
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An immunomodulatory encapsulation system to deliver human iPSC-derived dopaminergic neuron progenitors for Parkinson's disease treatment†
Emily A.
Atkinson
 *ab,
Holly N.
Gregory
ab,
Lara N.
Carter
ab,
Rachael E.
Evans
ab,
Victoria H.
Roberton
ab,
Rachael
Dickman
a and
James B.
Phillips
*ab
*ab,
Holly N.
Gregory
ab,
Lara N.
Carter
ab,
Rachael E.
Evans
ab,
Victoria H.
Roberton
ab,
Rachael
Dickman
a and
James B.
Phillips
*ab
aUCL School of Pharmacy, University College London, London, UK. E-mail: emily.atkinson@ucl.ac.uk; jb.phillips@ucl.ac.uk
bUCL Centre for Nerve Engineering, University College London, London, UK
First published on 21st February 2025
Abstract
Parkinson's disease is a neurodegenerative condition associated with the progressive loss of dopaminergic neurons. This leads to neurological impairments with heightening severity and is globally increasing in prevalence due to population ageing. Cell transplantation has demonstrated significant promise in altering the disease course in the clinic, and stem cell-derived grafts are being investigated. Current clinical protocols involve systemic immunosuppression to prevent graft rejection, which could potentially be avoided by encapsulating the therapeutic cells in a locally immunosuppressive biomaterial matrix before delivery. Here we report the progression of an immunomodulatory encapsulation system employing ultrapure alginate hydrogel beads alongside tacrolimus-loaded microparticles in the encapsulation of dopaminergic neuron progenitors derived from human induced pluripotent stem cells (hiPSCs). The hiPSC-derived progenitors were characterised and displayed robust viability after encapsulation within alginate beads, producing dopamine as they matured in vitro. The encapsulation system effectively reduced T cell activation (3-fold) and protected progenitors from cytotoxicity in vitro. The alginate bead diameter was optimised using microfluidics to yield spherical and monodisperse hydrogels with a median size of 215.6 ± 0.5 μm, suitable for delivery to the brain through a surgical cannula. This technology has the potential to advance cell transplantation by locally protecting grafts from the host immune system.
Introduction
Parkinson's disease is a progressive neurodegenerative condition affecting at least 1% of the population over 65 years old and is characterised by the irreversible focal degradation of A9 dopaminergic neurons of the substantia nigra pars compacta in the ventral midbrain. The resulting dopamine deficiency leads to motor impairments including bradykinesia, rigidity, postural instability and rest tremors.1 In addition, patients may present non-motor symptoms such as depression, anxiety, dementia, and insomnia.2 Cases of Parkinson's disease are predicted to rise to 13 million worldwide by 2040,3 emphasising the urgent need for effective therapies.Cell therapy remains at the forefront of clinical and preclinical research into Parkinson's disease treatment. Dopaminergic neurons or their precursors are transplanted directly into the brain, with the intention that they eventually innervate the striatum and produce dopamine.4 Clinical trials investigating this treatment have been undertaken since the 1980s, reporting a range of outcomes from graft-induced dyskinesias to patient benefits persisting for decades (reviewed by Barker et al.).5 In earlier trials, the source of these cells was often human foetal ventral mesencephalic tissue, which while efficacious in some patients, presents practical and ethical issues for widespread adoption. Alternative cell sources with wider availability and potential for standardised production have been sought, with a particular focus on dopaminergic neural progenitors derived from human induced pluripotent stem cells (hiPSCs).6
Robust protocols have been developed to differentiate hiPSCs into dopaminergic progenitors in vitro, encompassing both embryonic and induced pluripotent stem cells.7,8 Moreover, grafts derived from human pluripotent stem cells have produced functional improvements in rodent,9,10 and primate models of Parkinson's disease.11 A clinical trial of dopaminergic progenitors derived from hiPSCs for Parkinson's disease was initiated in 2018,12 and derived from human embryonic stem cells in 2022 (STEM-PD).13 In both cases, therapeutic cells were injected directly into the brain in suspension through a surgical cannula. Accordingly, this technology may advance Parkinson's disease treatment and alleviate the drawbacks of foetal tissue grafts.
Immunosuppression and graft protection are key aspects of cell grafting for Parkinson's disease, and this presents an opportunity for biomaterials to be explored as a way to improve the delivery of these therapeutics directly into the brain.14 Although the brain has a degree of immune privilege, at least short-term immunosuppression is likely to be beneficial for graft survival.15 Patients in major recent trials of foetal tissue have received systemic immunosuppression for approximately 12 months post-transplant.16 Participants receiving the first iPSC-based therapy for Parkinson's disease (clinical trial ID: UMIN000033565) will be immunosuppressed with tacrolimus for 12 months.
Participants in the STEM-PD trial will also undergo a 12-month immunosuppressive regime, with the systemic delivery of tacrolimus, alongside other immunosuppressants.13 Despite its necessity, the systemic administration of immunosuppressants (and tacrolimus in particular) in solid organ transplantation has been associated with substantial adverse effects including neuro- and nephrotoxicity, hypertension, and diabetogenesis.17 Rather than systemically delivering immunosuppressants, local delivery of tacrolimus within a controlled-release formulation could be explored. This may reduce the associated side effects by vastly reducing the dosage required to protect the transplanted cells from the host immune system and minimise side effects.
Moreover, this local immune isolation approach could be extended by encapsulating the therapeutic cells within a semi-permeable hydrogel. This could act as a physical barrier against host immune cells whilst allowing the diffusion of oxygen and nutrients into the hydrogel for the survival of the encapsulated cells and the diffusion of dopamine and therapeutic factors out. Alginate has been extensively investigated in this capacity for the transplantation of pancreatic islet cells and has yielded immune protection of functional islets in both primates and humans.18,19 Alginate hydrogels have been well characterised and can be formulated with the appropriate mechanical and physical properties for brain delivery.20–23 A recent clinical trial investigated the intra-striatal delivery of alginate-encapsulated porcine choroid plexus cells in patients with Parkinson's disease. Although the efficacy of the transplanted cells was not established, the technology was determined to be safe and well-tolerated in humans.24
We have previously reported an immunoprotective cell encapsulation system comprised of an alginate hydrogel and tacrolimus-loaded nanoparticles, which displayed appropriate mechanical properties and permeability while maintaining the viability of encapsulated SH-SY5Y neuroblastoma cells over two weeks in vitro.20 The present study progresses that technology by investigating the encapsulation of dopaminergic progenitors derived from hiPSCs, equivalent to those used in current clinical trials for Parkinson's disease. Furthermore, it aimed to optimise the volume and diameter of the alginate hydrogel beads for clinical translation by identifying a method of alginate bead production that maintains encapsulated cell viability, is suitable for passing through a surgical cannula for direct delivery into the brain (currently used in clinical trials) and has a diameter which avoids the formation of a necrotic core (<200 μm).14
In this work, dopaminergic neuron progenitors were incorporated into the alginate encapsulation system and combined with novel tacrolimus-loaded polycaprolactone microparticles manufactured using a more robust single emulsion technique. We hypothesised that encapsulating hiPSC-derived dopaminergic neuron progenitors in alginate beads with tacrolimus microparticles would modulate T cell responses and improve encapsulated cell survival in an in vitro model of T-cell mediated immune rejection. To test this hypothesis, we investigated the capacity of the biomaterial system to protect the dopaminergic neuron progenitors from a T cell response in vitro, alongside its ability to modulate T cell population number. The technology was compared with unencapsulated cells and cells encapsulated in alginate only. Finally, the production methodology for alginate beads was optimised using a coaxial flow reactor to reduce their size and improve homogeneity.
