated from 50 °C to 700 °C under N2 atmosphere with a constant heating rate of 10 °C min−1.
2.6. Hemocompatibility assay
Fresh rabbit blood samples for in vitro blood-related experiments were obtained from the ear arteries of New Zealand rabbits provided by Chengdu Dashuo Laboratory Animal Co. Citrate was used as an anticoagulant during sample processing. All animal experiments were conducted in accordance with the Guidelines for the Keeping and Use of Laboratory Animals of Sichuan University and approved by the Animal Keeping and Use Committee of Sichuan University (SCU42-2404-02).Erythrocytes were obtained by the centrifugation of fresh anticoagulated rabbit blood at 200g for 15 min and diluted twice with PBS to prepare a 5% (v/v) erythrocyte suspension. 1 mg of lyophilized assembled product was added to 2 mL of the erythrocyte suspension before incubation on a shaker at 37 °C for 1 h and then centrifuged at 3000 rpm for 10 min. The supernatant was measured for absorbance at 540 nm. Commercially available chitosan hemostatic powder (Celox™) has been used in various medical and military contexts, with its biosafety demonstrated through extensive research and practical uses.52,53 Consequently, Celox™ served as the control. Erythrocyte suspensions treated with deionized water and PBS were used as the positive and negative controls, respectively. The hemolysis rate was calculated as follows.
 | (3) |
2.7. Cytocompatibility in vitro
The viability of L929 fibroblasts was measured using the Cell Counting Kit-8 (CCK-8) method to assess the cytotoxicity of the samples.54 Briefly, each lyophilized assembled product after 24 h of UV radiation sterilization was immersed separately in the cell culture medium and incubated for 24 h at 37 °C in a CO2-humidified environment to obtain extracts. L929 fibroblasts were inoculated into 96-well plates at a density of 5000 cells per well for 4 h. The medium was then replaced with extracts and incubated for 1, 3 and 5 days (37 °C, 5% CO2). Then, CCK-8 working solution (10 μL per well) was added to the cultures and incubated at 37 °C for 4 h before measuring the optical density (OD) at 450 nm. The L929 cells incubated with commercially available chitosan hemostatic powder (Celox™) and without the samples were used as the control and blank control groups, respectively.Furthermore, the live/dead staining assay and morphological staining assay were carried out by dyeing L929 fibroblasts co-cultured with the above extracts in the ratio of 1:1 (V/V) for 3 days.14 AM/PI dye was used for live/dead staining assay following the reagent kit instructions. The morphology staining assay was performed according to the following instructions.55 The cultured cells were fixed with paraformaldehyde at 4 °C for 4 h, then stained with fluorescein isothiocyanate phalloidin (TRITC phalloidin) and co-incubated at 37 °C for 30 min in darkness. After rinsing three times with PBS, the cells on the surface were re-stained with 4,6-diamidino-2-phenylindole (DAPI) solution. The visualized indicator of cytotoxicity and the morphology of cell growth were observed with a fluorescence microscope (TI2-U, Nikon, Japan).
2.8. In vitro hemostasis
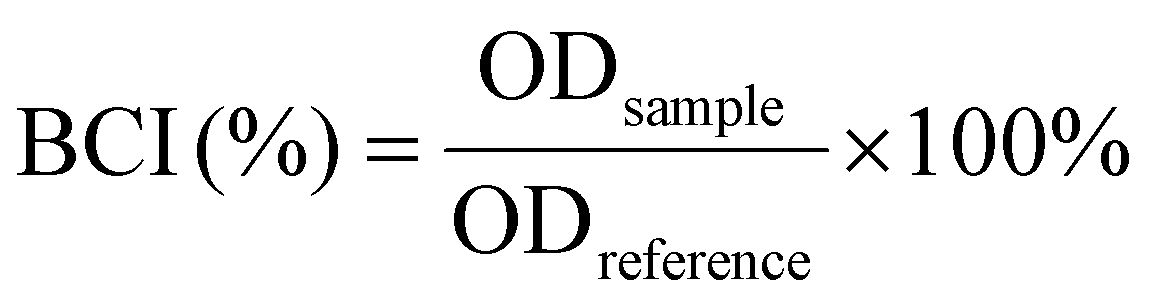 | (4) |
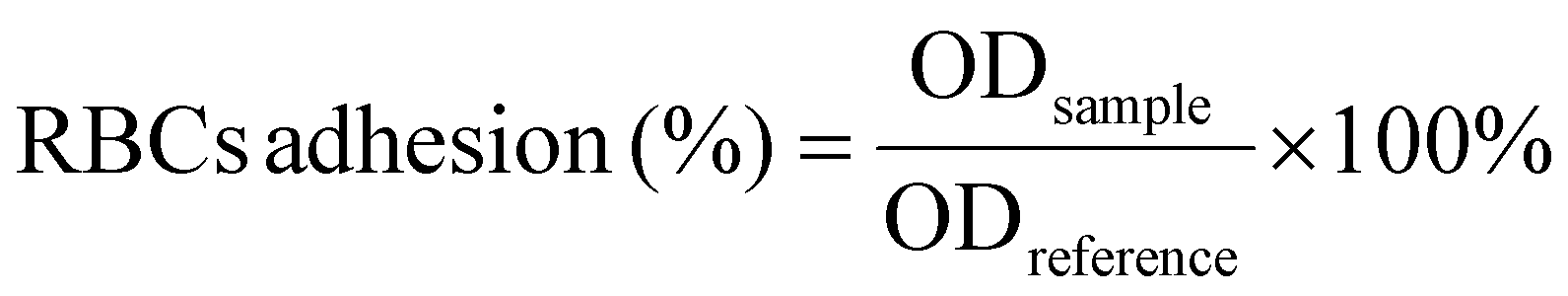 | (5) |
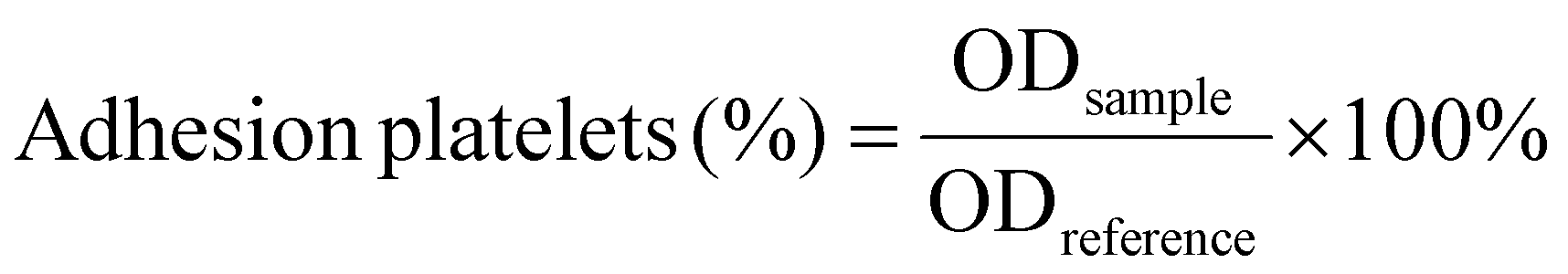 | (6) |
2.9. In vivo hemostasis
The hemostatic potential of collagen/chitosan co-assembled products was assessed using a normal rat liver injury model and a rat tail incision injury model. Pure collagen and commercially available Celox™ pellets were used as the control groups, and the group that did not receive hemostatic treatment was used as the blank group. Sprague Dawley (SD) rats used in all experiments were male and were provided by Chengdu Dashuo Laboratory Animal Co. The sex of the animals had no effect on the results of the study. The rats used for the hemostasis test weighed 250–300 g (7–8 weeks). All animal experiments were conducted in accordance with the Guidelines for the Keeping and Use of Laboratory Animals of Sichuan University and approved by the Animal Keeping and Use Committee of Sichuan University (SCU42-2404-02).2.10. Statistical analysis
