
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Matrix deformation and mechanotransduction as markers of breast cancer cell phenotype alteration at matrix interfaces†
Cornelia
Clemens
 ,
Rosa
Gehring
,
Philipp
Riedl
and
Tilo
Pompe
*
,
Rosa
Gehring
,
Philipp
Riedl
and
Tilo
Pompe
*
Institute of Biochemistry, Leipzig University, Johannisallee 21-23, 04103, Leipzig, Germany. E-mail: tilo.pompe@uni-leipzig.de
First published on 7th February 2025
Abstract
The dissemination of metastatic cells from the primary tumor into the surrounding tissue is a key event in the progression of cancer. This process involves the migration of cells across defined tissue interfaces that separate the dense tumor tissue from the adjacent healthy tissue. Prior research showed that cell transmigration across collagen I matrix interfaces induces a switch towards a more aggressive phenotype including a change in directionality of migration and chemosensitivity correlated to increased DNA damage during transmigration. Hence, mechanical forces acting at the nucleus during transmigration are hypothesized to trigger phenotype switching. Here, we present results from a particle image velocimetry (PIV) based live cell analysis of breast cancer cell transmigration across sharp matrix interfaces constituted of two collagen type I networks with different pore sizes. We found strong and highly localized collagen network deformation caused by cellular forces at the moment of crossing interfaces from dense into open matrices. Additionally, an increased contractility of transmigrated cells was determined for cells with the switch phenotype. Moreover, studies on mechanotransductive signaling at the nucleus, emerin translocation and YAP activation, indicated a misregulation of these signals for transmigrated cells with altered phenotype. These findings show that matrix interfaces between networks of different pore sizes mechanically challenge invasive breast cancer cells during transmigration by a strong asymmetry of contracting forces, impeding nuclear mechanotransduction pathways, with a subsequent trigger of more aggressive phenotypes.
1. Introduction
Throughout tumor progression, the tumor microenvironment (TME) undergoes significant changes that promote tumor growth and metastasis.1 The ongoing remodeling processes are characterized by increased deposition of matrix components and matrix crosslinking, as well as alterations in fiber alignment, that contribute to a denser topology and higher stiffness of the tumor tissue.2 In breast cancer, the tissue's elastic modulus can more than double as the tumor progresses. These changes in the tissue make the tumor palpable and serve as a basis for elastography methods in addition to conventional diagnostic techniques.3Notably, collagen type I, the most abundant protein in the extracellular matrix (ECM), appears to play a major role in tumor stiffening, with its quantity and organization significantly influencing the biomechanical properties of the TME.4 Specifically, collagen alignment and density were shown to enhance breast tumor aggressiveness as well as contribute to its metastatic potential.5,6 In a biomimetic matrix model of collagen I, increased matrix density correlated with a more invasive phenotype in breast carcinoma cells.7 Properties like collagen fibril diameter and fibril bending stiffness were additionally shown to regulate cell morphology, cluster formation and invasion as well as the overall behavior of invasive and non-invasive breast cancer cells.8,9 Recent studies have extensively investigated the impact of ECM stiffness variations on tumor progression, revealing that a stiff microenvironment within a breast tumor promotes the acquisition of malignant phenotypes in non-malignant mammary epithelial cells.10,11 Hence, ECM's mechanics and microstructure are not only accepted as defining characteristics but moreover as important modulators of phenotype and aggressiveness of breast cancer cells.
Cells are in bidirectional communication with the surrounding matrix, by physically anchoring themselves to ECM fibers like collagen via integrin heterodimers.12,13 Depending on the properties of the surrounding microenvironment, these adhesion sites sequentially recruit additional proteins to form multiprotein complexes that mature from nascent adhesions via focal complexes to focal adhesions and fibrillar adhesions. This maturation is frequently characterized by phosphorylation and activation of proteins like focal adhesion kinase, vinculin and paxillin.14 Through the application of traction forces transmitted by the cytoskeleton and the adhesion sites to the surrounding matrix, cells dynamically sense the mechanical properties of the ECM and adapt to them in a process known as mechanotransduction.15 Such significant forces can be visualized and quantified by methods like traction force microscopy16–19 and micropillar arrays20 combined with particle tracking velocimetry21 or particle image velocimetry (PIV)22 based algorithms to reveal information, e.g. on strength, long-range distance, and directionality.
Conformational changes in proteins within the focal adhesion complexes, such as vinculin, are induced depending on the stiffness of the ECM fibers. These changes activate mechanosensitive signaling pathways, ultimately modulating gene expression and cellular phenotype. Herein, especially the YAP/TAZ (yes-associated protein 1/transcriptional coactivator with PDZ-binding motif) protein pathway has received considerable attention, as many mechanotransduction pathways are regulated via this axis, leading to the periplasmatic or nuclear localization of activated YAP.23
The importance of ECM density and stiffness extends beyond the primary tumor to the metastatic cascade, the most life-threatening part of cancer. Metastasis is a process by which cancer cells detach from the primary tumor and subsequently colonize distant tissues and organs.24,25 Throughout these processes, the metastatic cells have to adapt to ECMs with varying topological and mechanical properties.26–28 The invasive front of breast cancer displays a further increase in stiffness and already exposes invasive cells to heterogeneous structures.29 Thus, one of the first obstacles for metastasizing cells leaving the primary tumor are defined interfaces that occur between the stiff and dense tumor tissue and the softer, more open porous healthy tissue. Hereby, research has already shown that invasion markers are notably more active at these boundaries.30,31
To better understand the dynamic interplay between cancer cells and their respective microenvironment during these first steps of metastasis, prior studies employed a biomimetic ECM model that recapitulates such tumor–tissue interfaces.32,33 By sequentially fibrillating two collagen I matrices with different pore sizes, and respective fibrillar density, a defined matrix interface was formed. Embedding the invasive breast cancer cell line MDA-MB-231 into the dense compartment revealed a transition of cells towards a more aggressive phenotype as the cells transmigrate across the interface into the more open porous matrix. In particular, the cells that crossed the interface exhibited a transition from random to more directed migration,32 increased proliferation, higher chemoresistance to doxorubicin, and upregulation in the expression of gene clusters linked to metastasis.34 Matrix-related phenomena in triggering directed cell migration like chemotaxis, durotaxis or haptotaxis were excluded in those previous studies.
Notably, in those reports, cells exhibited strong deformations of the nuclear membrane and a higher incidence of DNA damage directly at the matrix interface, suggesting that this boundary may serve as an activator of mechanical signals involving the nucleus, leading to the metastatic phenotype switch. In this study, we aimed to use this collagen I matrix interface model to explore in more detail the possible triggers of mechanotransduction in transmigrating breast cancer cells at the matrix interface. Using PIV analyses of ECM deformations, we provide functional evidence of changed contractility of MDA-MB-231 cells during transmigration, which reflects the previously observed phenotype switching. Moreover, we were able to gain a better understanding of the contractile forces acting directly at these interfaces during cell transmigration. In addition, our investigation of mechanotransduction pathways revealed misregulations in response to transmigration. These results improve our understanding of the activation of metastatic phenotypes of breast cancer cells at matrix interfaces and the importance of mechanotransduction in this process.
2. Materials and methods
2.1. Materials
Reagents were purchased as follows: glass coverslips (∅ 13 or 19 mm), acetic acid, 5-(and 6)-carboxytetramethylrhodamine succinimidyl ester (TAMRA-SE), Dulbecco's modified Eagle's medium (DMEM), Hank's Balanced Salt Solution (HBSS) (VWR, Germany); poly(styrene-alt-maleic anhydride) (PSMA, MW 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), monosodium phosphate and disodium phosphate, bovine serum albumin (BSA) (Sigma Aldrich, Germany); collagen I (Advanced Biomatrix, USA); FCS (Merck, Germany); Zellshield® (Minerva Biolabs, Germany); ROTI®Cell 10× PBS, and paraformaldehyde (PFA) (Carl Roth, Deutschland). For immunostaining, primary antibodies emerin (D3B9G, rabbit, 1:200, Cell Signaling Technology, USA) and YAP (63.7, mouse, 1:100, Santa Cruz Biotechnology, USA) were purchased as well as the secondary antibodies anti-rabbit IgG (H + L), F(ab′)2 fragment (goat, Cell Signaling Technology) and anti-mouse conjugated with Cy3 (donkey, Jackson Immuno Research, USA). Additional dyes used were 4′-6-diamidino-2-phenylindole-dihydrochloride (DAPI, 1:10000, Sigma Aldrich) and phalloidin conjugated with Alexa Fluor 488 (1:400, Invitrogen, USA). The cell line MDA-MB-231 was purchased from Leibniz Institute DSMZ German Collection of Microorganisms and Cell Cultures GmbH (Braunschweig, Germany).