Materials and methodology
Materials
The ESI (ESI Tables 1 and 2†) contains details on materials including suppliers and catalogue numbers.hiPSC differentiation and characterisation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 at 80% confluency. All cultures were maintained in a humidified incubator at 37 °C with 5% CO2. hiPSCs were differentiated into dopaminergic neuron progenitors (day 16), then mature dopaminergic neurons (day 35) adapting the protocol from Kirkeby et al. in Scheme 1 and Table 1.7,8
:4 at 80% confluency. All cultures were maintained in a humidified incubator at 37 °C with 5% CO2. hiPSCs were differentiated into dopaminergic neuron progenitors (day 16), then mature dopaminergic neurons (day 35) adapting the protocol from Kirkeby et al. in Scheme 1 and Table 1.7,8
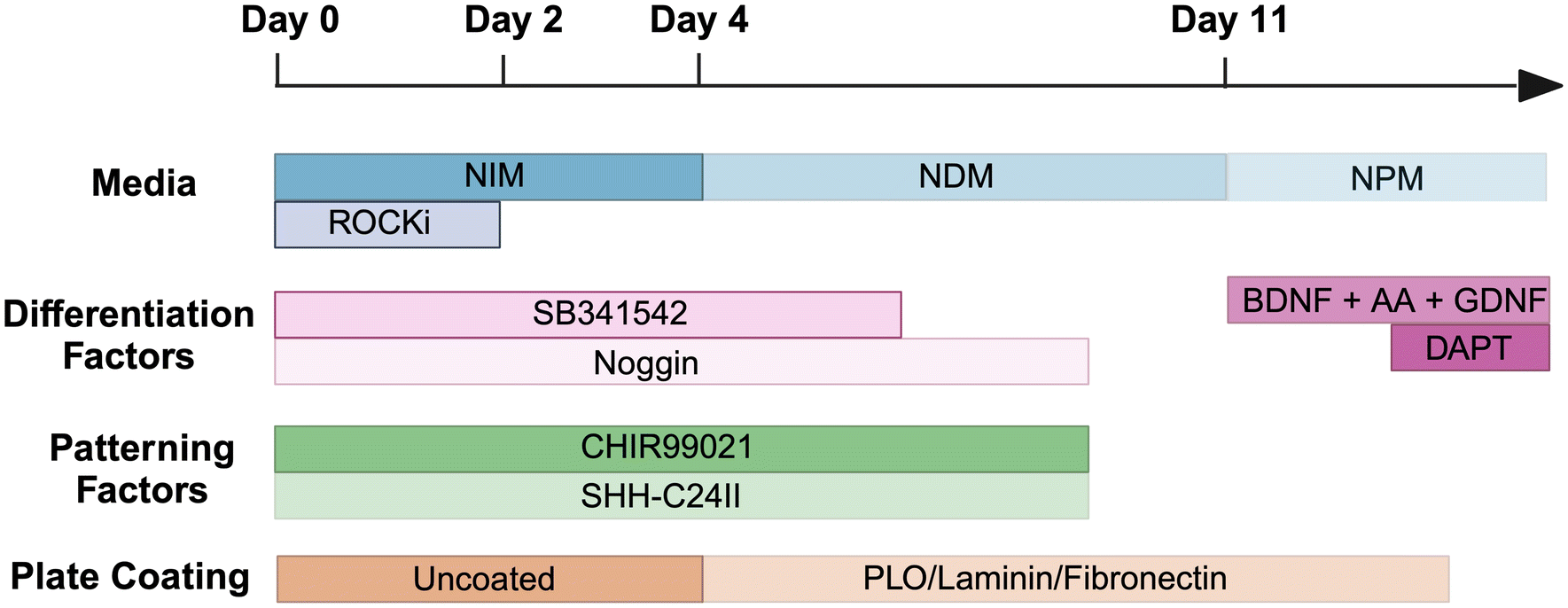 | ||
| Scheme 1 Schematic of differentiation methodology from hiPSCs to dopaminergic neurons, adapted from Kirkeby et al.7,8 Abbreviations include neural induction media (NIM), neural differentiation media (NDM), neural proliferation media (NPM), ROCK inhibitor (ROCKi), brain-derived neurotrophic factor (BDNF), ascorbic acid (AA), glial cell line-derived neurotrophic factor (GDNF), poly-L-ornithine (PLO). | ||
| Neural induction media (NIM) | Neural proliferation media (NPM) | Neural differentiation media (NDM) |
|---|---|---|
| DMEM/F12: Neurobasal (1:1) 1× N2 (1:100) |
DMEM/F12: Neurobasal (1:1) 0.5× N2 (1:200) |
Neurobasal |
| 1× B27 (1:50) |
0.5× B27 (1:100) |
1× B27 (1:50) |
| 2 mM L-glutamine (1:100) |
2 mM L-glutamine (1:100) |
2 mM L-glutamine (1:100) |
In summary, on day 0, hiPSCs were passaged with EDTA (0.5 mM) then cultured in Neural Induction Medium (NIM) (Table 1) and supplemented with Y-27632 (10 μM), SB431542 (10 μM), Noggin (100 ng mL−1), CHIR99021 (200 ng mL−1) and SHH-C24II (200 ng mL−1). On day 2, cells were suspended in the same media without Y-27632, and began to form embryoid bodies. During this time, plates were coated with poly-L-ornithine (PLO) in H2O (15 μg mL−1) and were incubated for 48 h at 37 °C. Wells were then washed with H2O thrice then fibronectin (0.5 mg mL−1) and laminin (5 μg mL−1) in PBS were added for a further 48 h at 37 °C. On day 4, cells were seeded onto the coated plates and cultured in Neural Proliferation Media (NPM) (Table 1) plus SB431542 (10 μM), Noggin (100 ng mL−1), CHIR99021 (200 ng mL−1) and SHH-C24II (200 ng mL−1). On day 11, cells were replated onto coated plates and media changed to Neural Differentiation Medium (NDM) (Table 1) plus GDNF (10 ng mL−1), BDNF (20 ng mL−1) and ascorbic acid (0.2 mM). On day 14 the medium was replaced, with the addition of DAPT (1 μM), and cells were maintained in this medium for future use (Scheme 1).
| Antibody | Dilution |
|---|---|
| β-III-Tubulin (raised in rabbit) | 1:300 |
| β-III-Tubulin (raised in mouse) | 1:300 |
| LMX1A (raised in rabbit) | 1:400 |
| Nestin (raised in mouse) | 1:400 |
| OCT3/4 (raised in goat) | 1:100 |
| Tyrosine Hydroxylase (raised in mouse) | 1:500 |
| Dylight 488 horse anti-mouse IgG | 1:400 |
| Dylight 594 goat anti-rabbit IgG | 1:400 |
| Dylight 594 horse anti-mouse IgG | 1:400 |
| Dylight 594 horse anti-goat IgG | 1:200 |
| Goat anti-mouse Alexa Fluor Plus 488 | 1:200 |
| Goat anti-rabbit Alexa Fluor Plus 594 | 1:200 |
Tacrolimus-loaded microparticles
000 rpm using a Silverson high-speed laboratory mixer. The solution containing polymer and drug was streamed into the emulsifier over 7–9 seconds, and then mixing continued for two minutes. The mixture was then stirred at 500 rpm for four hours, before being filtered through a 100 μm cell strainer. The microparticles were then washed with excess distilled water and collected via centrifugation (3 minutes, 3000 rpm). After repeating this step five times, the particles were frozen in liquid nitrogen and freeze-dried for 72 hours, prior to storage at −20 °C.
To assess the drug release profile, 20.0 mg of microparticles (n = 3) were added to 50 mM ammonium bicarbonate release buffer (1 mL) in microtubes and placed in a shaking incubator at 37 °C and 75 rpm. Every 2–3 days the microtubes were centrifuged at 12500 rpm for five minutes, the buffer completely removed and then replaced with fresh buffer. Release samples were stored at −20 °C until analysis.
The concentration of tacrolimus in the release buffer was determined using ultra-high performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS), with a Shimadzu Nexera X2 UHPLC/Shimadzu LCMS 8060 and a Phenomenex Kinetex C8 (50 × 2.1 mm) column with 5 μm pore size, with the column oven set at 50 °C. The mobile phase used for analysis comprised water with 0.1% formic acid (A) and acetonitrile:water (95:5) with 0.1% formic acid (B) under gradient elution according to Table 3, with a flow rate of 0.4 mL min−1. An internal standard of tolbutamide in acetonitrile (500 ng mL−1) was used in all calibration samples, standards and blanks.
| Time (min) | Solvent A: Water with 0.1% formic acid (%) | Solvent B: Acetonitrile: Water (95:5) with 0.1% formic acid: (%) |
|---|---|---|
| 0 | 98 | 2 |
| 0.3 | 98 | 2 |
| 1.1 | 5 | 95 |
| 1.75 | 5 | 95 |
| 1.8 | 98 | 2 |
| 2.5 | 98 | 2 |
Cell encapsulation system and in vitro efficacy
Live/Dead™ assay
At 24 h after encapsulation, cells were stained using a Live/Dead™ Cell Imaging Kit (Thermo Fisher #04511) as per the manufacturer's instructions to assess cell viability. In darkness, encapsulated cells were washed with PBS and incubated with the staining solution (1:1000) for 15 minutes at 37 °C. The staining solution was removed and cells were washed with culture media. Cells were counted from three planes in at least three alginate beads per experimental group using a fluorescence microscope (Zeiss Axiolab.A1).
T cell isolation, purification and culture
Human CD3+ T cells were isolated from human peripheral blood mononuclear cells (PBMCs). T cells were separated using the EasySep™ Human CD3 Positive Selection Kit II (STEMCELL™ Technologies) by following the manufacturer's instructions. In brief, the selection cocktail (100 μL) was added to PBMCs (1 × 108 cells per mL) and incubated at RT for three minutes. RapidSpheres™ (60 μL mL−1) were introduced to the PBMCs and incubated for a further three minutes at RT. RPMI-160 medium with 1% penicillin/streptomycin/S, 1% L-glutamine, 1% non-essential amino acids and 10% fetal bovine serum (to 2.5 mL) was added and the T cells were extracted using a magnet for three minutes. The supernatant was removed, and the extraction process was repeated thrice. Cells were cultured for up to two weeks in the RPMI-based media.T cell assay
T cells were seeded into 12-well plates (40000 cells per well) containing no cells, unencapsulated dopaminergic neuron progenitors (50000 cells per well), encapsulated dopaminergic neuron progenitors, or encapsulated dopaminergic neuron progenitors with tacrolimus-loaded PCL microparticles (0.25 mg). Cultures were monitored over five days using the Sartorius Incucyte® Live-Cell Analysis instrument and the number of T cells in each condition was determined at the end of the experiment using a haemocytometer. The viability of the dopaminergic neuron progenitors was measured using the LIVE/DEAD™ assay, as described above.