Statistics are expressed as mean ± standard deviation. Data were analyzed using one-way ANOVA followed by Tukey's multiple comparison test. Differences were considered significant if P < 0.05 (“ns” indicates nonsignificant difference, *P < 0.05, **P < 0.01, and ***P < 0.001).3. Results and discussion
3.1. Co-assembly of collagen and chitosan with varying DD
Turbidimetric assay is a typical method used to assess the self-assembly process of collagen, characterized by a typical ‘S’ shaped turbidity-time curve that encompasses three main phases: the lag phase, growth phase, and plateau phase. Fig. 1a illustrates the kinetic profile of collagen self-assembly in the presence of chitosan with varying DD. Similar to pure collagen, the collagen/chitosan blend systems showed a characteristic ‘S’ curve. Further analysis of the kinetic curves, as shown in Table 1, indicated that the introduction of chitosan resulted in a reduction of the lag time, increase of the growth rate, and increased change in the turbidity value (Δh). These results indicated that the addition of chitosan enhanced the formation of collagen fibrils. In addition, as the DD of chitosan increased, both the growth rate and Δh initially increased before subsequently decreasing, with optimal collagen fibril formation occurring at a chitosan DD of 70%. Previous literature has reported that charged polysaccharides significantly influence collagen fibril formation through electrostatic, hydrophobic, and hydrogen bonding interactions, with electrostatic interactions being particularly crucial.26,27,58 Variations in the degree of deacetylation of chitosan result in differences in the charge it carries.55 The effect of different DD of chitosan on collagen fibril formation was further elucidated by analyzing the zeta potential of the samples. As shown in Fig. 1b, the zeta potential of pure collagen was measured at −1.78 ± 0.14 m V under the assembled solvent conditions, indicating a negative charge. Upon the introduction of cationic chitosan, the zeta potential shifted towards a positive charge. Evidence suggests that minimizing the surface charge of collagen promotes the nucleation and growth processes of collagen self-assembly to increase the rate of fibril formation.28,58 Notably, the zeta potential of the blend system containing 70% DD chitosan was recorded at 0.37 ± 0.05 m V, which was closer to zero than that of the other systems, thereby maximizing collagen self-assembly.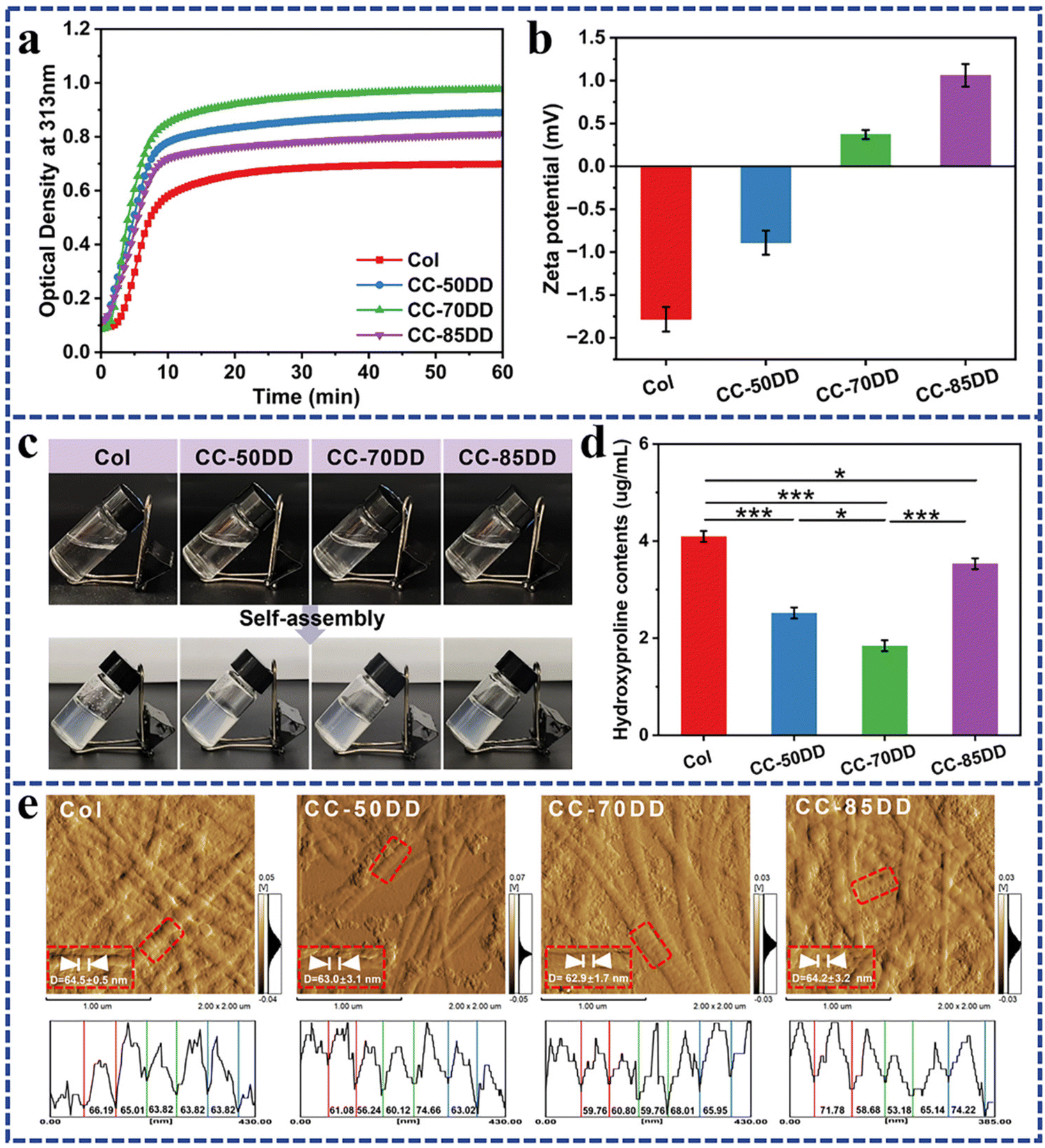 | ||
| Fig. 1 Collagen fibrils reconstitution processes in the presence of chitosan with different DD. (a) Turbidity curves. (b) Zeta potential measurement. (c) Photographs of sample appearance before and after self-assembly. (d) Content of Hyp in the supernatant. (e) AFM images of assembled products and amplitudes of their fibrils surface along the red frame. | ||
| Samples | Lag time (s) | Growth rate (ν × 10−3) | Δh | Assembly degree (%) |
|---|---|---|---|---|
| Col | 170.87 ± 8.99 | 1.41 ± 0.01 | 0.576 ± 0.011 | 98.49 ± 0.04 |
| CC-50DD | 52.36 ± 2.17 | 1.55 ± 0.02 | 0.764 ± 0.007 | 99.07 ± 0.04 |
| CC-70DD | 72.11 ± 7.34 | 2.05 ± 0.08 | 0.872 ± 0.011 | 99.32 ± 0.04 |
| CC-85DD | 81.57 ± 12.44 | 1.43 ± 0.01 | 0.663 ± 0.016 | 98.70 ± 0.04 |
Hydroxyproline (Hyp) is the characteristic amino acid of type I collagen and serves as a quantitative marker for collagen content in the composites.59 As shown in Fig. 1d, the Hyp content in the supernatants of the collagen/chitosan assemblies was consistently lower than that of Col, indicating that chitosan promoted collagen self-assembly within the composites. Furthermore, as the DD of chitosan increased, the Hyp content in the co-assemblies’ supernatant exhibited an initial decrease and then increase. Notably, the Hyp content in the supernatant of CC-70DD was the lowest, while the assembly degree reached a maximum value of 99.32% (Table 1). These findings were consistent with the results obtained from turbidity curve analysis.