000 g mol−1), monosodium phosphate and disodium phosphate, bovine serum albumin (BSA) (Sigma Aldrich, Germany); collagen I (Advanced Biomatrix, USA); FCS (Merck, Germany); Zellshield® (Minerva Biolabs, Germany); ROTI®Cell 10× PBS, and paraformaldehyde (PFA) (Carl Roth, Deutschland). For immunostaining, primary antibodies emerin (D3B9G, rabbit, 1:200, Cell Signaling Technology, USA) and YAP (63.7, mouse, 1:100, Santa Cruz Biotechnology, USA) were purchased as well as the secondary antibodies anti-rabbit IgG (H + L), F(ab′)2 fragment (goat, Cell Signaling Technology) and anti-mouse conjugated with Cy3 (donkey, Jackson Immuno Research, USA). Additional dyes used were 4′-6-diamidino-2-phenylindole-dihydrochloride (DAPI, 1:10000, Sigma Aldrich) and phalloidin conjugated with Alexa Fluor 488 (1:400, Invitrogen, USA). The cell line MDA-MB-231 was purchased from Leibniz Institute DSMZ German Collection of Microorganisms and Cell Cultures GmbH (Braunschweig, Germany).
2.2. Reconstitution and topological characterization of collagen I interface matrices
Collagen I matrices with distinct interfaces, separating two compartments with different fibril densities, were reconstituted, as previously described; see also ESI Fig. S1.†32,34 Briefly, for the covalent attachment of the collagen networks, glass coverslips (∅ 13 or 19 mm) were coated with 0.14% w/w poly(styrene-alt-maleic anhydride) (PSMA, MW 30000 g mol−1) after activation of glass surfaces by aminosilanization. Subsequently, the collagen I stock solution was mixed on ice with 0.02 N acetic acid and 500 mM phosphate buffer, containing monosodium phosphate and disodium phosphate. The first solution was then pipetted onto the coverslips (20 μL for 13 mm coverslips and 50 μL for 19 mm coverslips) and polymerized at 37 °C in a wet chamber. After a washing step, the second compartment was prepared accordingly using the other collagen I concentration (50 μL for 13 mm coverslips and 250 μL for 19 mm coverslips). Using two subsequent polymerization steps for two collagen I solutions created distinct interfaces mimicking the tumor–tissue boundary. As a result of this process, the first matrix compartment has a thickness of roughly 0.2 mm (for 13 mm coverslips) or 0.3 mm (for 19 mm coverslips), and the second matrix has a thickness of approximately 0.5 mm (for 13 mm coverslips) or 0.9 mm (for 19 mm coverslips).
To assess the topology of the aforementioned collagen networks, the matrices were stained using 50 μM 5-(and 6)-carboxytetramethylrhodamine succinimidyl ester (TAMRA-SE). In particular, the interface matrices were incubated with the staining solution at room temperature for 1 h, followed by washing using 1× PBS. With a confocal laser scanning microscope (LSM700, Carl Zeiss Microscopy, Germany), imaging of the network topology was performed at 40× magnification. This imaging recorded stacks over a total distance of 50 μm with individual xy-planes spaced 5 μm apart. The images had a resolution of 1024 × 1024 px, resulting in an xy-pixel size of 0.16 × 0.16 μm. Using a MATLAB image analysis tool described by Franke et al.,35 mean pore diameter and mean fibril diameter were analyzed.
2.3. Embedding of MDA-MB-231 breast cancer cells in collagen I matrices
For all steps, MDA-MB-231 cells were maintained as prescribed by the supplier using DMEM supplemented with 10% FCS and 1% Zellshield® in an incubator with 5% CO2, at 37 °C and with a humidity of 95%.For the analysis of breast cancer cell migration across matrix interfaces, MDA-MB-231 cells were embedded into the second compartment of the aforementioned interface matrices. To do so, we resuspended MDA-MB-231 cells in phosphate buffer, replaced a part of the collagen I solution with the cell suspension and mixed properly, before adding the solution onto the coverslips. Thereby, 104 cells per matrix were enclosed within the collagen networks and subsequently incubated for 7 d.
2.4. Analysis of matrix deformation based on particle image velocimetry
To analyze the matrix deformation in real time, the collagen I matrix interfaces with embedded MDA-MB-231 cells were pre-incubated at 37 °C with 5% CO2 and 95% humidity for 24 h. Afterwards, the samples were transferred to an inverted fluorescence microscope (Axio Observer.Z1, Carl Zeiss Microscopy) equipped with an incubation chamber set to 37 °C with 5% CO2. Over a period of seven days, image stacks were captured at 10 min intervals (Δt) using 10× magnification in phase-contrast mode. The size of each image was 692 × 520 px, with a voxel size of 0.806 × 0.806 × 5.000 μm. Depending on the matrix thickness, the total stack size was set to 175–225 μm. Quantitative analysis of matrix deformation was performed using a custom-built Python algorithm based on particle image velocimetry as described previously.22By cross correlating the images between consecutive time points, a two-dimensional vector field was generated for each interval Δt, which is described hereinafter with the parameter's deformation and convergence. The deformation is described by the mean vector length calculated for each interval Δt representing the amount of overall matrix deformation. High convergence values represent regions with high local deformations and for each interval Δt, the convergence values were summed up. The mean ratio between convergence and deformation for each interval Δt represents the cell number independent contractility of the cells. Furthermore, the vector fields are plotted in an xy-plot with the plotting area colored depending on either the deformation or convergence values.
2.5. Quantitative analysis of nuclear mechanotransduction
Following the embedding of breast cancer cells within interface matrices, we analyzed the nuclear proteins emerin and YAP by immunostaining. Therefore, the enclosed cells were rinsed with Hank's Balanced Salt Solution (HBSS), before being fixed by incubating with a 4% PFA solution at room temperature for 15 min. Afterwards, the interface matrices with cells were washed three times for 5 min with 1× PBS and were then blocked at room temperature for 1 h with 1.5% bovine serum albumin (BSA) in 1× PBS. The cells were stained with the primary antibodies emerin (D3B9G, rabbit, 1:200) and YAP (63.7, mouse, 1:100) as well as the secondary antibodies anti-rabbit IgG (H + L), F(ab′)2 fragment (goat) and anti-mouse conjugated with Cy3 (donkey). All antibodies were diluted in 1.5% BSA in 1× PBS, whereby the secondary antibodies were diluted at a ratio of 1:800. Additionally, the staining solution contained either 4′-6-diamidino-2-phenylindole-dihydrochloride (DAPI, 1:10000) or DAPI in combination with phalloidin conjugated with Alexa Fluor 488 (1:400). Using a confocal laser scanning microscope (Leica TCS SP8 LIA FALCON, Leica Microsystems, Germany), the stained cells were documented with 40× magnification, an image size of 2048 × 2048 px and an xy-pixel size of 0.142 μm. At least 20 cells per condition were imaged.
The total emerin amount per cell was quantified using ImageJ.36 The microscopy images of the channels of DAPI and the protein of interest were exported to TIFF-format and converted to 8-bit. A Z-project was generated by summing all slices. Subsequently, the contrast was enhanced to a saturation of 0.15 and binarized using the default thresholding of ImageJ. Next, the cell shape was selected using the “Analyze Particles”-function. These selections were then used to quantify the integrated density in the channel of the protein of interest.
To quantify nuclear localized YAP, the images were exported likewise using ImageJ. The DAPI images were converted to 8-bit and with the default thresholding of ImageJ, binary images were generated. The nuclear shape was selected using the “Analyze Particles”-function. These selections were then overlayed onto the YAP channel and the integrated YAP density for the nuclear region was measured.