Alginate bead size optimisation
:3–1:20) and flow rates of both alginate (10–40 μL min−1) and sunflower oil (100–400 μL min−1) were optimised. The alginate beads were then washed with hexane (3 × 1 mL) and PBS (3 × 1 mL). The alginate bead size was measured by laser diffraction (Mastersizer). Images were taken at ×10 using a phase contrast light microscope (EVOS), and alginate beads were measured with ImageJ.25
Data analysis
Normality was determined using the Shapiro–Wilk test. If normal distribution was shown then either a 1- or 2-way statistical analysis of variance (ANOVA) was used as appropriate with a post-hoc test by either a Dunnett's, Tukey's or Bonferroni multiple comparison test. Degrees of significance were assessed by four different rating values: *p < 0.05, **p < 0.01 ***p < 0.001 and ****p < 0.0001.Results
CGT-RCiB-10 hiPSCs were differentiated first into dopaminergic progenitors (day 14–16) then dopaminergic neurons (day 35) using an established protocol.7 Cell phenotype was assessed using phase contrast imaging and immunocytochemistry, alongside dopamine production and cytokine release. Cell morphology changed markedly over 16 days in the presence of various patterning factors (Fig. 1). Day 0 hiPSCs formed small colonies with hexagonal cell morphology (yellow arrow), and by day 2–4, floating embryoid bodies were formed (red arrow). On day 4, these cell clusters were triturated and attached to a laminin and PLO-coated plate. By day 14–16, through exposure to various factors, the cells displayed a spiny morphology with neural network formation (purple arrows) which are typical of dopaminergic progenitors. | ||
| Fig. 1 Differentiation of hiPSCs into dopaminergic progenitors. Phase contrast micrographs displaying hiPSCs (yellow arrow) formed embryoid bodies (red arrow) between days 1 and 4, and dopaminergic progenitors formed a network of interconnecting neurites (purple arrows) by day 14. Scale bars = 100 μm. | ||
Differentiation was also assessed using immunocytochemistry, comparing undifferentiated hiPSCs with hiPSC-derived dopaminergic progenitors (day 16) and mature dopaminergic neurons (day 30–35). hiPSCs expressed the pluripotency marker Oct4 alongside the neural stem cell marker Nestin, and day 16 dopaminergic progenitors were positive for the neuronal cytoskeletal protein βIII-tubulin. At day 30–35 of differentiation, cells also expressed βIII-tubulin and LMX1A (Fig. 2A). Further immunocytochemistry on day 16 revealed that dopaminergic progenitors also expressed the midbrain dopaminergic neuron marker LMX1A with nuclear localisation at this time point, as well as tyrosine hydroxylase (TH) (Fig. 2B).
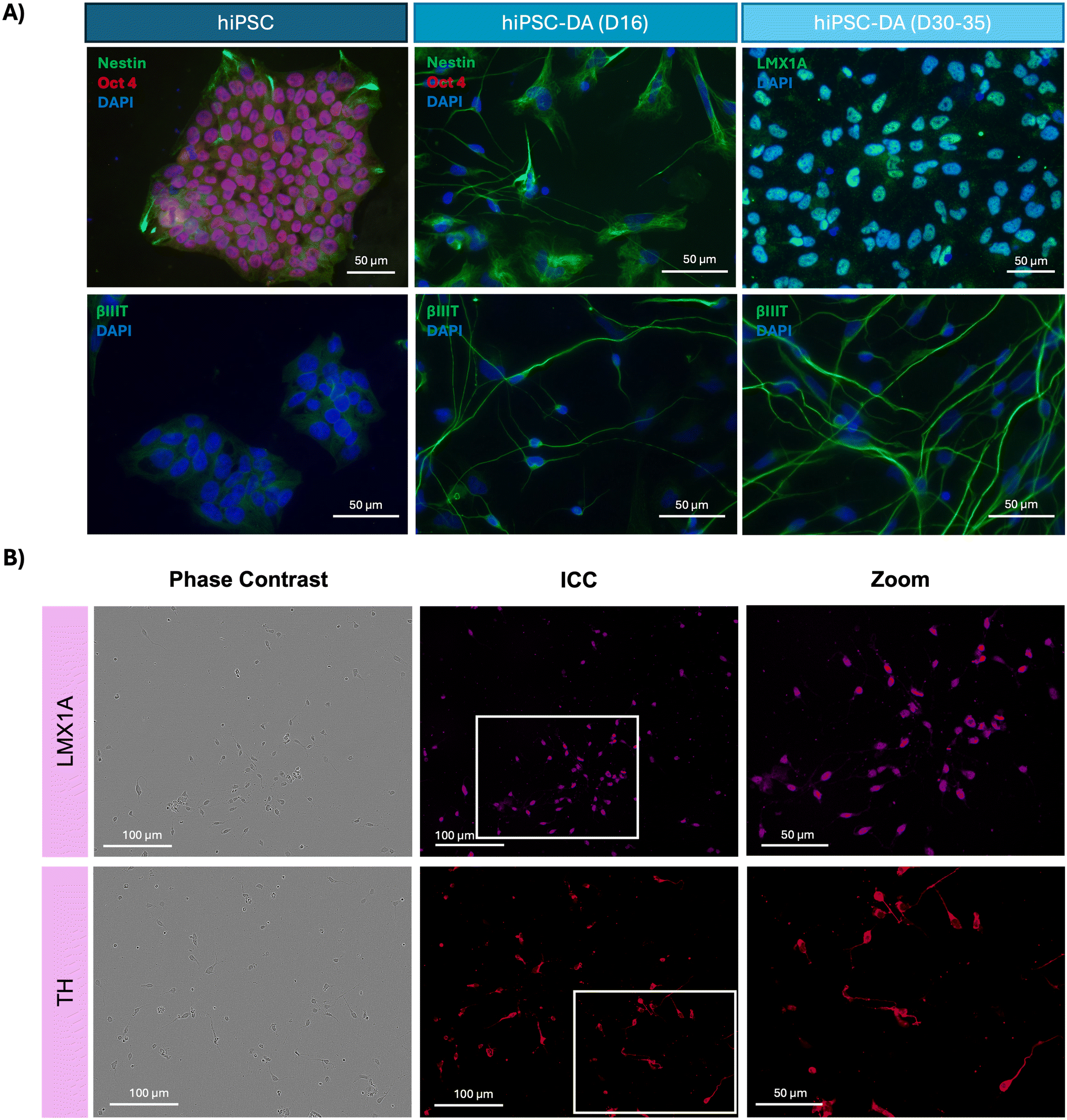 | ||
| Fig. 2 Immunocytochemistry analysis of hiPSCs, dopaminergic progenitors and dopaminergic neurons. (A) Fluorescence micrographs demonstrating hiPSCs express the pluripotency marker Oct4 and neural stem cell marker Nestin, day 16 hiPSC-DAs express neuronal marker βIII-tubulin, and day 35 hiPSC-DAs express βIII-tubulin and midbrain dopaminergic neuron marker LMX1A. (B) Phase contrast images and fluorescence micrographs displaying day 16 hiPSC-DAs express and midbrain dopaminergic neuron marker LMX1A and the dopaminergic neuron marker tyrosine hydroxylase (TH). | ||
Thereafter, the concentration of dopamine in the secretome of these cultures was determined by ELISA to confirm the dopaminergic functionality of the neurons generated by this protocol (Fig. 3A). The day 35 hiPSC-derived dopaminergic neuron secretome contained 217 ± 31.7 pg mL−1 dopamine, amounting to a seven-fold increase (p < 0.0001) over the 29.1 ± 6.1 pg mL−1 dopamine detected from undifferentiated hiPSCs. The secretome from each cell population was also analysed for the presence of 36 individual human cytokines and chemokines, of which four were detected (Fig. 3B and Fig. S1†). CCL5, CXCL12, IL-1β and CD40 were all present in hiPSC secretome, where levels of CCL5 were highest. The concentration of this chemokine was reduced in both the day 16 dopaminergic progenitor and day 35 dopaminergic neuron secretome, and IL-1β was undetectable in these cultures. In comparison, levels of CXCL12 were elevated in the differentiated cell types and CD40, while present in day 16 progenitor culture media, was absent in day 35 dopaminergic neuron media.