The formation of D-periodicity is a hallmark of successful biomimetic reconstitution in vitro mimicking native collagen, which plays an important role in the hemostatic, mechanical and biological properties of the collagen matrix.58,60 The AFM images presented in Fig. 1e demonstrated that collagen fibrils in samples Col, CC-50DD, CC-70DD and CC-85DD exhibited distinct D-periodicity, with average widths of 64.5 ± 0.5 nm, 63.0 ± 3.1 nm, 62.9 ± 1.7 nm and 64.2 ± 3.2 nm, respectively. There were no significant differences among these measurements (Fig. S3†), and they aligned with the typical D-periodicity observed in native collagen fibrils, which ranges from 62 to 67 nm. These results indicated that a biomimetic composite fibril could be fabricated by the co-assembly of collagen and chitosan, and 70% DD of chitosan could maximize the biomimetic fibrillation of collagen.
3.2. SEM analysis
The microstructures of collagen/chitosan assemblies prepared with varying DD of chitosan were observed using SEM, and the average diameter of collagen fibrils across all the samples was further analyzed, as illustrated in Fig. 2. Statistical comparisons of fibril diameters among the samples are presented in Fig. S4.† The average diameter of fibrils in pure collagen and collagen/chitosan composites remained similar, while the structural characteristics of the fibrils were influenced by chitosan. Specifically, chitosan beads were tightly bound to the collagen fibril backbone, suggesting strong interactions between the two materials. Furthermore, several collagen fibrils or different portions of a single fibril seem to be woven and interconnected through chitosan, leading to a denser distribution of collagen fibrils and enhanced structural integrity of the composites. As the DD value of chitosan increased, a greater number of collagen fibrils were sequestered together, leading to the formation of larger clumps and knobs. In the CC-70DD sample, these clumps were relatively uniformly distributed, which contributed to a more stable fibrous structure. In contrast, the CC-85DD sample exhibited a locally dispersed fibrous network. This phenomenon could be attributed to the fact that a higher DD in chitosan led to stronger intermolecular interactions, which facilitated the formation of larger fibril aggregates.61 These aggregates might locally widen the distance between collagen fibrils, consequently disrupting the overall homogeneity of the composite.62 | ||
| Fig. 2 (a–d) Digital images. (a1–d1) and (a2–d2) SEM images at 10000× and 50000× magnifications. (a3–d3) Average diameter of collagen fibrils across all samples. | ||
3.3. Physicochemical characterization
The chemical structures of collagen fibrils and collagen/chitosan assemblies were characterized by FT-IR spectroscopy, with the results shown in Fig. 3a and b. For the native collagen fibril, the absorption peaks at 3287 cm−1 (amide A) and 2937 cm−1 (amide B) were attributed to the N–H stretching and CH2 asymmetric stretching vibration, respectively.63 The amide I band at 1634 cm−1, primarily attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, and the amide II band at 1549 cm−1, linked to N–H bending vibrations and C–N stretching vibrations, were critical for assessing the secondary structure of collagen.13 Additionally, the amide III band at 1239 cm−1 further supported the presence of the triple helical conformation as it related to C–N stretching vibration and CH2 wagging vibration.64 Importantly, these typical characteristic bands of collagen were also clearly observed in all the samples, suggesting that the co-assembly of chitosan with collagen did not disrupt the triple helical structure of collagen.
O stretching, and the amide II band at 1549 cm−1, linked to N–H bending vibrations and C–N stretching vibrations, were critical for assessing the secondary structure of collagen.13 Additionally, the amide III band at 1239 cm−1 further supported the presence of the triple helical conformation as it related to C–N stretching vibration and CH2 wagging vibration.64 Importantly, these typical characteristic bands of collagen were also clearly observed in all the samples, suggesting that the co-assembly of chitosan with collagen did not disrupt the triple helical structure of collagen.
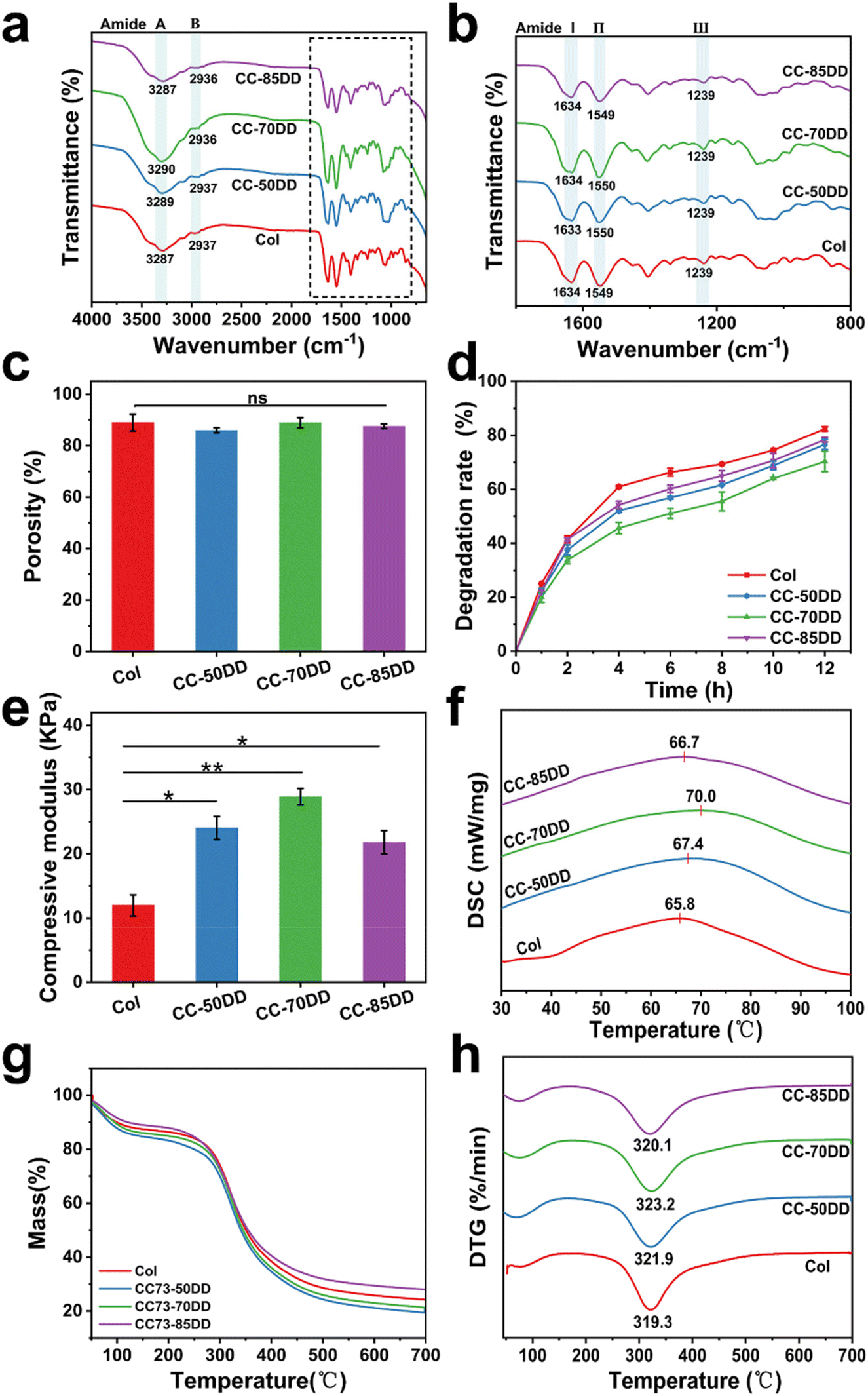 | ||
| Fig. 3 (a) FT-IR spectra in the range of 4000–600 cm−1, (b) FT-IR spectra in the range of 2000–750 cm−1, (c) porosity, (d) degradation rate, (e) compression modulus, (f) DSC curves, (g) TGA curves and (h) DTG curves of Col, CC-50DD, CC-70DD and CC-85DD fibrillar assemblies. | ||
Fig. 3c shows the porosity of Col and collagen/chitosan composites. The porosity of all the samples was similar with no significant difference, indicating that the DD of chitosan had little effect on the porosity of the composites. In addition, the porosity of the samples ranged between 86.01% and 89.03%, which met the requirement of an ideal hemostatic material.65
The degradation behavior of the composites was evaluated using type I collagenase under physiological conditions. As shown in Fig. 3d, Col exhibited rapid degradation, with an 82% degradation rate observed within 12 hours. In comparison, the degradation rate of collagen/chitosan composites was lower than that of Col, indicating that chitosan increased the resistance of collagen to collagenase action. Furthermore, the DD of chitosan also affected the degradability of the composites. CC-70DD consistently had the lowest degradation rate compared to CC-50DD and CC-85DD. This phenomenon might be attributed to the fibrillar structure of the composites through the electrostatic interaction that enhanced the integrity.43,47,66 Higher DD resulted in stronger interactions, decreased local molecular connectivity, and consequently decreased the resistance to enzymatic degradation.62,67
The compressive modulus of Col and collagen/chitosan assemblies are shown in Fig. 3e. The compressive modulus of composites exhibited a notable enhancement compared to that of pure collagen (11.97 ± 1.65 kPa). This improvement could be attributed to the synergistic interactions between collagen and chitosan, which contribute to the overall structural integrity of the composite.47 As the DD of chitosan varied from 50% to 85%, the modulus initially increased to 28.90 ± 1.29 kPa for the composite containing 70% DD chitosan (CC-70DD), before subsequently declining to 21.78 ± 1.80 kPa. This was because excessive interactions might hinder the maintenance of mechanical integrity, ultimately leading to reduced performance.68 Therefore, CC-70DD had the best mechanical properties.