2.6. Statistical analysis
Each experiment was carried out in triplicate, unless stated otherwise. Error bars depict the standard deviation (SD). Statistical significance was determined with a Kruskal–Wallis test using GraphPad Prism 9 (GraphPad Software, USA) or R 4.4.0 (The R Foundation, Austria).37 The significance level was set at p < 0.05 (*: p < 0.05; **: p < 0.01; ***: p < 0.001).3. Results and discussion
3.1. Engineering sharp collagen I matrix interfaces to mimic tumor–tissue boundaries
Breast cancer tissue and healthy tissue are distinguishable based on their mechanical properties and densities. Cancerous tissue often shows increased stiffness and reduced porosity, whereas healthy tissue displays softer characteristics and a lower density. Previous research studies, including our own studies and those of others, have demonstrated an impact of the boundary that emerges during tumor progression between the dense tumor tissue and the surrounding healthy tissue, influencing breast cancer cell behavior.32–34 In studies using biomimics of such boundaries, a phenotype switching towards a more aggressive behavior and alterations in gene expression were observed.32,34To recapitulate such tumor–tissue boundaries in vitro in lab experiments, a three-dimensional collagen I model was established in our previous studies, comprising two distinct collagen matrices with varying porosities.32 This model was used to mimic the invasion from the primary tumor into the surrounding healthy tissue by embedding the highly invasive breast cancer cell line MDA-MB-231 into the denser compartment, allowing transmigration across the interface into the open porous compartment (Fig. 1A, ESI Fig. S1†). Importantly, previous studies attributed the observed changes in the cellular phenotype upon transmigration across a dense to open interface exclusively to the sharp change in porosity, rather than the alteration in stiffness. This was confirmed by systematically varying collagen I concentration and crosslinking states. Directed migration occurred only during dense-to-open migration and was unaffected by crosslinking or matrix stiffness, highlighting pore size differences as the key regulatory factor. A reversed configuration where the MDA-MB-231 cells migrated from the open porous compartment to the denser compartment served as a control condition.32,34
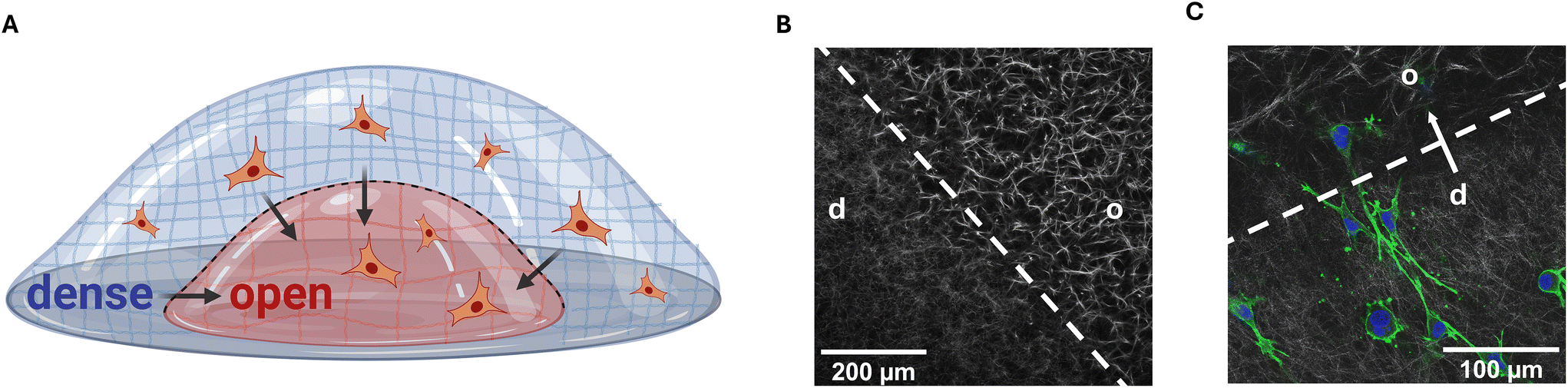 | ||
| Fig. 1 Experimental setup to mimic the sharp decrease in matrix density at the tumor–tissue boundary in vitro. (A) Schematic representation of matrix interfaces on a polymer-coated coverslip with an open porous and a denser compartment separated by a defined interface indicated by the dashed line. Highly invasive MDA-MB-231 breast cancer cells are embedded in the denser matrix compartment and can transmigrate across the matrix interface during a seven-day incubation period. (B) An in vitro matrix interface stained with TAMRA-SE and visualized via cLSM. (C) Confocal laser scanning microscopy (cLSM) images of transmigrating MDA-MB-231 breast cancer cells across a biomimetic tumor–tissue interface. Cells were stained using DAPI (blue) and AlexaFluor 488 coupled phalloidin (green). The matrix was imaged using reflection mode (grey). d – dense, o – open. | ||
In this study, we similarly prepared collagen I matrices with two compartments featuring different porosities separated by a defined interface (Fig. 1B), where the porosities were around 5 μm for the open porous compartments and 4 μm for the dense compartments. The fibril diameter remained constant across all conditions at around 0.95 μm (for details, see ESI Table S1†). Such matrices were frequently shown in our previous studies to exhibit matrix stiffness with the elastic modulus in the range of 100 Pa. The highly invasive MDA-MB-231 breast cancer cells were embedded into the denser compartment and the setup was incubated for seven days to enable transmigration across the interface (Fig. 1C, ESI Fig. S1†). Subsequently, dense matrices are abbreviated as ‘d’ and open-porous matrices as ‘o’. The transmigration across a matrix interface is indicated by an arrow (‘d → o’ or for control conditions ‘o → d’).
3.2. Strong and highly localized contractile forces at matrix interfaces during cell transmigration
This study builds on our previous reports showing a strongly altered phenotype of the breast cancer cells MDA-MB-231 after transmigration across a matrix interface from a dense to a porous compartment, leading to enhanced aggressiveness of the already invasive breast cancer cells. Initial analyses trying to unravel the molecular mechanisms leading to phenotype switching have shown that crossing the d → o interface causes nuclear deformation and an increased probability of DNA damage at the interface. These findings suggest a pivotal role of the nucleus during transmigration.32,34We hypothesize that the biomimetic d → o interface has a mechanical influence on the breast cancer cells by uneven topological properties sensed by the cell during the transmigration process resulting in mechanical stress on the cell and the nucleus. Cells sense mechanical and topological features of the microenvironment by contraction induced force transmission whereby the cells actively deform the surrounding matrix fibers. To get an insight into the dynamic interplay of cells and the ECM at matrix interfaces, a previously published PIV-based algorithm by Riedl et al.22 was used to analyze cell-induced matrix deformations of cells during transmigration.
For the PIV-analysis, phase contrast images of three-dimensionally cultured cells are captured over a period of several days at a specific time interval, which should be chosen depending on the research question and the cell type. In the past, the differentiation stages of fibroblasts were analyzed over a period of seven days with a specifically optimized interval of 15 min. Even though the migration speeds of fibroblasts and the breast cancer cells MDA-MB-231 are comparable in collagen networks,32,38 we chose an interval of 10 min to get a more detailed view of the cells transmigrating across the interface and to closely monitor the process of interface crossing. The algorithm itself involves the generation of two-dimensional vector fields by calculating displacement vectors between consecutive time points using cross correlation. Subsequently, the cell-induced matrix deformation is quantified by measuring mean displacement vector lengths (deformation) and their convergence sum per time interval, allowing the estimation of cellular contractility, and effectively identifying regions of high contraction (Fig. 2). Three time points were selected from an experiment over seven days in which a single migrating breast cancer cell was imaged before, during and after interface crossing (t0, t0 +120 min, t0 +750 min).