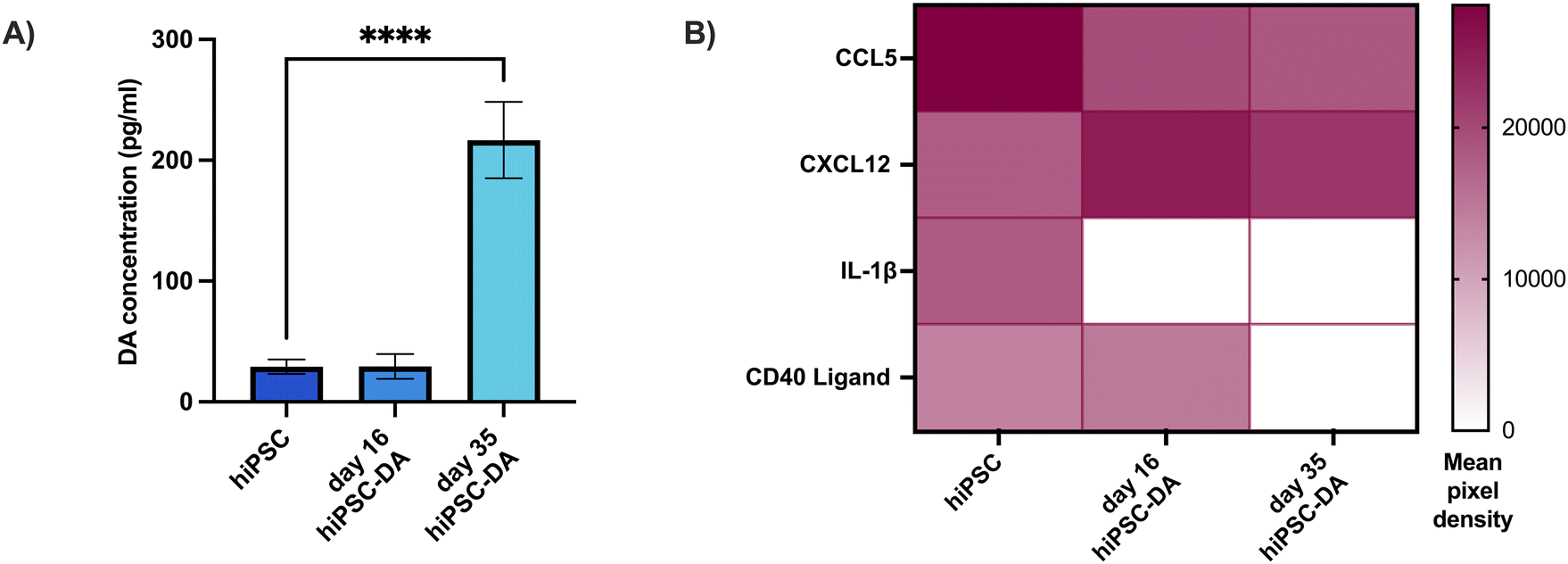 | ||
| Fig. 3 Dopamine production and cytokine array for hiPSCs, dopaminergic progenitors and functional dopaminergic neurons. (A) In vitro dopamine production was significantly increased in day 35 hiPSC-DAs compared with undifferentiated hiPSCs (one-way ANOVA with Dunnett's post-hoc test, n = 3, mean ± SD, **** p < 0.0001). (B) Heat map illustrating the detection of cytokines in the secretome throughout differentiation towards dopaminergic neurons. Data expressed as the mean of the two spots on the array for each cytokine (n = 1). | ||
To protect the grafted progenitors from the host immune system pharmacologically, polycaprolactone microparticles loaded with the immunosuppressant tacrolimus were manufactured, for use in conjunction with the encapsulation system (Fig. 4). A single emulsion fabrication technique was employed, which generated discrete and spherical microparticles (Fig. 4A), with an average diameter of 6.98 ± 0.33 μm (mean ± SEM) and a maximum diameter of 29.41 μm (Fig. 4B). Tacrolimus was eluted from the microparticles in a sustained manner, reaching a cumulative total of 900 ng of immunosuppressant released per milligram of material over three weeks (Fig. 4C).
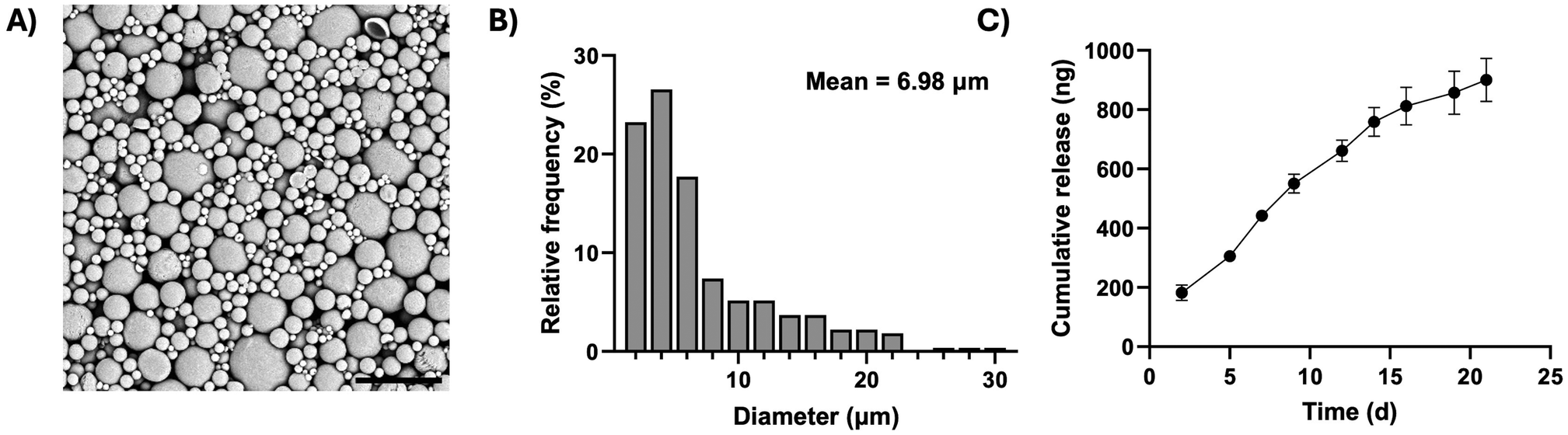 | ||
| Fig. 4 Synthesis and characterisation of tacrolimus-loaded microparticles. (A) Scanning electron micrograph of tacrolimus-loaded microparticles, scale bar = 30 μm. (B) Frequency distribution of microparticle diameter from scanning electron micrographs (n = 3 images, ∼90 measurements per image). (C) Cumulative release of tacrolimus per mg of microparticles over three weeks (n = 3 samples ± SD). | ||
The effect of encapsulation within alginate beads on the viability of dopaminergic progenitors was explored, alongside the capacity of the immunomodulatory encapsulation system to protect these cells in an in vitro model of immune response (Fig. 5). Day 16 hiPSC-derived dopaminergic progenitors were encapsulated within alginate beads, generated by dropwise release from a 31 G needle and crosslinking in a CaCl2 bath (Fig. 5A). The viability of the progenitors 24 hours after encapsulation was 79.58 ± 1.4%, a small but significant reduction compared to unencapsulated cells at 91.31 ± 0.92% (p < 0.0001) (Fig. 5B).