The thermal stability and thermal degradation behavior of Col and the composite fibrils were investigated using DSC and TGA. The DSC curves, as depicted in Fig. 3f, showed that the thermal denaturation temperature (Td) of the samples increased from 65.8 °C for Col to 67.4 °C, 70.0 °C and 66.7 °C for CC-50DD, CC-70DD, and CC-85DD, respectively. All the composite fibrils showed higher Td than pure collagen fibril, and the Td values increased with the increasing DD of chitosan. When the DD of chitosan reached 70%, the collagen/chitosan exhibited the highest Td, which was 4.2 °C higher than that of the pure collagen fibril. However, as the DD increased to 85%, the Td of the composite showed a slight decrease. The enhanced thermal stability of the composites could be attributed to the electrostatic interactions between collagen and chitosan.69 Nonetheless, an excessive degree of electrostatic interactions might compromise the structural integrity, leading to a reduction in thermal stability at higher DD values. The TGA curves presented in Fig. 3g indicate that the weight loss of all the samples occurred in two distinct stages. The initial stage, occurring at approximately 100 °C, primarily resulted from the evaporation of both free and bound water present in the samples.68 The second stage of weight loss, which transpired between 290 °C and 330 °C, was associated with the thermal degradation of the collagen and chitosan components within the samples, respectively.70,71 From the DTG curves (Fig. 3h), it was evident that the thermal degradation temperatures (Tm) of the composites were consistently higher than that of Col (319.3 °C). This observation further confirmed that the co-assembly of collagen and chitosan enhanced the thermal stability of the materials. Moreover, as the DD of chitosan increases, the Tm of the composites exhibited a slight but variable increase. Notably, the highest Tm recorded for the composites was 323.3 °C, achieved at a chitosan DD of 70%. This trend was consistent with the findings from the DSC results.
3.4. In vitro hemocompatibility and cytocompatibility assessment
Biosafety is a crucial prerequisite for the application of biomedical materials in treating bleeding wounds, with hemolytic safety and cytotoxicity being the main considerations.2,57 The hemocompatibility of collagen fibrils and collagen/chitosan composites was evaluated through hemolysis tests, with the results presented in Fig. 4a. The hemolysis rates for Col, CC-50DD, CC-70DD, and CC-85DD samples were all below 1%, which was lower than that of the commercially available chitosan hemostatic material, Celox™, which recorded a hemolysis rate of 1.5%. These results complied with the international standard for hemolysis rates in hemostatic materials (<5%), indicating that the samples exhibited excellent hemocompatibility. Furthermore, CCK8 assays and live/dead staining were conducted to assess the cytotoxicity of the materials, utilizing L929 fibroblast cells as the model cells. As illustrated in Fig. 4b, all the samples demonstrated a slight enhancement in cell proliferation and growth compared to the blank control and Celox™, suggesting that the collagen fibrils and collagen/chitosan composites were non-toxic to cells and possessed good cytocompatibility. Fluorescence staining (Fig. 4c) further confirmed the absence of cytotoxicity in these materials. In addition, cytoskeletal staining was performed to observe the growth state of the cells (Fig. 4d), revealing that the cell morphology of all the samples predominantly exhibited a spindle-shape. In summary, collagen/chitosan composites were safe for use in wound hemostasis, demonstrating good biocompatibility and suitability for applications in the field of biomedical materials.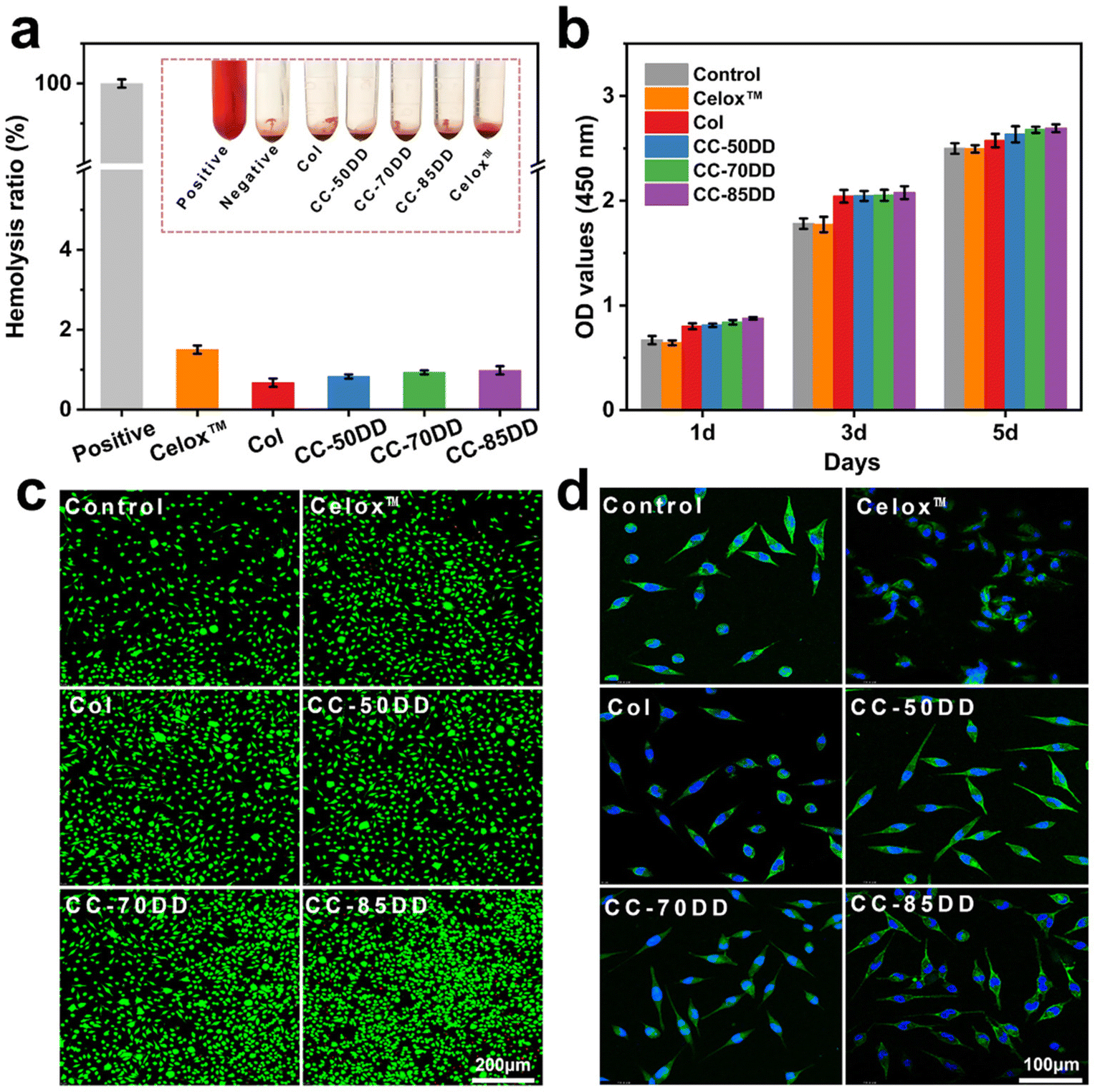 | ||
| Fig. 4 Hemocompatibility and cytocompatibility assessment of various materials. (a) Hemolysis rate. (b) OD values indicating cell number (c) live/dead staining. (d) Growth status of cells cultured with each sample extract for 3 days. | ||
3.5. In vitro hemostatic evaluation