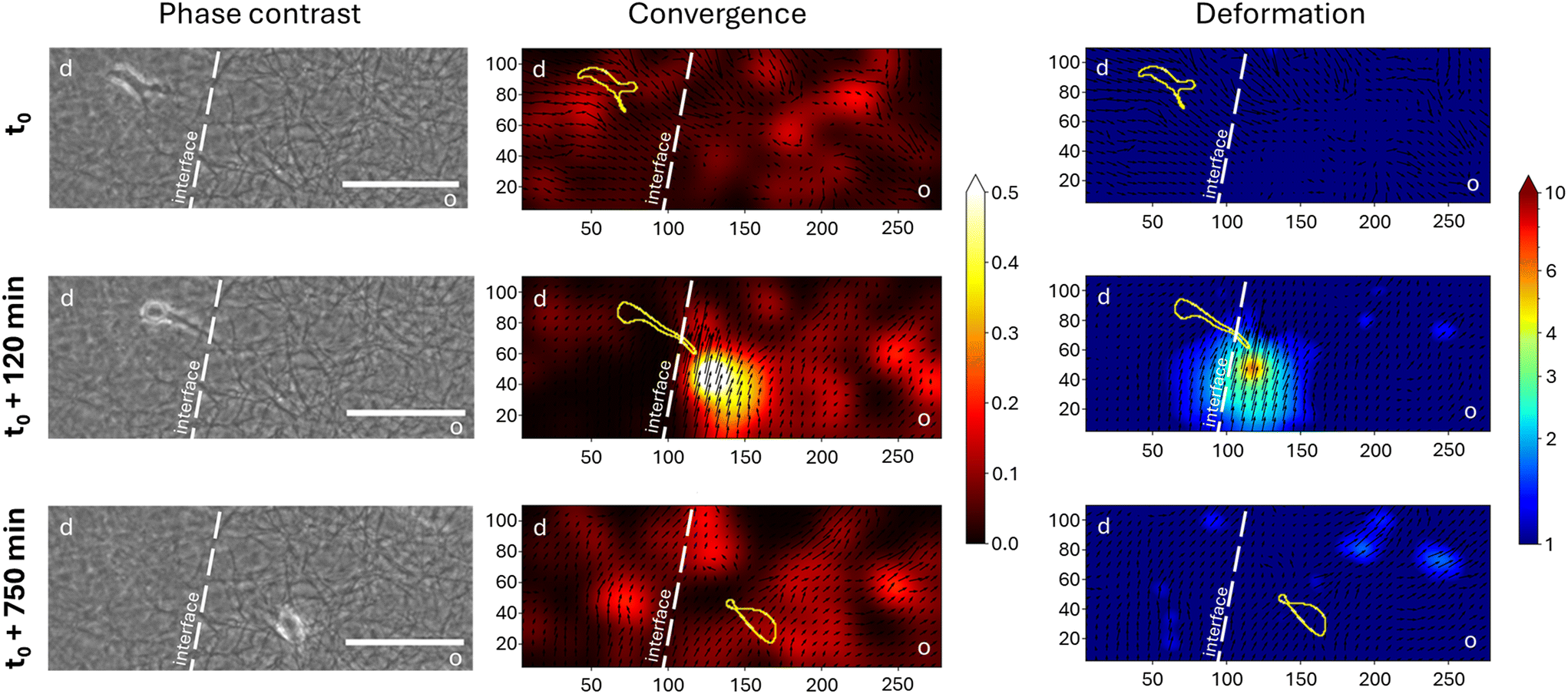 | ||
| Fig. 2 Matrix deformation analysis of a transmigrating cell across a d → o interface. Depicted are the raw phase contrast images (left column) as well as the calculated displacement vector field using a PIV-based algorithm colored according to the local convergence (middle column) or deformation (right column). The transmigration of the cell is shown for three time points (top, center, and bottom). The cell shape is indicated in the PIV images by a yellow line based on phase contrast image information. The matrix interface is shown by a white dashed line. Scale bar depicts 100 μm. d – dense, o – open. | ||
Before the cell encounters the interface, the vector field is quite uniform and does not show regions of high deformation or convergence. As the MDA-MB-231 cell elongates and forms a cell protrusion across the interface into the open porous compartment, the cell deforms the collagen fibers at the interface in the encountered compartment way more than at other locations indicated by the strong local deformation and convergence at the tip of the cell within the open porous compartment. After the entire cell body crosses the interface and the cell is completely in the open porous compartment, no region of high convergence and deformation can be detected.
The differing porosity and thus matrix stiffness at the interface therefore lead to different deformations of the matrix. It can be assumed that the cell perceives different mechanical signals at the same time from the migrating front in the open porous compartment compared to the rear of the cell, which is still embedded in the dense compartment. A build-up of high contractility in the open porous compartments of the invasive front seems to be balanced by the remaining part of the cell in the dense compartment. Such asymmetric cellular force distributions are transmitted directly to the cell nucleus within a few milliseconds and can directly influence the nucleus shape. This may lead to nuclear rupture in the short term, and/or adaptation of its mechanics in the long term.39 Hence, we conclude that the observed strong and highly localized contractility of the cells can be correlated to the previously observed strong nucleus deformation and DNA damage occurring during transmigration of MDA-MB-231 breast cancer cells at matrix interfaces.34 Furthermore, our results support the previous findings of Bordeleau et al. of induced front–rear polarity of malignant and non-malignant cells at d → o interfaces.33 It was shown that cells in contact with the interface exhibit an uneven distribution of focal adhesion sites promoting the required cellular polarity for directional migration after interface crossing.
3.3. Matrix deformation as a marker of the altered contractile phenotype of MDA-MB-231 cells after transmigration of matrix interfaces
Following the observation that transmigrating cells exhibit highly localized contractility during transmigration of the matrix interface, we investigated whether the PIV analysis of matrix deformation can also functionally detect the altered phenotype of transmigrated cells in terms of contractility. Their previously observed directed migration as well as elongated and polarized morphology suggests changes in the contractile behavior, which might be correlated to differences in nuclear mechanotransduction and gene expression.Given the differences in porosity and bulk network elasticity between the dense and open compartments, it is expected that these variations lead to differences in matrix deformation, assuming a constant contractile phenotype. At first, we therefore studied contractile MDA-MB-231 behavior in control experiments, using homogeneous collagen I matrices with different densities by altering the collagen I concentration using the same experimental PIV setup. The correlation between matrix deformation and collagen concentration was evaluated using the averaged deformation at day 3 after matrix embedding. This decision was based on the observation that the cells had fully adapted to the culture conditions by day 3. Additionally, the differences between the conditions were pronounced and consistent, as seen in time-dependent deformation and convergence over the full experimental duration, shown in ESI Fig. S2A and B.† The matrix deformation as well as the convergence averaged over 24 h at day 3 shows a decrease with increasing matrix density, which is expected as stiffer matrices should be less deformable by similarly contractile cells. (The constant linear increase in deformation over time is attributed to the increase in cell number by proliferation as discussed previously.22) Moreover, we can see that matrix deformation scales with collagen I concentration ∝ c−1.8 (ESI Fig. S2C†). This scaling of matrix deformation with collagen concentration proves that our PIV algorithm can quantitatively and functionally measure contractility of cells within 3D matrices. Within linear elasticity theory, one can assume matrix deformation to be linearly related to cellular forces and inversely proportional to the elastic modulus of the matrices. The elastic modulus of collagen matrices was frequently reported with a similar scaling with collagen concentration of c1.5 to c1.7, which fits our finding.40 Hence, our results show that matrix deformation directly relates to the network bulk elasticity and indicates that MDA-MB-231 cells exhibit similar contractile forces within different collagen matrices. While both matrix deformation and convergence depend on matrix stiffness, the ratio of convergence and deformation should not. Therefore, we plotted this ratio over time for the full experimental period (Fig. 3A). (The ratio also allows us to eliminate the cell dependence of deformation and convergence as mentioned above.) With this analysis, we can more precisely demonstrate that MDA-MB-231 cells within different matrix densities exhibit a non-altered contractile phenotype.