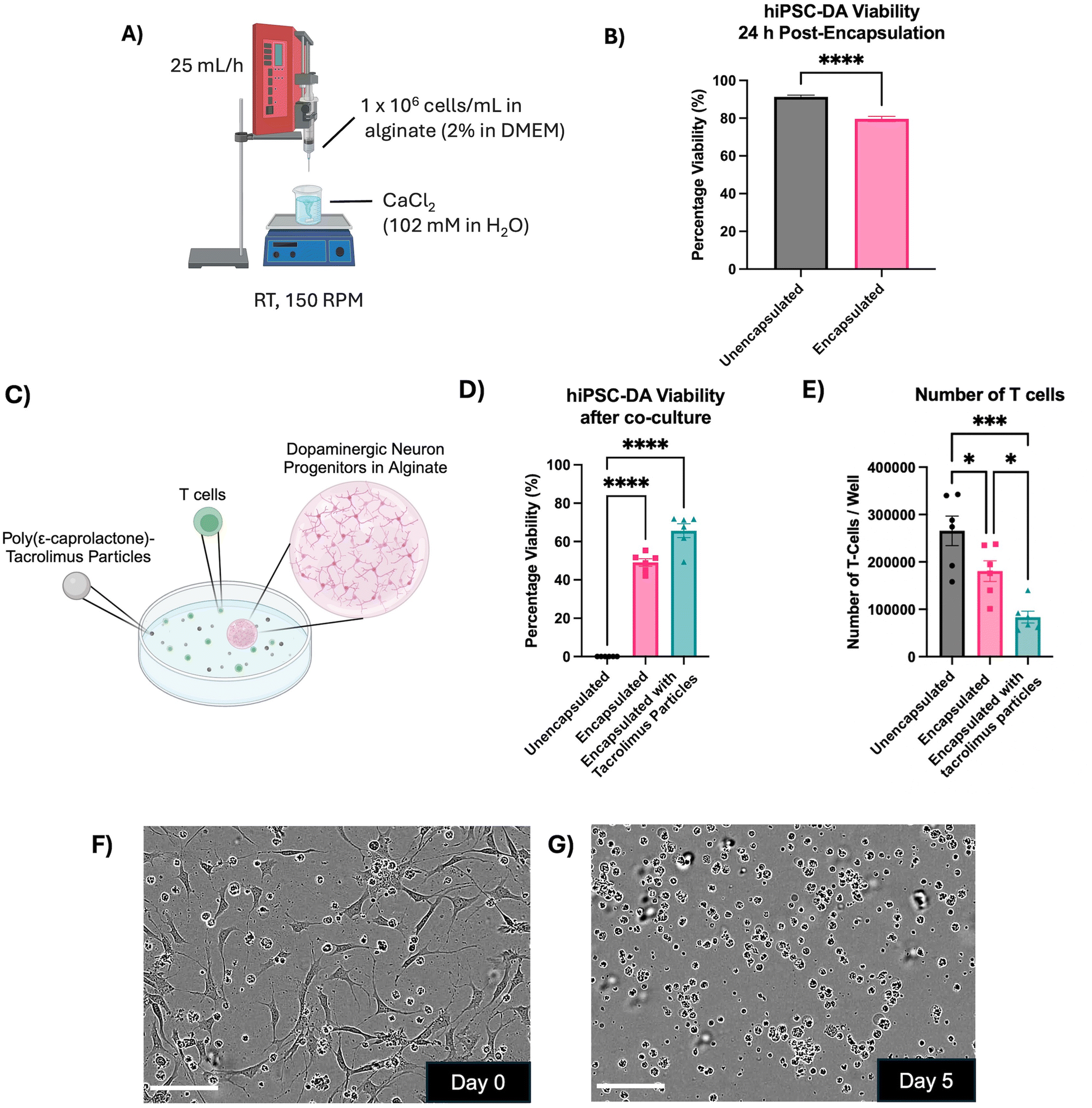 | ||
| Fig. 5 Immunomodulatory encapsulation system protects dopaminergic progenitors and reduces T cell activation in an in vitro model of immune response. (A) Schematic of the method for encapsulation of day 16 dopaminergic progenitors in alginate beads. (B) Viability of day 16 hiPSC-derived dopaminergic progenitors 24 h after encapsulation in alginate beads (two-tailed T test, n = 15 alginate beads or n = 6 wells from three independent experiments, mean ± SEM, **** p < 0.0001). (C) Schematic of in vitro immune response assay where dopaminergic neuron progenitors were encapsulated in alginate and cultured with tacrolimus-loaded microparticles (25 μg mL−1) and reactive T cells (40000 per well) for five days. (D) Viability of dopaminergic progenitors after five-day exposure to reactive human T cells (one-way ANOVA with Dunnett's multiple comparisons test, n = 6 alginate beads from two independent experiments, mean ± SEM, **** p < 0.0001). (E) Number of T cells per well after five days in culture (one-way ANOVA with Tukey's post-hoc test, n = 6 wells per condition from two independent experiments, mean ± SEM, * p < 0.05 and *** p < 0.001). (F) and (G) Phase contrast micrographs of the unencapsulated dopaminergic neuron progenitor and T cell coculture under ×20 magnification on day 0 and day 5. Scale bar = 100 μm. | ||
Cultures of mismatched human T cells were then introduced to either unencapsulated day 16 dopaminergic progenitors, progenitors encapsulated in alginate beads, or progenitors encapsulated in alginate beads alongside tacrolimus microparticles (Fig. 5C). After five days, unencapsulated progenitors had 0% viability. Progenitors encapsulated in alginate displayed 49.08 ± 1.97% viability (±SEM), which significantly increased to 65.7 ± 3.61% with the addition of tacrolimus microparticles (p < 0.0005) (Fig. 5D). The number of T cells cocultured with the unencapsulated dopaminergic neuron progenitors increased 6.6-fold from 40000 cells per well at the start of the experiment to 265750 ± 31100 (±SEM). The number of T cells increased to a lesser 4.5-fold extent, 180717 ± 21912 (±SEM), when cells were encapsulated in alginate and the increase was only 2-fold, 83583 ± 12569 (±SEM), with the addition of tacrolimus-loaded microparticles (Fig. 5E). Phase contrast images showed the co-culture of unencapsulated T cells and unencapsulated dopaminergic neuron progenitors on day 0 of the assay (Fig. 5F), and day 5 of the assay (Fig. 5G) at which point no dopaminergic neurons were observed to survive.
To enhance the clinical relevance of this technology, the encapsulation process was optimised to produce smaller and more consistent alginate beads (Fig. 6). Initially, the same syringe pump system and associated conditions were employed, and the alginate solution was extruded through needles of decreasing diameter (Fig. 6A). A reduction in median alginate bead size was observed with reducing needle diameter (Fig. 6B). The median alginate bead diameter decreased almost three-fold from 2475 μm using an 838 μm (internal diameter) needle, to 895 μm using a 133 μm needle. However, all needles produced a wide range of bead diameters – the smallest range was 45 μm, obtained with the 133 μm needle, and the largest was 1950 μm, using the 413 μm needle. To explore how far the correlation between needle diameter and bead size continued, pulled pipettes with smaller, decreasing internal diameters were investigated (Fig. 6C), showing a gradual decrease in alginate bead diameter from 1957 ± 197 μm with a 100 μm pipette to 1111 ± 322 μm with a 10 μm pipette (Fig. 6D).
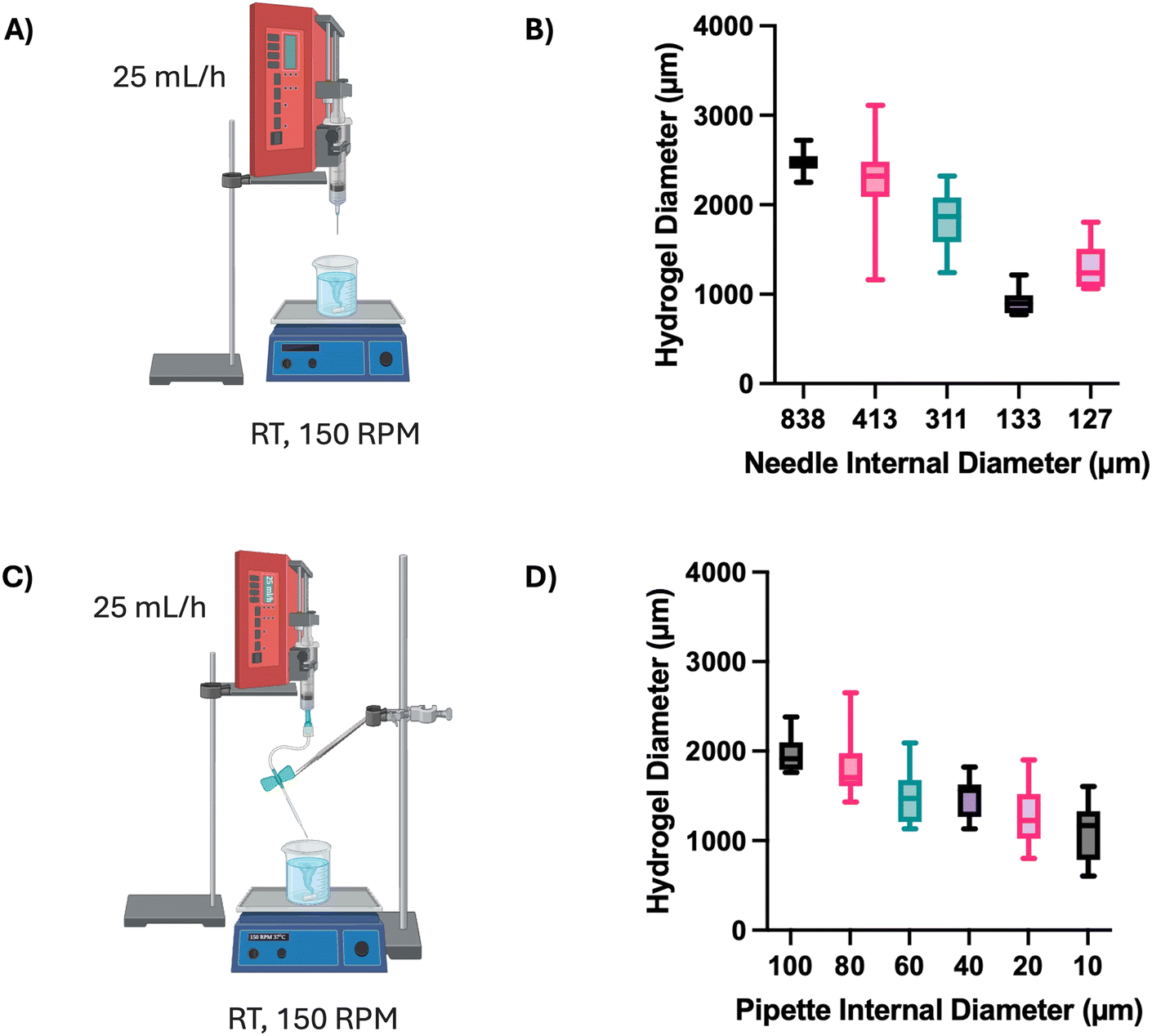 | ||
| Fig. 6 Development of alginate bead encapsulation system. (A) Schematic of alginate bead production with dropwise release from a needle. (B) Effect of needle internal diameter on alginate bead size (n = 20 beads per condition, central line = median, box = upper and lower quartiles). (C) Schematic of alginate bead production with dropwise release from a pulled glass pipette. (D) Effect of pipette internal diameter on alginate bead size (n = 10 beads per condition). | ||
To further decrease the alginate bead size and improve homogeneity, a coaxial flow reactor was created to generate hydrogel beads using microfluidics (Fig. 7). The reactor utilised a continuous phase of sunflower oil and a disperse phase of 2% alginate at various flow rates and flow rate ratios (Fig. 7A). At the outset, the flow rate ratio between disperse and continuous phases was varied. Increasing the flow rate ratio to or above 1:5 (alginate:sunflower oil) produced visibly discrete spherical beads (Fig. 7B). The hydrogel beads formed at lower ratios of 1:3 and 1:4 were interconnected, whereas ratios of 1:5 and 1:10 formed consistently distinct alginate beads throughout the process. By testing four conditions with varying flow rates of both the continuous and disperse phases, the flow rate ratio of 1:10 was selected for further optimisation of bead size and homogeneity (Fig. 7C). The hydrogel beads formed at each flow rate were measured by laser diffraction, and the D10, D50 and D90 percentiles by volume were calculated (Fig. 7D and E). Increasing flow rate proportionally increased the D50 value of the resulting alginate beads, with those formed at a total rate of 110 μL min−1 averaging 55.70 ± 18.42 μm compared with those at 440 μL min−1 at 215.56 ± 0.52 μm. Further, while the low flow rate in Condition 1 produced a large distribution in particle diameter with three distinct size classes, the higher flow rate in Condition 4 generated beads with a narrow diameter distribution and a single distinct size class. Increasing the total flow rate considerably impacted alginate bead diameter, where the mean particle size as measured by microscopy ranged from 312.55 μm ± 122.56 (20 μL min−1:200 μL min−1) to 134.20 μm ± 65.50 (30 μL min−1:300 μL min−1) (Fig. 7F). Notably, the lower flow rates in conditions 1 and 2 generated alginate beads with a cone-like morphology, whereas beads produced at higher flow rates were spherical.