The coagulation index (BCI) is a crucial indicator for assessing in vitro hemostatic capacity, reflecting a material's ability to form thrombus formations upon contact with blood. A lower BCI value signifies enhanced coagulation capacity.72 To quantitatively evaluate the coagulation ability of the collagen/chitosan composites, the BCI of the samples was determined at various time points using a dynamic whole blood coagulation assay, with commercially available chitosan hemostatic material (Celox™) and collagen fibril (Col) as the controls. As shown in Fig. 5a, the BCI values for the composites consistently remained lower than those of the control materials across all time points, with values increasing in the order of CC-70DD < CC-85DD < CC-50DD. This indicated that the composites possessed superior procoagulant properties compared to Col and Celox™. This might be related to the fact that the hemostatic mechanisms of collagen fibrils and chitosan are synergistic.39 The cationic nature of chitosan in the composites facilitated the attraction of negatively charged erythrocytes and platelets via electrostatic interactions, resulting in their adhesion and aggregation on the surface of the material.73 Furthermore, collagen fibrils featuring a D-periodic functional domain offered activated adhesion sites for the platelets, which further enhanced their activation and aggregation, ultimately accelerating the blood coagulation process.21,22 Notably, the procoagulant ability of the composites was influenced by the DD of chitosan. The hemostatic properties of CC-70DD are better than those of CC-50DD because the higher the DD, the stronger the electrostatic interactions and the more favorable the activation and aggregation of platelets and erythrocytes. However, the coagulation efficacy of CC-85DD decreased compared to CC-70DD. This decrease might be due to the localized loosening of its fibrous structure, which was not conducive to blood cell adhesion.47 In addition, the competition for chitosan's positively charged sites by collagen, platelets, and erythrocytes might increase when the DD was elevated, further reducing its hemostatic potential. Consequently, the composite CC-70DD containing 70% deacetylated chitosan demonstrated the highest potential for hemostasis. Fig. 5b illustrates the blood coagulation process, where red blood cells (RBCs) not involved in coagulation were lysed with distilled water after a 5 minutes contact period with blood. Visual observations revealed that the composites exhibited a noticeably lighter color compared to the control materials, particularly in the cases of CC-70DD and CC-85DD. This color change serves as a visual indicator of coagulation efficiency, with lighter colors correlating to faster coagulation.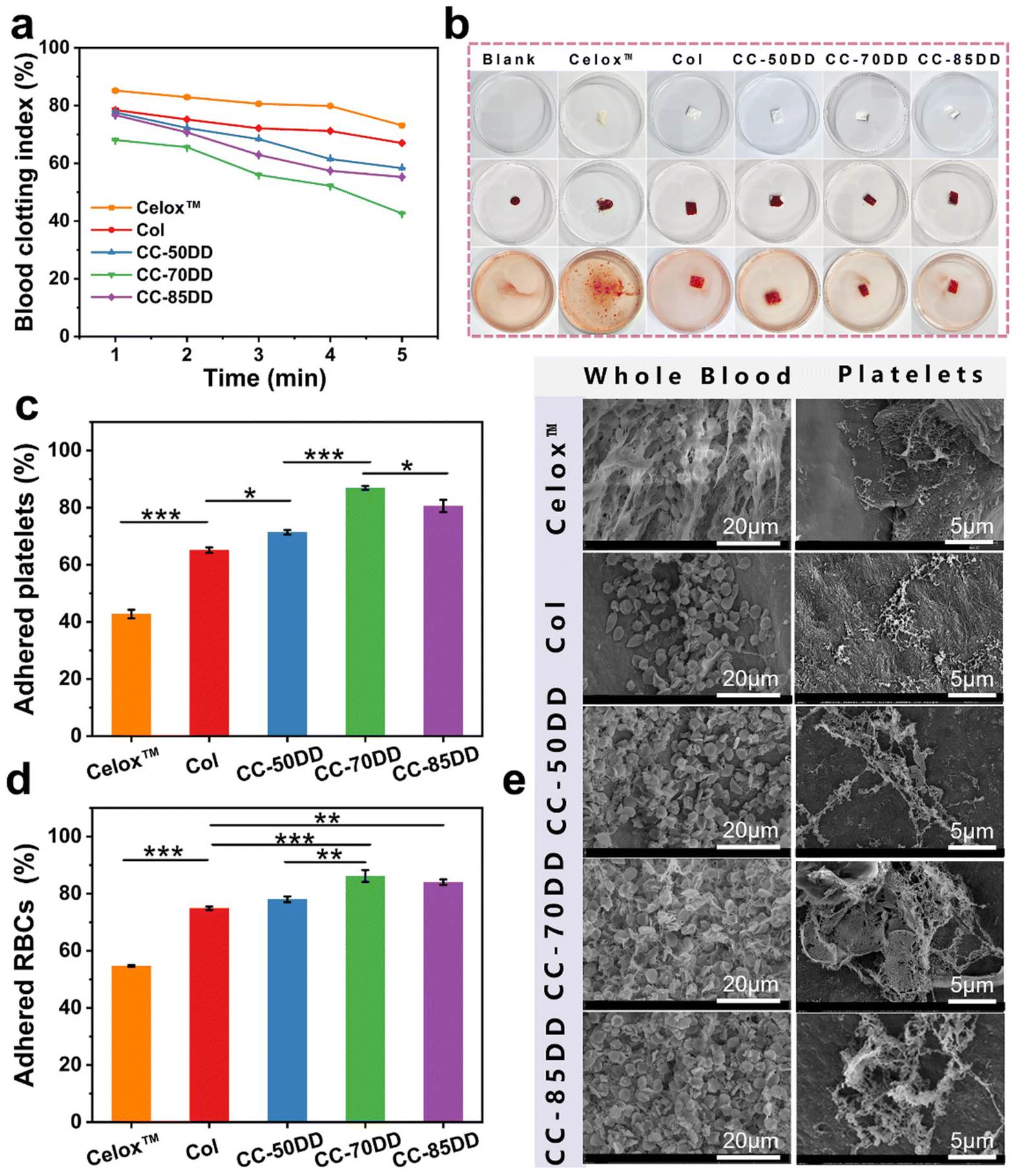 | ||
| Fig. 5 In vitro coagulation of different materials. (a) Blood clotting index curves. (b) Representative photographs of the clotting process. (c) Platelets adhesion ratio. (d) RBCs adhesion ratio. (e) SEM images showing the adhesion of whole blood and platelets on the surface of different materials. | ||
The coagulation effect of the different materials was further investigated by platelet and red blood cell adhesion tests.57 As illustrated in Fig. 5c, the platelet adhesion rate of the composites was significantly superior to that of Col (65.14%) and Celox™ (42.76%). This was attributed to the synergistic effect of collagen fibrils and chitosan on platelet adhesion and aggregation.11 The platelet adhesion rate of the composites varied with different DD values of chitosan and the differences were significant, with the CC-70DD composite exhibiting the highest platelet adhesion rate of 86.94%. This might be due to the fact that chitosan with a DD value of 70% is most favorable for promoting collagen fibrillogenesis and more collagen fibrils with functional domains of the D-periodic might be produced in the composites, thus providing more sites for platelet adhesion and activation.19,22 In addition, at this DD, the interaction of collagen fibrils with chitosan might not hinder the adhesion and activation of chitosan to the platelets. In terms of erythrocyte adhesion, as shown in Fig. 5d, the composites also outperformed the controls, with CC-70DD achieving an adhesion rate of 86.21%, exceeding that of CC-50DD (78.05%) and CC-85DD (84.03%). The difference in the erythrocyte adhesion rates of the composites may be mainly related to the electrostatic effect of cationic chitosan.73 This was attributed to the high erythrocyte adhesion rate of both CC-70DD and CC-85DD. Secondly, the adhesion rate of CC-85DD to erythrocytes was lower than that of CC-70DD, suggesting that the adhesion of the composites may also be related to their fibril network structure. Overall, the CC-70-DD composite effectively integrated the hemostatic properties of chitosan and collagen.