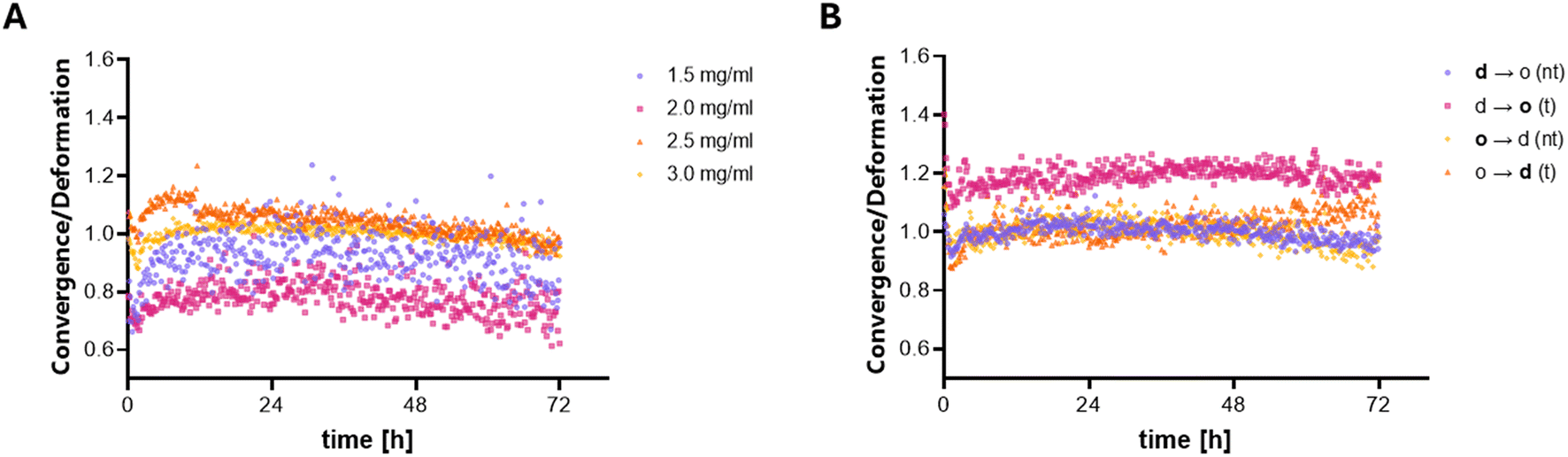 | ||
| Fig. 3 Results of the PIV-based matrix deformation analysis. (A) Ratio of convergence to deformation determined from live imaged MDA-MB-231 cells embedded in collagen I matrices with different densities. The mean ratio of 3.0 mg mL−1 was set to 1. (B) Influence of the crossing of MDA-MB-231 cells over defined interfaces on the ratio of convergence to deformation. The ratio of the transmigrated cells was normalized to the mean ratio of the corresponding non-transmigrated cells. d – dense, o – open, d → o – dense to open, o → d – open to dense, nt – non-transmigrated, t – transmigrated. Data at each time point represent mean deformation or convergence values of 12 different image positions. | ||
Based on those control experiments, which proved the functional determination of MDA-MB-231 contractility within collagen matrices of different pore sizes (and densities), we examined cell contractility of MDA-MB-231 cells before and after transmigration of sharp matrix interfaces. Cells were analyzed in parallel in both compartments in the d → o configuration as well as under the control condition of o → d compartments. As transmigration leads to a steady increase in the number of cells in the second compartment, it was important to analyze the cell number independent and pore size independent ratio of convergence to deformation; see the discussion above. With transmigration across d → o interfaces, a higher ratio of contractility to deformation was observed, indicating a higher contractility of the transmigrated cells (Fig. 3B). As expected, the control experiments of o → d interfaces showed no differences in contractility after transmigration from the open to the dense compartment. However, with increasing incubation time, cell proliferation and continued transmigration may still influence the results, particularly toward the end of the measurement.
Previous studies have demonstrated that cell–substrate forces are directly linked to the invasion of cancer cells in three-dimensional matrices, with for example the metastatic breast cancer cell line MDA-MB-231 generating significantly higher traction stresses than their non-metastatic counterparts.41,42 Therefore, our results support our previous findings of a more aggressive and more invasive phenotype upon interface crossing, and provide functional evidence of a more contractile phenotype and altered mechanotransduction of the transmigrated cells.32,34
3.4. Altered nuclear mechanotransductive signals by transmigration of MDA-MB-231 cells across matrix interfaces
The nucleus plays a major role in cancer cell migration. In particular, movement through topologically challenging structures like confinements has been shown to induce nuclear envelope integrity loss, nucleocytoplasmic content exchange, and DNA damage.43 We hypothesize that, in addition to the widely studied migration in confined microenvironments,43–45 transmigration across defined d → o matrix interfaces is mechanically challenging for invasive breast cancer cells facilitating a more aggressive phenotype and can lead to DNA damage and subsequent alterations of gene expression as previously observed.34Given the strong mechanical challenges and high asymmetric contractility at the matrix interface during MDA-MB-231 cell transmigration, we aimed to further relate these findings to mechanotransduction processes associated with the nucleus. We therefore analyzed the nuclear envelope protein emerin, which is crucial for regulating mechanotransduction signaling pathways originating from integrin-mediated adhesions.46 As mechanical signals propagate onto the nuclear envelope, emerin localization and phosphorylation are modulated.47–49 This, in turn, leads to remodeling of the nuclear shape and the overall structural integrity of the nuclear envelope, thereby protecting the chromatin in response to mechanical stimuli.50
Immunostaining for emerin and quantification of the total emerin signal per cell revealed a high dependence of the cellular emerin amount on the matrix density in controls of homogeneous matrices (Fig. 4A and B; ESI Fig. S3†). Embedding MDA-MB-231 cancer cells into homogeneous open porous collagen I resulted in more than twice the amount of emerin compared to cells embedded in denser matrices. This behavior is in line with other reports showing an increase in emerin on softer substrates.50
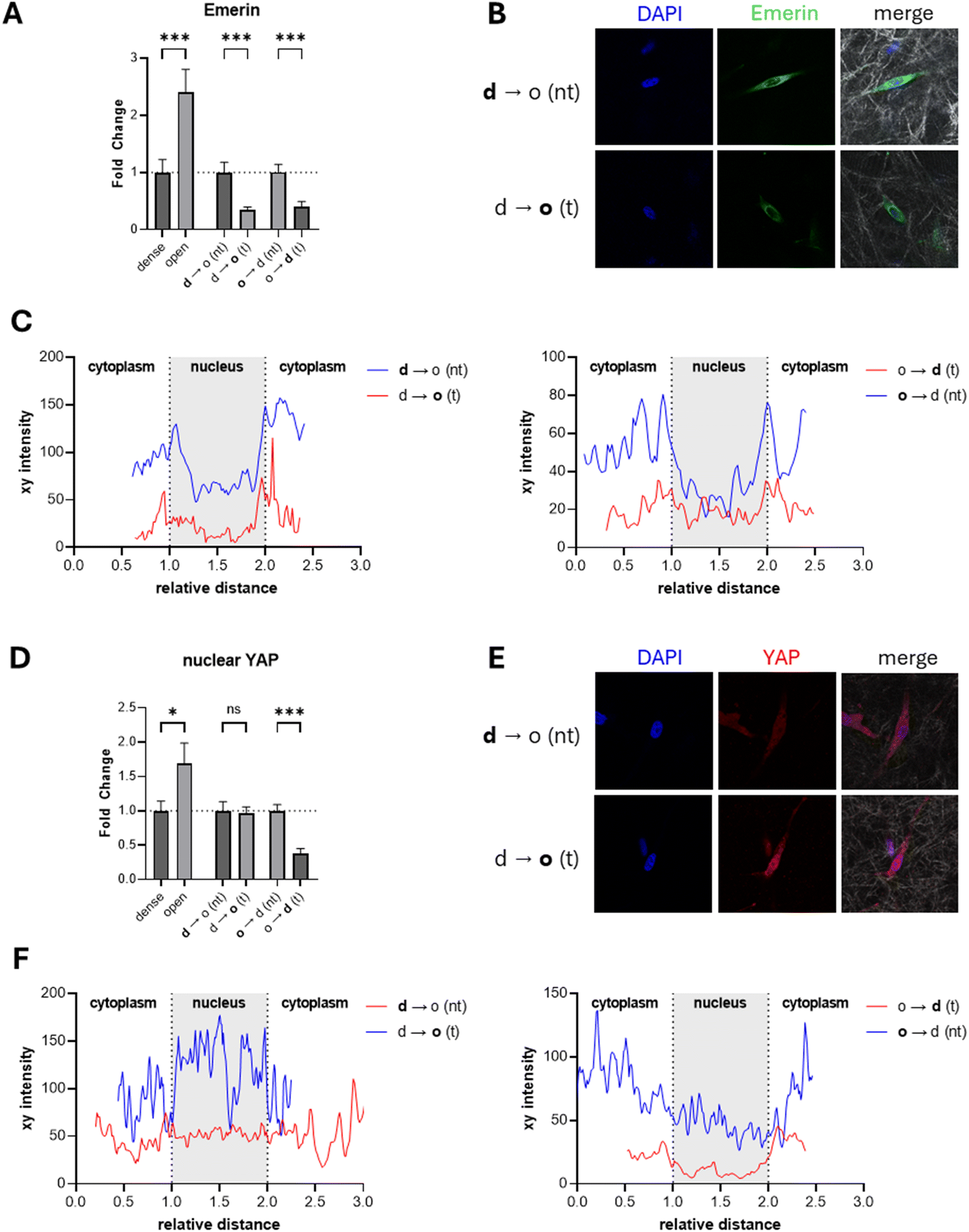 | ||
| Fig. 4 Immunofluorescence of emerin and YAP in matrices with different densities as well as before and after transmigration across defined collagen I matrix interfaces. (A) Fold change of the nuclear membrane protein emerin normalized either to the dense matrix condition or the non-transmigrated cells in the interface matrices (n > 20). (B) Confocal images of emerin (green) in MDA-MB-231 breast cancer cells cultivated in d → o matrix interfaces after 7 d of cultivation. DAPI staining is shown in blue; greyscale shows reflection mode images of collagen matrices. (C) Fluorescence intensity plot along a cross section of the nucleus shown in (B) or ESI Fig. S3.† The xy distance was normalized according to the position of the nucleus assessed by the DAPI channel. (D) Fold change of nuclear localized YAP normalized either to the dense matrix condition or the non-transmigrated cells in the interface matrices (n > 20). (E) Confocal images of YAP (red) in MDA-MB-231 breast cancer cells cultivated in d → o matrix interfaces after 7 d of cultivation. (F) Fluorescence intensity plot along a cross section of the nucleus shown in (E) or ESI Fig. S4.† DAPI staining is shown in blue; greyscale shows reflection mode images of collagen matrices. Data are presented as the mean ± SD. *, ** and *** represent p < 0.05, p < 0.01 and p < 0.001. n.s. states nonsignificant differences. Image size: 100 μm × 100 μm. d – dense, o – open, d → o – dense to open, o → d – open to dense, nt – non-transmigrated, t – transmigrated. | ||