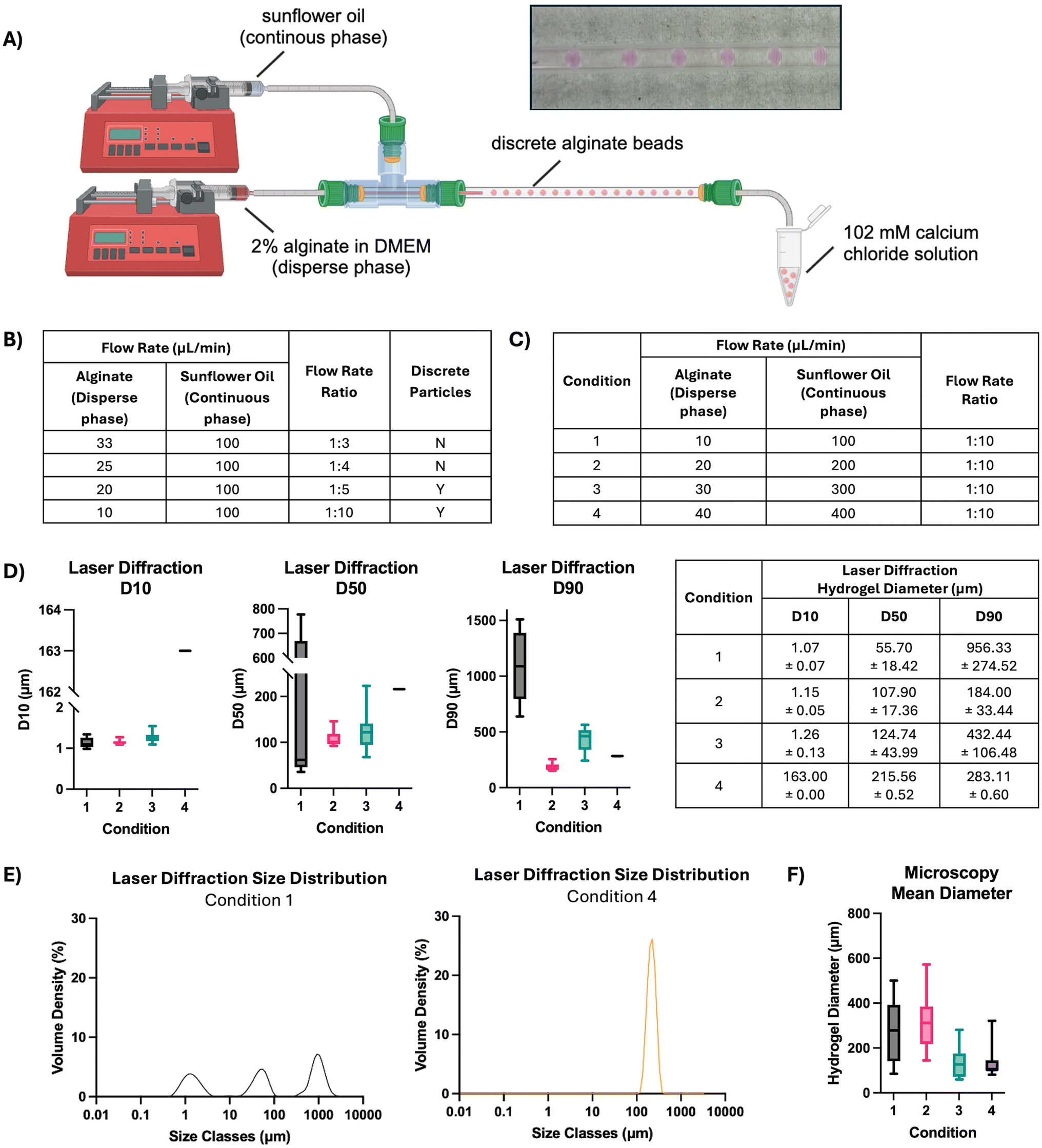 | ||
| Fig. 7 Development of coaxial reactor conditions to yield monodisperse alginate beads. (A) Schematic of alginate bead production using a coaxial flow reactor developed in-house. Image of distinct alginate beads in glass capillary. (B) Table detailing the effect of flow rate ratio between disperse and continuous phases on formation of discrete beads. (C) Table detailing the flow rates used to optimise the diameter of the alginate beads. (D) D10, D50 and D90 measurements and (E) size distribution of alginate beads, as determined by laser diffraction (n = 9 measurements). (F) Effect of total flow rate on alginate bead size determined by microscopy (n = 10 beads per condition, central line = median, box = upper and lower quartiles and whiskers represent the minimum and maximum values). | ||
Discussion
Day 16 dopaminergic progenitors were used in this study since equivalent cells have been shown to improve outcomes following neural transplantation in rat models of Parkinson's disease,26 and are currently being tested in clinical trials to treat Parkinson's disease patients.27,28 An established protocol was adapted to differentiate hiPSCs into day 16 dopaminergic progenitors and these were also further differentiated into dopaminergic neurons (day 35) for in vitro confirmation of functionality.7,8 This method was based on passing the cells through the floor plate stage, critical in the embryonic development of midbrain dopaminergic neurons.29–31 Immunocytochemical characterisation demonstrated that the hiPSCs expressed the transcription factor Oct4, which confirmed that the cells were pluripotent prior to differentiation.32 The cells also expressed Nestin, a cytoplasmic intermediate filament protein involved in the stabilisation of cytoskeletal architecture. Nestin is predominantly expressed by neural progenitors in the human brain, although it has also been detected elsewhere in both the brain,33 and other body tissues (reviewed in ref. 34). By day 16 the cells extended projections and were positive for βIII-tubulin,35 indicating that these day 16 progenitors were of neuronal phenotype. Furthermore, the dopaminergic neuron progenitors also displayed expression of tyrosine hydroxylase, an enzyme critical for the synthesis of dopamine, and LMX1A, a marker commonly used to identify mesencephalic dopaminergic progenitors,7 with nuclear localisation. At day 35, mature dopaminergic neurons were also positive for βIII-tubulin and LMX1A, and were confirmed to release dopamine in vitro. This is consistent with other reports demonstrating dopamine production in hiPSC-derived neurons at this timepoint.36,37Cytokine and chemokine levels were compared across the three differentiation stages. Secretion of the pro-inflammatory cytokine CCL5 was greatest in hiPSCs compared with differentiated cells, in line with a previous study.38 The homeostatic cytokine CXCL12 was elevated in the secretome of day 16 progenitors. This cytokine is expressed in the developing midbrain, the adult brain, and plays a role in the migration of A9 and A10 dopaminergic neurons.39,40 Secretion of the pro-inflammatory cytokine IL-1β was detected from the hiPSCs, which is also reported elsewhere,41 but was not detected in the differentiated cells. The CD40 ligand was elevated in the secretome of the hiPSCs and day 16 differentiated cells but not in the secretome of day 35 hiPSC-derived neurons. This was of particular note in this study as CD40 is involved in T cell regulation.42 Future studies could explore how cytokines from transplanted progenitors influence the host immune response, informing immunosuppression requirements.