Fig. 5e depicts the microscopic morphology of different materials after contact with whole blood and platelets, respectively. A greater number of blood cells adhered and aggregated on the surface of the collagen/chitosan co-assembled composites compared to Col and Celox™. In addition, the composites had a deeper stimulus activation and formed a partial fiber network around the blood cells, particularly evident in CC-70DD. Platelet adhesion showed similar results. Specifically, Celox™ promoted platelet aggregation activation to some extent. Platelets adhering to Col were activated, and their morphology transitioned from a slightly protruding disc shape to an irregular form with extended pseudopods.20 In contrast, platelets on the composites were numerous and showed higher levels of activation. CC-50DD and CC-70DD formed a fibrin fiber network and CC-70DD formed irregular platelet aggregates.55 These results further confirmed that the synergistic effect of collagen and chitosan significantly enhanced the hemostatic properties of the composites, with CC-70DD demonstrating an excellent capacity to promote blood coagulation.
3.6. In vivo hemostasis evaluation
The CC-70DD composite was selected as an optimized sample for animal experiments because of its notable advantages in mechanical properties, in vitro biosafety and hemostatic capacity. The in vivo hemostatic effect of CC-70DD was assessed using a rat amputation model and a rat liver trauma model (Fig. 6). For comparison, commercially available chitosan hemostatic material (Celox™) and the pure collagen fibrils (Col) were used as the controls because Celox™ is a commonly used hemostat in prehospital and hospital scenarios. Moreover, CC-70DD was made of collagen fibrils and chitosan granules. Fig. 6b and c illustrate the blood loss and hemostasis time in the rat amputation model. The blood loss in untreated group reached 947.5 ± 11.3 mg. Wounds treated with CC-70DD experienced the least blood loss at 177.8 ± 57.1 mg, in contrast to those treated with Celox™ (611.3 ± 38.6 mg) and Col (320.0 ± 15.3 mg). Furthermore, CC-70DD effectively halted bleeding within 1.53 ± 0.08 min, representing a substantial improvement over the blank group (5.15 ± 0.14 min), Celox™ (4.03 ± 0.19 min), and Col (2.39 ± 0.20 min). In the rat liver trauma model, Celox™ exhibited a non-significant hemostatic effect, with blood loss recorded at 693.4 ± 36.3 mg and a hemostasis time of 4.64 ± 0.19 min, which are close to those of the untreated group (906.3 ± 27.4 mg and 6.34 ± 0.20 min). CC-70DD demonstrated superior hemostatic capabilities, achieving a blood loss of 64.1 ± 8.0 mg and a hemostatic time of 1.17 ± 0.06 min, which was more effective than that of the pure collagen fibril (130.7 ± 11.6 mg and 2.00 ± 0.06 min). These findings were consistent with the results of in vitro hemostatic assays, underscoring the remarkable procoagulant properties of CC-70DD. The composite effectively reduced both blood loss and bleeding time, confirming that composites containing 70% DD of chitosan possessed excellent hemostatic effects, as anticipated. These promising results emphasized the potential of collagen/chitosan co-assembled materials in managing hemorrhage and facilitating wound healing in clinical settings.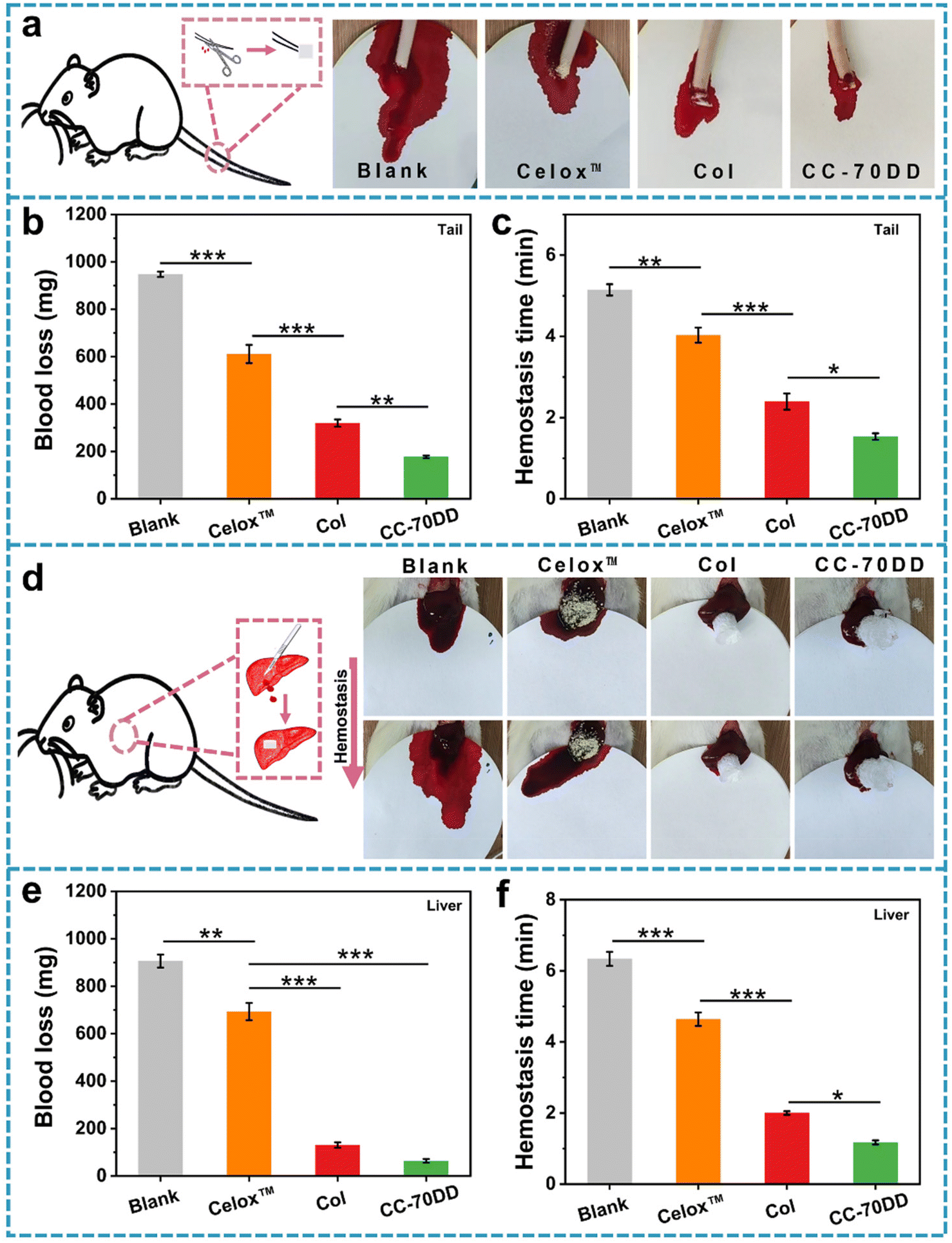 | ||
| Fig. 6 Different wound models for hemostasis. (a) Schematic and images of the rat tail amputation model. (b) Blood loss of various materials in the rat tail amputation model. (c) Hemostasis time of various materials in the rat tail amputation model. (d) Schematic and images of the rat liver trauma model. (e) Blood loss of various materials in the rat liver trauma model. (f) Hemostasis time of various materials in the rat liver trauma model. | ||
4. Conclusion
In this study, collagen/chitosan co-assembled fibrils were developed for hemostatic applications by modulating the fibrillogenesis of collagen through variations in the DD of chitosan. The incorporation of positively-charged chitosan into the collagen system facilitated collagen self-assembly through intermolecular electrostatic interactions. This enhancement in the self-assembly of collagen exhibited a trend of initial increase followed by a decrease as the DD of chitosan increased. Also, the collagen fibrils in all the samples displayed typical D-periodicity similar to those of native collagen. Following the introduction of chitosan, the collagen fibrils became tighter, resulting in a more stable structure for the composites. The fibrillar composites effectively stimulated platelet aggregation and activation, induced erythrocyte adhesion, and promoted blood coagulation. In particular, the fibrillar composites containing 70% DD chitosan demonstrated the best mechanical properties, thermal stability and in vitro hemostatic ability, along with good biocompatibility. The hemostatic properties of CC-70DD were significantly better than those of the pure collagen fibrils (Col) and Celox™, as evidenced in both the rat tail-break model and the rat liver injury model. This study provides valuable references and options for the further development of effective clinical hemostatic materials derived from collagen and chitosan.Author contributions
Xingling Zeng: methodology, investigation, data curation, and writing – original draft. Zhaohui Sun: methodology. Lidan Chen: formal analysis. Xiaoxia Zhang: formal analysis. Xin Guo: validation. Guoying Li: supervision, conceptualization, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22078206).References
- Y. Guo, M. Wang, Q. Liu, G. Liu, S. Wang and J. Li, Theranostics, 2023, 13, 161–196 CrossRef PubMed
.