Subsequently, we examined emerin in cells before and after transmigration of matrix interfaces. Here, we observed a decrease in total emerin by more than half after transmigration across the d → o interface, contrasting the behavior in the homogeneous matrices. The loss of emerin indicates a change in cellular phenotype and not the expected response to a lower stiffness or matrix density. MDA-MB-231 cells transmigrating in the control configuration o → d showed a comparable significant decrease in emerin after transmigration, recapitulating the cells’ response as observed in the homogeneous controls.
Mechanical stress induces the transport of emerin from the nuclear interior to the cytoplasm and facilitates the mislocalization from the inner nuclear membrane to the outer nuclear membrane.48,49 Besides total emerin intensity analysis, we additionally examined emerin distribution at the nucleus by plotting intensity cross sections of the nucleus (Fig. 4C). For the dense and open homogeneous controls we observed a peak incidence of emerin at the nuclear membrane with overall more nuclear emerin for the open porous condition. With increasing matrix density, emerin appeared to be more present in the cytoplasm than in the nuclear interior (ESI Fig. S3†). This recapitulates previous findings from colorectal adenocarcinoma cells that showed an increased amount of emerin when cultured on soft polyacrylamide gels compared to stiff ones.49 Additionally, the colorectal adenocarcinoma cells had a higher emerin localization in the nuclear interior and at the nuclear membrane. This altered emerin localization facilitates the mislocalization of chromosome territories leading to an altered transcriptome. For transmigrated MDA-MB-231 breast cancer cells from a dense into an open matrix, we observed an enrichment of emerin at the nuclear membrane only and a more similar distribution in the nucleus interior and the cytoplasm, contrasting expectations for softer matrices and results of homogeneous matrices.48 This contrasting behavior of the transmigrated cells in the d → o configuration indicates that the relocalization of emerin into the nucleus during transmigration may be a crucial factor in the observed phenotypic changes, suggesting potential misregulation of its mechanotransductive function.
To demonstrate that additional mechanosignaling pathways are activated by transmigration across d → o interfaces and might modulate gene expression upon mechanical stress, we stained the transcriptional coactivator YAP in MDA-MB-231 cells. The YAP/TAZ pathway is known to be activated under high mechanical forces like an increased stiffness of the surrounding matrix.23 Upon activation, the transcription factors YAP and TAZ are relocalized from the cytoplasm into the nucleus, where they induce the expression of a wide range of genes involved in cell proliferation, survival and migration.51 Inactivation leads to relocalization in the cytoplasm, phosphorylation and subsequent degradation.23,52
We determined the activity of the YAP/TAZ signaling pathway similar to emerin by immunostaining YAP. In contrast to previous studies, where the ratio between nuclear and cytoplasmic YAP was quantified in two-dimensional cultured cells,53,54 we quantified the total amount of nuclear YAP (Fig. 4D and E, ESI Fig. S4†). The three-dimensional spreading of cells within the collagen I matrix and the use of mid-range resolution to cover a large number of cells for analysis make nuclear-to-cytoplasmic ratio profiling an inaccurate approach in the context of our experimental settings. The nuclear localized YAP exhibited a significant increase with decreasing matrix density, in contrast to previous reports of a decrease in nuclear YAP with decreasing matrix stiffness (or density).23,53 A direct comparison of previous findings with our data is not straightforward, as the regulation of YAP has been studied only in environments in the kPa range. The elastic modulus of our collagen I matrices was previously examined in the range of 100 Pa; hence, similar elasticity-dependent activation to that for matrices with elastic moduli in the range of kPa cannot be expected.35 However, the localization of YAP is not only regulated by the elasticity of the ECM but also by the morphology and spreading of the cells. The YAP protein was reported to be preferentially located in the cell nucleus when human mesenchymal stem cells spread over a large area or micropillars, contrary to spreading over smaller areas.23 According to our immunostaining experiment, we can assume that in overall soft microenvironments, such as collagen I matrices, the density of the surrounding network has a stronger influence than its elasticity. Following this line of argumentation, the found YAP deactivation at high matrix density in our experiments can be correlated to a less spread cell morphology. Details of this complex regulatory mechanism of YAP activation depending on matrix elasticity and cell morphology in three dimensional matrices in overall soft matrices need to be investigated in the future.
When analyzing YAP localization before and after transmigration across d → o interfaces, we observe no alterations in YAP localization (Fig. 4D–F), contrasting the behavior in homogeneous matrices of different densities. We would expect an increase in transmigrated cells into the open porous compartment. While d → o transmigration is not in line with the expected YAP signaling, the control condition of o → d transmigration agrees with the behavior in homogeneous matrices with a decrease in the YAP signal for the transmigrated cells into the denser compartment. This result shows mislocalization of YAP in transmigrated cells in the d → o configuration, which could indicate a misregulation of the YAP/TAZ pathway, aligning with alterations in gene expression and cellular phenotype.34
Hence, the analysis of nuclear signaling pathways of mechanotransduction, namely emerin and YAP/TAZ, shows an altered nuclear mechanotransduction to be involved in the phenotypic switch of MDA-MB-231 breast cancer cells during transmigration of matrix interfaces.