As an approach to provide local immunosuppression to improve transplanted cell survival, polymeric microparticles loaded with tacrolimus were generated. Tacrolimus is widely used for immunosuppression, where it suppresses the activation and proliferation of T lymphocytes through calcineurin inhibition.43 It may also offer other beneficial effects in the context of treating Parkinson's disease, having been linked with protection and improved survival of dopaminergic neurons.44,45 The drug-loaded microparticles had an average diameter of 7 μm, which would allow them to readily pass through the delivery needles with diameters around 1000 μm employed in recent clinical trials.13,16,27 Steady release of tacrolimus was sustained from the microparticles for at least three weeks, which tends towards zero-order kinetics. This formulation has advantages over the use of tacrolimus-loaded nanoparticles that exhibited an initial burst release.20 The optimal dose of tacrolimus required for local immunosuppression is yet to be determined. Most studies using systemic immunosuppression aim to maintain a plasma concentration of 5–15 ng mL−1 but do not report the concentration of tacrolimus at a tissue level.46 Tacrolimus and other immunosuppressants are commonly delivered for 6–12 months post-transplantation in clinical studies, however, in vitro assessment of the immunogenicity of the cell therapy product used in the STEM-PD clinical trial has suggested that aggressive, long-term immunosuppression of this nature may not be required.47
As an approach to provide physical protection to improve cell transplantation, hiPSC-derived dopaminergic progenitors were encapsulated within alginate hydrogel beads, where they maintained high viability over 24 hours. Cell survival within the beads suggested that the encapsulation process was not overtly detrimental to viability and that the formed hydrogel structure permitted the exchange of oxygen and nutrients in culture. Crosslinked alginate has previously been shown to improve the viability of other injected cells when used as a carrier versus media alone.48 This feature may be valuable for improving the outcomes of cell transplantation through physical protection during the procedure as well as immunoisolation of the graft.
To model key aspects of the host immune response to allogeneic grafts, the dopaminergic progenitors were exposed to cultures of human T cells. Encapsulation in alginate beads appeared to protect the progenitors from immune cell-mediated death and the addition of tacrolimus microparticles further bolstered their survival. This demonstrated that the beads effectively provided a barrier between encapsulated cells and the T cells, a major principle of immunoisolation by microencapsulation.49 The increase in the number of T cells was considerably reduced in conditions with alginate-encapsulated progenitors. This finding is in line with previous work illustrating that alginate microencapsulation prevents the direct activation of cytotoxic CD8+ T cells.50 In our study this action was compounded in the presence of tacrolimus microparticles, an outcome in line with the expected pharmacological action of tacrolimus released from the polymer matrix. This confirmed that tacrolimus was released from the microparticles at a therapeutic concentration.
Having demonstrated the utility of encapsulating hiPSC-derived dopaminergic progenitors in alginate beads, the manufacturing process was optimised to improve the consistency and reduce the size of the hydrogel beads. Employing needles and pulled pipettes of decreasing diameter yielded a decrease in hydrogel bead diameter, down to a minimum size of ∼800 μm. This restricted minimum diameter may be explained by Tate's law,51 which implies a limit to droplet size based on the force balance between the droplet's gravity and the capillary force, and can be calculated using approximated values (eqn (S1)–(4) and Table S3†). Indeed, the predicted minimum hydrogel diameter under this law in this system is 775 μm, a bead size which may be too large for intracranial transplantation.
Using a coaxial flow reactor with sunflower oil as the continuous phase,52 alginate bead size was substantially reduced and homogeneity improved, yielding beads with a clinically relevant diameter of ∼100–200 μm. Other researchers have made similar-sized particles using alternative microfluidic devices and other reagents,53,54 but the approach developed here uses milder reagents. The current method included a washing step that used hexane, but this could be avoided by substituting the sunflower oil with a biocompatible oil such as Fluo-Oil 7500. This oil is commonly used in microfluidic droplet formation and would not need to be washed away following particle production.55 This would potentially streamline the manufacturing process of the particles. Although challenges may be faced when scaling up microfluidic particle production, the mass production of microscopic particles for biological delivery is feasible and has been reviewed in depth.56
Future work should explore the scale-up of the components within the immunomodulatory encapsulation system for clinical translation using Good Manufacturing Process (GMP) materials and processes. Moving forward, the biomaterial technology described here should be tested in an in vivo model to explore the effects on secreted factors, host immune response and transplanted cell survival. Though we developed this immunomodulatory encapsulation system with Parkinson's disease in mind, the technology has the potential to be adapted to other applications in regenerative medicine, for example, cell transplantation to treat Huntington's disease.57,58 The technology can be applied by tailoring the stiffness of the hydrogel for the desired delivery location, selecting a clinically relevant cell type and optimising the tacrolimus-loaded particles for the desired release profile.
Conclusion
This study reported the further development and refinement of a cell encapsulation system based on alginate and immunosuppressant-loaded microparticles, for the delivery and protection of therapeutic cells in Parkinson's disease. The technology was designed to produce alginate hydrogel beads loaded with dopaminergic neuron progenitors and tacrolimus-loaded microparticles. The aim was to simultaneously shield donor cells from the host immune system and potentially circumvent the current requirement for systemic immunosuppression in patients receiving therapeutic cell transplants. The efficacy of the immunomodulatory system was assessed in vitro through exposure to T cells, where significantly greater survival of dopaminergic progenitors was observed with alginate encapsulation and the presence of tacrolimus microparticles. The T cell response was 3-fold lower with the inclusion of the encapsulation system. In addition, a coaxial flow reactor was used to manufacture highly consistent alginate hydrogel beads with a median diameter of 215.6 ± 0.5 μm, potentially suitable for the delivery of therapeutic cells with local immunosuppression in humans.Author contributions
EAA: conceptualisation, methodology, formal analysis, investigation, writing – original draft, writing – review and editing, vsualization. HG: methodology, formal analysis, investigation, writing – original draft. REE: conceptualisation, methodology, investigation. LNC: investigation, methodology. JBP: conceptualisation, supervision, writing – review and editing. RD: supervision, writing – review and editing, methodology. VHR: conceptualisation, supervision, writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.† Any further data can be provided upon request.Conflicts of interest
None of the authors have any conflict of interest to declare.Acknowledgements
EA was supported by funding from EPSRC through the Centre for Doctoral Training in Transformative Pharmaceutical Technologies (EP/S023054/1) and REE was supported by funding from EPSRC through the Centre for Doctoral Training in Advanced Therapeutics & Nanomedicines (EP/L01646X/1). VR and HG were partly supported by funding from the 2018 UCL Rosetrees Stoneygate Prize and the Rosetrees Trust.The authors are grateful to Shuting Li for her assistance in setting up the microfluidics system and John Frost, Senior Mechanical Workshop Technician, for providing some of the parts. Valuable advice about microparticle formulation was kindly provided by Dr I-Ning Lee and Dr Lisa White at the University of Nottingham.
References
- B. R. Bloem, M. S. Okun and C. Klein, Lancet, 2021, 397, 2284–2303 Search PubMed.
- K. R. Chaudhuri, D. G. Healy, A. H. Schapira and E. National Institute for Clinical, Lancet Neurol., 2006, 5, 235–245 Search PubMed.
- E. R. Dorsey and B. R. Bloem, JAMA Neurol., 2018, 75, 9–10 Search PubMed.
- T. B. Stoker, N. F. Blair and R. A. Barker, Neural Regener. Res., 2017, 12, 389–392 Search PubMed.
- R. A. Barker, A. Björklund and M. Parmar, BioEssays, 2024, 2400118, DOI:10.1002/bies.202400118.
- M. Parmar, S. Grealish and C. Henchcliffe, Nat. Rev. Neurosci., 2020, 21, 103–115 Search PubMed.
- A. Kirkeby, S. Grealish, D. A. Wolf, J. Nelander, J. Wood, M. Lundblad, O. Lindvall and M. Parmar, Cell Rep., 2012, 1, 703–714 Search PubMed.
- S. Nolbrant, A. Heuer, M. Parmar and A. Kirkeby, Nat. Protoc., 2017, 12, 1962–1979 Search PubMed.
- Z. Alekseenko, J. M. Dias, A. F. Adler, M. Kozhevnikova, J. A. van Lunteren, S. Nolbrant, A. Jeggari, S. Vasylovska, T. Yoshitake, J. Kehr, M. Carlen, A. Alexeyenko, M. Parmar and J. Ericson, Nat. Commun., 2022, 13, 3046 Search PubMed.
- S. Grealish, E. Diguet, A. Kirkeby, B. Mattsson, A. Heuer, Y. Bramoulle, N. Van Camp, A. L. Perrier, P. Hantraye, A. Bjorklund and M. Parmar, Cell Stem Cell, 2014, 15, 653–665 Search PubMed.
- T. Kikuchi, A. Morizane, D. Doi, H. Magotani, H. Onoe, T. Hayashi, H. Mizuma, S. Takara, R. Takahashi, H. Inoue, S. Morita, M. Yamamoto, K. Okita, M. Nakagawa, M. Parmar and J. Takahashi, Nature, 2017, 548, 592–596 Search PubMed.
- D. Doi, H. Magotani, T. Kikuchi, M. Ikeda, S. Hiramatsu, K. Yoshida, N. Amano, M. Nomura, M. Umekage, A. Morizane and J. Takahashi, Nat. Commun., 2020, 11, 3369 Search PubMed.
- A. Kirkeby, J. Nelander, D. B. Hoban, N. Rogelius, H. Bjartmarz, R. Novo Nordisk Cell Therapy, P. Storm, A. Fiorenzano, A. F. Adler, S. Vale, J. Mudannayake, Y. Zhang, T. Cardoso, B. Mattsson, A. M. Landau, A. N. Glud, J. C. Sorensen, T. P. Lillethorup, M. Lowdell, C. Carvalho, O. Bain, T. van Vliet, O. Lindvall, A. Bjorklund, B. Harry, E. Cutting, H. Widner, G. Paul, R. A. Barker and M. Parmar, Cell Stem Cell, 2023, 30, 1299–1314 Search PubMed.