- Y. Yang, Y. Zhang, Y. Min and J. Chen, Int. J. Biol. Macromol., 2022, 222, 30–40 CrossRef PubMed
- M. Zhou, J. Liao, G. Li, Z. Yu, D. Xie, H. Zhou, F. Wang, Y. Ren, R. Xu, Y. Dai, J. Wang, J. Huang and R. Zhang, Carbohydr. Polym., 2022, 294, 119805 CrossRef CAS PubMed
- S. Zhang, X. Lei, Y. Lv, L. Wang and L.-N. Wang, Carbohydr. Polym., 2024, 327, 121673 CrossRef CAS
- H. Huang, S. Liao, D. Zhang, W. Liang, K. Xu, Y. Zhang and M. Lang, Carbohydr. Polym., 2024, 338, 122148 CrossRef CAS PubMed
- Y. Liu, Y. Zhang, W. Yao, P. Chen, Y. Cao, M. Shan, S. Yu, L. Zhang, B. Bao and F.-F. Cheng, ACS Appl. Bio Mater., 2024, 7, 1362–1380 CrossRef CAS PubMed
- Y. Guo, N. Cheng, H. Sun, J. Hou, Y. Zhang, D. Wang, W. Zhang and Z. Chen, Front. Bioeng. Biotechnol., 2023, 10, 1062676 CrossRef PubMed
- X. Yan, Y. Chen, W. Dan, N. Dan and Z. Li, J. Leather Sci. Eng., 2022, 4, 5 CrossRef
- B. Guo, R. Dong, Y. Liang and M. Li, Nat. Rev. Chem., 2021, 5, 773–791 CrossRef PubMed
- M. Mecwan, J. Li, N. Falcone, M. Ermis, E. Torres, R. Morales, A. Hassani, R. Haghniaz, K. Mandal, S. Sharma, S. Maity, F. Zehtabi, B. Zamanian, R. Herculano, M. Akbari, J. V. John and A. Khademhosseini, Regener. Biomater., 2022, 9, rbac063 CrossRef PubMed
- C. Zheng, X. Liu, X. Luo, M. Zheng, X. Wang, W. Dan and H. Jiang, J. Mater. Chem. B, 2019, 7, 7338–7350 RSC
- X. Cheng, Z. Shao, C. Li, L. Yu, M. A. Raja and C. Liu, PLoS One, 2017, 12, e0169731 CrossRef
- Y. Luo, F. Tao, J. Wang, Y. Chai, C. Ren, Y. Wang, T. Wu and Z. Chen, Int. J. Biol. Macromol., 2023, 253, 127014 CrossRef PubMed
- X. Zhang, C. Yang, X. Guo, C. Yang and G. Li, Biomater. Sci., 2023, 11, 7408–7422 RSC
- D. M. Darvish, Mater. Today Bio, 2022, 15, 100322 CrossRef PubMed
- S. Zhu, Q. Yuan, T. Yin, J. You, Z. Gu, S. Xiong and Y. Hu, J. Mater. Chem. B, 2018, 6, 2650–2676 RSC
- Y. He, J. Wang, Y. Si, X. Wang, H. Deng, Z. Sheng, Y. Li, J. Liu and J. Zhao, Int. J. Biol. Macromol., 2021, 178, 296–305 CrossRef PubMed
- T. Manon-Jensen, N. G. Kjeld and M. A. Karsdal, J. Thromb. Haemostasis, 2016, 14, 438–448 CrossRef PubMed
- Z. Zhao, F. Li, Q. Guo, Y. Zhou, Y. Miao, Y. Li, Z. Wang, R. Jiang, J.-F. Dong, X. Liu, J. Zhang and Y. Zhang, DNA Cell Biol., 2019, 38, 367–373 CrossRef CAS PubMed
- C. Ding, K. Cheng, Y. Wang, Y. Yi, X. Chen, J. Li, K. Liang and M. Zhang, Mater. Today Bio, 2024, 24, 100946 CrossRef
- X. Liu, M. Zheng, X. Wang, X. Luo, M. Hou and O. Yue, ACS Biomater. Sci. Eng., 2020, 6, 739–748 CrossRef PubMed
- A. Bernardo, A. L. Bergeron, C. W. Sun, P. Guchhait, M. A. Cruz, J. A. López and J. F. Dong, J. Thromb. Haemostasis, 2004, 2, 660–669 CrossRef PubMed
- C. Ding, M. Zhang, H. Tian and G. Li, Int. J. Biol. Macromol., 2013, 52, 319–326 CrossRef PubMed
- H. Tian, C. Li, W. Liu, J. Li and G. Li, Colloids Surf., B, 2013, 105, 259–266 CrossRef PubMed
- M. Yan, X. Jiang, G. Wang, A. Wang, X. Wang, X. Wang, X. Zhao, H. Xu, X. An and Y. Li, Carbohydr. Polym., 2020, 233, 115831 CrossRef PubMed
- S.-W. Tsai, R.-L. Liu, F.-Y. Hsu and C.-C. Chen, Biopolymers, 2006, 83, 381–388 CrossRef PubMed
- Y. Ran, W. Su, L. Ma, X. Wang and X. Li, Int. J. Biol. Macromol., 2021, 166, 1480–1490 CrossRef PubMed
- X. Wang, L. Sang, D. Luo and X. Li, Colloids Surf., B, 2011, 82, 233–240 CrossRef PubMed
- Z. Hu, D.-Y. Zhang, S.-T. Lu, P.-W. Li and S.-D. Li, Mar. Drugs, 2018, 16, 273 CrossRef
- Z. Hu, S. Lu, Y. Cheng, S. Kong, S. Li, C. Li and L. Yang, Molecules, 2018, 23, 3147 CrossRef
- B. Lorkowska-Zawicka, K. Kamiński, J. Ciejka, K. Szczubiałka, M. Białas, K. Okoń, D. Adamek, M. Nowakowska, J. Jawień, R. Olszanecki and R. Korbut, Mar. Drugs, 2014, 12, 3953–3969 CrossRef
- J. Weißpflog, D. Vehlow, M. Müller, B. Kohn, U. Scheler, S. Boye and S. Schwarz, Int. J. Biol. Macromol., 2021, 171, 242–261 CrossRef
- X. Song, Y. Zhao, Y. Liu, W. Zhang, X. Yuan, L. Xu and J. Zhang, Carbohydr. Polym., 2021, 273, 118615 CrossRef CAS PubMed
- L. Ding, Y. Huang, X. Cai and S. Wang, Carbohydr. Polym., 2019, 208, 133–141 CrossRef CAS
- J. B. M. Rocha Neto, G. G. Lima, A. Fiamingo, L. G. L. Germiniani, T. B. Taketa, R. A. Bataglioli, G. A. T. da Silveira, J. V. L. da Silva, S. P. Campana-Filho, O. N. Oliveira and M. M. Beppu, Int. J. Biol. Macromol., 2021, 172, 154–161 CrossRef CAS