3.5. General discussion
In prior work, we and others showed that MDA-MB-231 breast cancer cells that migrated across sharp interfaces between two differently porous collagen I matrices altered their migratory phenotype, suggesting a direct influence of tissue boundaries on the tumor cell behavior.32,33 Further investigations revealed a general phenotype switching towards a more invasive and aggressive behavior of MDA-MB-231 breast cancer cells after crossing a d → o matrix interface, including increased proliferation and chemoresistance accompanied by an overall altered transcriptome. In addition, transmigration induced deformed or fragmented nuclei together with an increased probability of DNA damage at the interface. It was hypothesized that a molecular mechanism leading to phenotype switching involves mechanical stress acting on the transmigrating cell due to high forward stress caused by the steep decrease in matrix density at the interface.34Here, we demonstrated that cells transmigrating from dense to open compartments exhibit a strong and highly localized contraction at the moment of interface crossing, as indicated by a region of high deformation and convergence in the open porous compartment. These results suggest that the cells, including the nucleus, are exposed to mechanical stress during this process of transmigration and experience differential mechanical signals along their migration direction. The pronounced and highly localized matrix deformation and convergence found only at d → o interfaces highlight the significant influence of interface microtopology on cell behavior. Additionally, this result implies that cells leaving a primary tumor into the surrounding healthy tissue, crossing an interface between differently porous matrices, may experience similar influences. In line with our previous results, one can conclude that under such conditions, the metastatic properties of cells can increase due to the steep decrease in matrix density. These findings support the critical role of the tumor microenvironment's topology in shaping tumor cell phenotypes, promoting a more invasive and aggressive behavior, and facilitating metastasis to distant organs, and an important role of mechanical signals and stresses.
Furthermore, the direction of the steep topological changes appears to be important as cells migrating from an open matrix into a denser matrix do not experience such an influence of the interface. We propose that cells migrating from the denser compartment into the more open porous matrix tend to strongly stretch into the open compartment with the migrating front, while the main cell body with the nucleus is still surrounded and constrained by the dense matrix compartment, leading to asymmetric contractions and mechanical stress to the cell body and the nucleus.
Together with the finding of asymmetric cellular forces along the cell body during the transmigration process, we functionally proved that the previously observed phenotype switch in migration, proliferation, and chemoresistance is accompanied by an increase in the contractility of the cells. Both findings reinforce our hypothesis that cell mechanics play a major role in the interfacial crossing process and the altered cellular phenotype, as higher contractility is closely associated with increased invasiveness.
Our investigations of nuclear mechanotransduction pathways additionally support the involvement of mechanical signals in the molecular mechanisms of phenotype switching at the tumor–tissue interface. The examination of the nuclear envelope protein emerin revealed a general mislocalization in transmigrated cells. Although the downregulation of emerin and its increased localization at the nuclear membrane after transmigration across d → o interfaces do not clearly indicate activation or deactivation of the signaling pathway, these changes suggest a misregulation of emerin by the respective mechanotransductive processes. This misregulation contributes to the observed phenotypic changes, further supporting the altered gene expression profile reported earlier. Previous work has already shown that the regulation of emerin is very diverse and already differs in soft matrices compared to stiff matrices.48,49 While the precise effects of emerin localization remain unclear, it is consistently involved in nuclear mechanotransduction, with its regulation varying depending on the cellular context and mechanical environment. The molecular mechanism by which emerin is regulated in nuclear mechanotransduction is currently not known and needs to be further investigated in the future. The YAP pathway is the main signaling pathway usually analyzed to determine whether mechanotransduction processes are activated in cells.55 This pathway additionally exhibited misregulation after transmigration of MDA-MB-231 cells across d → o interfaces, supporting the findings for emerin. Even though the cells migrate in our matrix interface model from a dense matrix into an open porous compartment, we observed an unaltered localization of YAP in the nucleus.
4. Conclusions
Taken together, our results revealed that MDA-MB-231 breast cancer cells experience strong and highly localized contractile forces while migrating across biomimetic tumor–tissue interfaces. This mechanical stress leads to deformations of the cell body and nucleus, DNA damage, and subsequently to misregulation of mechanotransductive pathways and a phenotype switch towards more invasive and metastatic breast cancer cells. Our study deepens the understanding of how topological features and spatial disparities within the extracellular matrix microenvironment influence the metastatic potential of tumor cells.Although we gained deeper insights into the involvement of mechanical signals during the transmigration of breast cancer cells at matrix interfaces, as well as the mechanical state of the cells throughout and after this process, follow-up research is needed. Exploration of the molecular details of mechanotransductive processes happening as a consequence of the strong and highly localized contractile cellular forces is necessary to understand these mechanisms. In addition, the use of a single breast cancer cell line represents a model limitation in our study. Future experiments should incorporate additional breast cancer cell lines and other cell types to gain a more comprehensive understanding of how mechanical stress at matrix interfaces influences cellular properties and phenotypic changes.
Our new findings highlight that targeting mechanotransductive pathways such as YAP or the activation of strong contractile forces involved in the found processes of altered cellular phenotype during transmigration of matrix interfaces may offer promising therapeutic strategies for managing metastatic breast cancer by preventing or reversing phenotypic changes associated with increased invasiveness.
Author contributions
Cornelia Clemens: Conceptualization, investigation, formal analysis, writing – original draft, and editing. Rosa Gehring: investigation and formal analysis. Philipp Riedl: conceptualization, investigation, and formal analysis. Tilo Pompe: Conceptualization, writing – original draft, review, and editing.Data availability
All data and home-built MATLAB and Python scripts reported in this work are available from the corresponding author upon reasonable request. The MATLAB script for network topology analysis is freely available at https://git.sc.uni-leipzig.de/pe695hoje/ topology-analysis.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by grants from EFRE and Free State of Saxony (SAB, grant: 100144684) and Deutsche Krebshilfe (grant: 70113998). The usage of the BioImaging Core Facility of the Faculty of Life Sciences of Leipzig University, supported by a grant from Deutsche Forschungsgemeinschaft INST 268/293-1 FUGG and INST 268/394-1 FUGG to Tilo Pompe, is gratefully acknowledged.References
- M. Najafi, B. Farhood and K. Mortezaee, J. Cell. Biochem., 2019, 120, 2782–2790 CrossRef CAS PubMed.
- H. Mohammadi and E. Sahai, Nat. Cell Biol., 2018, 20, 766–774 CrossRef CAS PubMed.
- D. T. Butcher, T. Alliston and V. M. Weaver, Nat. Rev. Cancer, 2009, 9, 108–122 CrossRef CAS PubMed.
- J. Sapudom and T. Pompe, Biomater. Sci., 2018, 6, 2009–2024 RSC.
- P. P. Provenzano, D. R. Inman, K. W. Eliceiri, J. G. Knittel, L. Yan, C. T. Rueden, J. G. White and P. J. Keely, BMC Med., 2008, 6, 11 CrossRef PubMed.
- P. P. Provenzano, K. W. Eliceiri, J. M. Campbell, D. R. Inman, J. G. White and P. J. Keely, BMC Med., 2006, 4, 38 CrossRef PubMed.
- P. P. Provenzano, D. R. Inman, K. W. Eliceiri and P. J. Keely, Oncogene, 2009, 28, 4326–4343 CrossRef CAS PubMed.
- J. Sapudom, S. Rubner, S. Martin, T. Kurth, S. Riedel, C. T. Mierke and T. Pompe, Biomaterials, 2015, 52, 367–375 CrossRef CAS PubMed.
- J. Sapudom, L. Kalbitzer, X. Wu, S. Martin, K. Kroy and T. Pompe, Biomaterials, 2019, 193, 47–57 CrossRef CAS PubMed.
- M. J. Paszek, N. Zahir, K. R. Johnson, J. N. Lakins, G. I. Rozenberg, A. Gefen, C. A. Reinhart-King, S. S. Margulies, M. Dembo, D. Boettiger, D. A. Hammer and V. M. Weaver, Cancer Cell, 2005, 8, 241–254 CrossRef CAS PubMed.
- O. Chaudhuri, S. T. Koshy, C. Da Branco Cunha, J.-W. Shin, C. S. Verbeke, K. H. Allison and D. J. Mooney, Nat. Mater., 2014, 13, 970–978 CrossRef CAS PubMed.
- F. Bordeleau, T. A. Alcoser and C. A. Reinhart-King, Am. J. Physiol.: Cell Physiol., 2014, 306, 110–120 CrossRef PubMed.
- K. M. Yamada, A. D. Doyle and J. Lu, Trends Cell Biol., 2022, 32, 883–895 CrossRef CAS PubMed.
- B. Zimerman, T. Volberg and B. Geiger, Cell Motil. Cytoskeleton, 2004, 58, 143–159 CrossRef CAS PubMed.
- J. D. Humphrey, E. R. Dufresne and M. A. Schwartz, Nat. Rev. Mol. Cell Biol., 2014, 15, 802–812 CrossRef CAS PubMed.