- V. H. Roberton and J. B. Phillips, in International Review of Neurobiology, ed. E. L. Lane, C. J. G. Drew and M. J. Lelos, Academic Press, 2022, vol. 166, pp. 191–205 Search PubMed.
- S. Qarin, S. K. Howlett, J. L. Jones and R. A. Barker, Neuronal Signaling, 2021, 5, NS20200083 Search PubMed.
- R. A. Barker and T. Consortium, Nat. Med., 2019, 25, 1045–1053 Search PubMed.
- C. E. Staatz and S. E. Tett, Clin. Pharmacokinet., 2004, 43, 623–653 Search PubMed.
- M. A. Bochenek, O. Veiseh, A. J. Vegas, J. J. McGarrigle, M. Qi, E. Marchese, M. Omami, J. C. Doloff, J. Mendoza-Elias, M. Nourmohammadzadeh, A. Khan, C. C. Yeh, Y. Xing, D. Isa, S. Ghani, J. Li, C. Landry, A. R. Bader, K. Olejnik, M. Chen, J. Hollister-Lock, Y. Wang, D. L. Greiner, G. C. Weir, B. L. Strand, A. M. A. Rokstad, I. Lacik, R. Langer, D. G. Anderson and J. Oberholzer, Nat. Biomed. Eng., 2018, 2, 810–821 Search PubMed.
- G. Basta, P. Montanucci, G. Luca, C. Boselli, G. Noya, B. Barbaro, M. Qi, K. P. Kinzer, J. Oberholzer and R. Calafiore, Diabetes Care, 2011, 34, 2406–2409 Search PubMed.
- D. Eleftheriadou, R. E. Evans, E. Atkinson, A. Abdalla, F. K. H. Gavins, A. S. Boyd, G. R. Williams, J. C. Knowles, V. H. Roberton and J. B. Phillips, RSC Adv., 2022, 12, 4005–4015 Search PubMed.
- R. D. Bartlett, D. Eleftheriadou, R. Evans, D. Choi and J. B. Phillips, Biomaterials, 2020, 258, 120303 Search PubMed.
- F. Brandl, F. Sommer and A. Goepferich, Biomaterials, 2007, 28, 134–146 Search PubMed.
- M. C. Catoira, L. Fusaro, D. Di Francesco, M. Ramella and F. Boccafoschi, J. Mater. Sci.: Mater. Med., 2019, 30, 115 Search PubMed.
- B. Snow, E. Mulroy, A. Bok, M. Simpson, A. Smith, K. Taylor, M. Lockhart, B. J. Lam, C. Frampton, P. Schweder, B. Chen, G. Finucane, A. McMahon and L. Macdonald, Parkinsonism Relat. Disord., 2019, 61, 88–93 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 Search PubMed.
- B. M. Hiller, D. J. Marmion, C. A. Thompson, N. A. Elliott, H. Federoff, P. Brundin, V. B. Mattis, C. W. McMahon and J. H. Kordower, npj Regener. Med., 2022, 7, 24 Search PubMed.
- J. Takahashi, Regener. Ther., 2020, 13, 18–22 Search PubMed.
- Nat. Biotechnol., 2023, 41, 1183 Search PubMed.
- S. Mesman and M. P. Smidt, Int. J. Mol. Sci., 2020, 21, 4638 Search PubMed.
- Y. Ono, T. Nakatani, Y. Sakamoto, E. Mizuhara, Y. Minaki, M. Kumai, A. Hamaguchi, M. Nishimura, Y. Inoue, H. Hayashi, J. Takahashi and T. Imai, Development, 2007, 134, 3213–3225 Search PubMed.
- A. Kirkeby, J. Nelander and M. Parmar, Front. Cell. Neurosci., 2012, 6, 64 Search PubMed.
- Z. Simandi, A. Horvath, L. C. Wright, I. Cuaranta-Monroy, I. De Luca, K. Karolyi, S. Sauer, J. F. Deleuze, L. J. Gudas, S. M. Cowley and L. Nagy, Mol. Cell, 2016, 63, 647–661 Search PubMed.
- M. L. Hendrickson, A. J. Rao, O. N. Demerdash and R. E. Kalil, PLoS One, 2011, 6, e18535 Search PubMed.
- A. Bernal and L. Arranz, Cell. Mol. Life Sci., 2018, 75, 2177–2195 Search PubMed.
- A. J. I. Roskams, X. Cai and G. V. Ronnett, Neuroscience, 1998, 83, 191–200 Search PubMed.
- S. Mahajani, A. Raina, C. Fokken, S. Kugler and M. Bahr, Cell Death Dis., 2019, 10, 898 Search PubMed.
- E. M. Hartfield, M. Yamasaki-Mann, H. J. Ribeiro Fernandes, J. Vowles, W. S. James, S. A. Cowley and R. Wade-Martins, PLoS One, 2014, 9, e87388 Search PubMed.
- M. Gao, H. Yao, Q. Dong, H. Zhang, Z. Yang, Y. Yang, J. Zhu, M. Xu and R. Xu, Sci. Rep., 2016, 6, 29955 Search PubMed.
- S. Yang, L. C. Edman, J. A. Sánchez-Alcañiz, N. Fritz, S. Bonilla, J. Hecht, P. Uhlén, S. J. Pleasure, J. C. Villaescusa, O. Marín and E. Arenas, Development, 2013, 140, 4554–4564 Search PubMed.
- G. O. Bodea, J.-H. Spille, P. Abe, A. S. Andersson, A. Acker-Palmer, R. Stumm, U. Kubitscheck and S. Blaess, Development, 2014, 141, 661–673 Search PubMed.
- L. Tamò, K. Fytianos, F. Caldana, C. Simillion, A. Feki, I. Nita, M. Heller, T. Geiser and A. Gazdhar, Int. J. Mol. Sci., 2021, 22, 958 Search PubMed.
- H. Choi, H.-J. Lee, H.-J. Sohn and T.-G. Kim, BMC Immunol., 2023, 24, 15 Search PubMed.
- O. M. Noceti, J. B. Woillard, A. Boumediene, P. Esperon, J. L. Taupin, S. Gerona, M. Valverde, C. Tourino and P. Marquet, Clin. Chem., 2014, 60, 1336–1345 Search PubMed.
- A. K. Wright, C. Miller, M. Williams and G. Arbuthnott, Brain Res., 2008, 1216, 78–86 Search PubMed.
- R. F. Castilho, O. Hansson and P. Brundin, Exp. Neurol., 2000, 164, 94–101 Search PubMed.
- E. C. Toll, A. M. Seifalian and M. A. Birchall, Regener. Med., 2011, 6, 635–652 Search PubMed.
- A. J. Curle, S. V. Fazal, S. Qarin, S. K. Howlett, X. He, R. A. Barker and J. L. Jones, bioRxiv, 2024, 2024.2001.2023.576826, DOI:10.1101/2024.01.23.576826.
- B. A. Aguado, W. Mulyasasmita, J. Su, K. J. Lampe and S. C. Heilshorn, Tissue Eng., Part A, 2012, 18, 806–815 Search PubMed.
- G. A. Paredes Juarez, M. Spasojevic, M. M. Faas and P. de Vos, Front. Bioeng. Biotechnol., 2014, 2, 26 Search PubMed.
- Y. Li, A. W. Frei, E. Y. Yang, I. Labrada-Miravet, C. Sun, Y. Rong, M. M. Samojlik, A. L. Bayer and C. L. Stabler, Biomaterials, 2020, 256, 120182 Search PubMed.
- T. Tate, London, Edinburgh Dublin Philos. Mag. J. Sci., 1864, 27, 176–180 Search PubMed.
- A. A. Dias Meirelles, A. L. Rodrigues Costa, M. Michelon, J. Viganó, M. S. Carvalho and R. L. Cunha, J. Food Eng., 2022, 315, 3385 Search PubMed.
- S. Utech, R. Prodanovic, A. S. Mao, R. Ostafe, D. J. Mooney and D. A. Weitz, Adv. Healthcare Mater., 2015, 4, 1628–1633 Search PubMed.
- V. L. Workman, S. B. Dunnett, P. Kille and D. D. Palmer, Biomicrofluidics, 2007, 1, 14105 Search PubMed.
- T. Tang, H. Zhao, S. Shen, L. Yang and C. T. Lim, Microsyst. Nanoeng., 2024, 10, 3 Search PubMed.
- H. Lin, J. Leng, P. Fan, Z. Xu and G. Ruan, Mater. Adv., 2023, 4, 2885–2908 Search PubMed.
- A. E. Rosser, C. M. Kelly and S. B. Dunnett, Future Neurol., 2010, 6, 45–62 Search PubMed.
- M. Csobonyeiova, S. Polak and L. Danisovic, Int. J. Mol. Sci., 2020, 21, 2239 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4bm01566e |
| This journal is © The Royal Society of Chemistry 2025 |