- T. U. Wani, A. H. Pandith and F. A. Sheikh, J. Drug Delivery Sci. Technol., 2021, 65, 102730 CrossRef CAS
- H. Hattori and M. Ishihara, Biomed. Mater., 2015, 10, 015014 CrossRef PubMed
- J. Yang, F. Tian, Z. Wang, Q. Wang, Y.-J. Zeng and S.-Q. Chen, J. Biomed. Mater. Res., Part B, 2008, 84B, 131–137 CrossRef CAS PubMed
- Z. Zhang, X. Zhao, Z. Song, L. Wang and J. Gao, Biomed. Mater., 2024, 19, 055024 CrossRef CAS PubMed
- D. Tripathi, A. Sharma, P. Tyagi, C. S. Beniwal, G. Mittal, A. Jamini, H. Singh and A. Tyagi, AAPS PharmSciTech, 2021, 22, 138 CrossRef CAS
- H. Yan, Q. Wang, W. Li, N. Li, P. Huang and J. Xiao, J. Mater. Chem. B, 2024, 12, 8757–8766 RSC
- R. M. Abdel-Rahman, A. M. Abdel-Mohsen, J. Frankova, F. Piana, L. Kalina, V. Gajdosova, L. Kapralkova, M. A. Thottappali and J. Jancar, Biomacromolecules, 2024, 25, 3449–3463 CrossRef CAS PubMed
- M. Andonegi, K. L. Heras, E. Santos-Vizcaíno, M. Igartua, R. M. Hernandez, K. de la Caba and P. Guerrero, Carbohydr. Polym., 2020, 237, 116159 CrossRef PubMed
- E. Devernois and T. Coradin, Gels, 2023, 9, 518 CrossRef PubMed
- F. Chicatun, C. E. Pedraza, C. E. Ghezzi, B. Marelli, M. T. Kaartinen, M. D. McKee and S. N. Nazhat, Biomacromolecules, 2011, 12, 2946–2956 CrossRef PubMed
- F. Chicatun, E. Rezabeigi, N. Muja, M. T. Kaartinen, M. D. McKee and S. N. Nazhat, Emergent Mater., 2019, 2, 245–262 CrossRef
- W. Tan, R. Krishnaraj and T. A. Desai, Tissue Eng., 2001, 7, 203–210 CrossRef PubMed
- L. Shen, H. Bu, Y. Zhang, P. Tang and G. Li, Polym. Compos., 2021, 42, 4448–4460 CrossRef
- Y. Zhou, W. Liu, B. Gan, Y. Wang, Z. Fan, Y. Yang, X. Xiong, Y. Li, H. Chen, M. Yu, X. Peng and Y. Zhou, J. Mater. Sci., 2022, 57, 13570–13585 CrossRef
- H. Cheng, X. Pan, Z. Shi, X. Huang, Q. Zhong, H. Liu, Y. Chen, Q. Lian, J. Wang and Z. Shi, Carbohydr. Polym., 2022, 284, 118953 CrossRef PubMed
- Y. Zhang, C. Yang, M. Gu, X. Zhang, X. Zhang and G. Li, Collagen Leather, 2023, 5, 29 CrossRef
- M. Pozza and R. W. J. Millner, Eur. J. Emerg. Med., 2011, 18, 31–33 CrossRef PubMed
- M. Mardani, H. R. Eftekharian, M. Naseri, S. M. H. Hosseini, H. Mohammadi, H. Danesteh, N. Ghadimi and S. Fazel, Surgery, 2022, 172, 1007–1014 CrossRef
- C. Yang, Y. Zhang, X. Zhang, P. Tang, T. Zheng, R. Ran and G. Li, Carbohydr. Polym., 2023, 320, 121231 CrossRef
- X. Zhang, C. Yang, X. Zeng and G. Li, Carbohydr. Polym., 2024, 343, 122409 CrossRef
- X. Du, L. Wu, H. Yan, Z. Jiang, S. Li, W. Li, Y. Bai, H. Wang, Z. Cheng, D. Kong, L. Wang and M. Zhu, Nat. Commun., 2021, 12, 4733 CrossRef PubMed
- Y. Du, Y. Bai, S. Lang, D. Xing, L. Ma, K. Li, J. Peng, X. Li and G. Liu, Biomacromolecules, 2023, 24, 5313–5327 CrossRef
- X. Liu, N. Dan and W. Dan, Mater. Sci. Eng., C, 2017, 70, 689–700 CrossRef PubMed
- C. Yang and G. Li, J. Appl. Polym. Sci., 2024, 141, e55567 CrossRef
- Y. Ran, W. Su, L. Ma, Y. Tan, Z. Yi and X. Li, Int. J. Biol. Macromol., 2021, 189, 380–390 CrossRef PubMed
- L. A. Reis, L. L. Y. Chiu, Y. Liang, K. Hyunh, A. Momen and M. Radisic, Acta Biomater., 2012, 8, 1022–1036 CrossRef
- Y. Li, M. Cai, H. Liu and X. Liu, Int. J. Biol. Macromol., 2023, 238, 124138 CrossRef
- P. Tang, T. Zheng, L. Shen and G. Li, Polym. Degrad. Stab., 2021, 189, 109614 CrossRef
- R. Ran, Y. Xiong, T. Zheng, P. Tang, Y. Zhang, C. Yang and G. Li, Food Hydrocolloids, 2024, 147, 109326 CrossRef
- Y. Fan, Q. Lu, W. Liang, Y. Wang, Y. Zhou and M. Lang, Eur. Polym. J., 2021, 157, 110619 CrossRef
- J. Si, Y. Yang, X. Xing, F. Yang and P. Shan, Polym. Degrad. Stab., 2019, 166, 73–85 CrossRef
- Y. Yang, A. C. Ritchie and N. M. Everitt, Mater. Sci. Eng., C, 2021, 121, 111846 CrossRef PubMed
- Y. Zhang, L. Shen, Y. Cheng and G. Li, Polym. Degrad. Stab., 2021, 193, 109742 CrossRef CAS
- C. Hou, L. Gao, Z. Wang, W. Rao, M. Du and D. Zhang, J. Food Process Eng., 2020, 43, e13086 CrossRef
- T. Zheng, P. Tang and G. Li, J. Appl. Polym. Sci., 2022, 139, e52995 CrossRef
- I. Leceta, P. Guerrero, I. Ibarburu, M. T. Dueñas and K. de la Caba, J. Food Eng., 2013, 116, 889–899 CrossRef
- S. Lv, M. Cai, F. Leng and X. Jiang, Carbohydr. Polym., 2022, 288, 119369 CrossRef PubMed
- R. Cassano, P. Perri, E. Scarcello, P. Piro, R. Sole, F. Curcio and S. Trombino, Polymers, 2024, 16, 1770 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4bm01211a |
| This journal is © The Royal Society of Chemistry 2025 |