- N. Q. Balaban, U. S. Schwarz, D. Riveline, P. Goichberg, G. Tzur, I. Sabanay, D. Mahalu, S. Safran, A. Bershadsky, L. Addadi and B. Geiger, Nat. Cell Biol., 2001, 3, 466–472 CrossRef CAS PubMed.
- C.-M. Lo, H.-B. Wang, M. Dembo and Y. Wang, Biophys. J., 2000, 79, 144–152 CrossRef CAS PubMed.
- J. Steinwachs, C. Metzner, K. Skodzek, N. Lang, I. Thievessen, C. Mark, S. Münster, K. E. Aifantis and B. Fabry, Nat. Methods, 2016, 13, 171–176 CrossRef CAS PubMed.
- D. Shakiba, F. Alisafaei, A. Savadipour, R. A. Rowe, Z. Liu, K. M. Pryse, V. B. Shenoy, E. L. Elson and G. M. Genin, ACS Nano, 2020, 14, 7868–7879 CrossRef CAS PubMed.
- J. L. Tan, J. Tien, D. M. Pirone, D. S. Gray, K. Bhadriraju and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1484–1489 CrossRef CAS PubMed.
- C. Mark, T. J. Grundy, P. L. Strissel, D. Böhringer, N. Grummel, R. Gerum, J. Steinwachs, C. C. Hack, M. W. Beckmann, M. Eckstein, R. Strick, G. M. O'Neill and B. Fabry, eLife, 2020, 9, e51912 CrossRef CAS PubMed.
- P. Riedl and T. Pompe, Biomater. Sci., 2021, 9, 5917–5927 RSC.
- S. Dupont, L. Morsut, M. Aragona, E. Enzo, S. Giulitti, M. Cordenonsi, F. Zanconato, J. Le Digabel, M. Forcato, S. Bicciato, N. Elvassore and S. Piccolo, Nature, 2011, 474, 179–183 CrossRef CAS PubMed.
- M. Liu, J. Yang, B. Xu and X. Zhang, MedComm, 2021, 2, 587–617 CrossRef PubMed.
- I. J. Fidler and M. L. Kripke, Cancer Metastasis Rev., 2015, 34, 635–641 CrossRef CAS PubMed.
- J. A. Eble and S. Niland, Clin. Exp. Metastasis, 2019, 36, 171–198 CrossRef CAS PubMed.
- F. Kai, A. P. Drain and V. M. Weaver, Dev. Cell, 2019, 49, 332–346 CrossRef CAS PubMed.
- D. M. Gilkes, G. L. Semenza and D. Wirtz, Nat. Rev. Cancer, 2014, 14, 430–439 CrossRef CAS PubMed.
- I. Acerbi, L. Cassereau, I. Dean, Q. Shi, A. Au, C. Park, Y. Y. Chen, J. Liphardt, E. S. Hwang and V. M. Weaver, Integr. Biol., 2015, 7, 1120–1134 CrossRef CAS PubMed.
- S. Kang, H. S. Shim, J. S. Lee, D. S. Kim, H. Y. Kim, S. H. Hong, P. S. Kim, J. H. Yoon and N. H. Cho, J. Proteome Res., 2010, 9, 1157–1164 CrossRef CAS PubMed.
- H.-C. Hsu, L.-C. Liu, H.-Y. Wang, C.-M. Hung, Y.-C. Lin, C.-T. Ho and T.-D. Way, PLoS One, 2017, 12, 0164661 Search PubMed.
- J. Sapudom, S. Rubner, S. Martin and T. Pompe, Adv. Healthcare Mater., 2016, 5, 1861–1867 CrossRef CAS PubMed.
- F. Bordeleau, L. N. Tang and C. A. Reinhart-King, Phys. Biol., 2013, 10, 065004 CrossRef CAS PubMed.
- P. Riedl, J. Sapudom, C. Clemens, L. Orgus, A. Pröger, J. C. M. Teo and T. Pompe, ACS Appl. Mater. Interfaces, 2023, 15, 24059–24070 CrossRef CAS PubMed.
- K. Franke, J. Sapudom, L. Kalbitzer, U. Anderegg and T. Pompe, Acta Biomater., 2014, 10, 2693–2702 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- R: The R Project for Statistical Computing, https://www.r-project.org/, (accessed November 28, 2024).
- P. Pakshir, M. Alizadehgiashi, B. Wong, N. M. Coelho, X. Chen, Z. Gong, V. B. Shenoy, C. A. McCulloch and B. Hinz, Nat. Commun., 2019, 10, 1850 CrossRef PubMed.
- N. Wang, J. D. Tytell and D. E. Ingber, Nat. Rev. Mol. Cell Biol., 2009, 10, 75–82 CrossRef CAS PubMed.
- L. Kalbitzer, K. Franke, S. Möller, M. Schnabelrauch and T. Pompe, J. Mater. Chem. B, 2015, 3, 8902–8910 RSC.
- C. M. Kraning-Rush, J. P. Califano and C. A. Reinhart-King, PLoS One, 2012, 7, e32572 CrossRef CAS PubMed.
- A. Aung, Y. N. Seo, S. Lu, Y. Wang, C. Jamora, J. C. del Álamo and S. Varghese, Biophys. J., 2014, 107, 2528–2537 CrossRef CAS PubMed.
- C. M. Denais, R. M. Gilbert, P. Isermann, A. L. McGregor, M. te Lindert, B. Weigelin, P. M. Davidson, P. Friedl, K. Wolf and J. Lammerding, Science, 2016, 352, 353–358 CrossRef CAS PubMed.
- R. Golloshi, C. Playter, T. F. Freeman, P. Das, T. I. Raines, J. H. Garretson, D. Thurston and R. P. McCord, EMBO Rep., 2022, 23, 52149 CrossRef PubMed.
- D. Fanfone, Z. Wu, J. Mammi, K. Berthenet, D. Neves, K. Weber, A. Halaburkova, F. Virard, F. Bunel, C. Jamard, H. Hernandez-Vargas, S. W. G. Tait, A. Hennino and G. Ichim, eLife, 2022, 11, 73150 CrossRef PubMed.
- Y. Tang, L. Zhu, J.-S. Cho, X.-Y. Li and S. J. Weiss, Dev. Cell, 2022, 57, 480–495 CrossRef CAS PubMed.
- C. Guilluy, L. D. Osborne, L. van Landeghem, L. Sharek, R. Superfine, R. Garcia-Mata and K. Burridge, Nat. Cell Biol., 2014, 16, 376–381 CrossRef CAS PubMed.
- H. Q. Le, S. Ghatak, C.-Y. C. Yeung, F. Tellkamp, C. Günschmann, C. Dieterich, A. Yeroslaviz, B. Habermann, A. Pombo, C. M. Niessen and S. A. Wickström, Nat. Cell Biol., 2016, 18, 864–875 CrossRef CAS PubMed.
- R. Pradhan, D. Ranade and K. Sengupta, Nucleic Acids Res., 2018, 46, 5561–5586 CrossRef CAS PubMed.
- A. Fernandez, M. Bautista, L. Wu and F. Pinaud, J. Cell Sci., 2022, 135, jcs258969 CrossRef CAS PubMed.
- Z. Meng, T. Moroishi and K.-L. Guan, Genes Dev., 2016, 30, 1–17 CrossRef CAS PubMed.
- S. Dupont and S. A. Wickström, Nat. Rev. Genet., 2022, 23, 624–643 CrossRef CAS PubMed.
- A. Elosegui-Artola, I. Andreu, A. E. M. Beedle, A. Lezamiz, M. Uroz, A. J. Kosmalska, R. Oria, J. Z. Kechagia, P. Rico-Lastres, A.-L. Le Roux, C. M. Shanahan, X. Trepat, D. Navajas, S. Garcia-Manyes and P. Roca-Cusachs, Cell, 2017, 171, 1397–141014 CrossRef CAS PubMed.
- T. P. Driscoll, B. D. Cosgrove, S.-J. Heo, Z. E. Shurden and R. L. Mauck, Biophys. J., 2015, 108, 2783–2793 CrossRef CAS PubMed.
- X. Cai, K.-C. Wang and Z. Meng, Front. Cell Dev. Biol., 2021, 9, 673599 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4bm01589d |
| This journal is © The Royal Society of Chemistry 2025 |