
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pentablock thermoresponsive hydrogels for chemotherapeutic delivery in a pancreatic cancer model†
Amr
Elsherbeny‡
 abc,
Hulya
Bayraktutan
acd,
Nurcan
Gumus
acef,
Phoebe
McCrorie
c,
Andres
Garcia-Sampedro
g,
Shreeya
Parmar
b,
Alison A.
Ritchie
b,
Marian
Meakin
b,
Umut Can
Oz
ah,
Ruman
Rahman
c,
Jennifer C.
Ashworth
ci,
Anna M.
Grabowska
bc,
Cara
Moloney‡
ac and
Cameron
Alexander
*a
abc,
Hulya
Bayraktutan
acd,
Nurcan
Gumus
acef,
Phoebe
McCrorie
c,
Andres
Garcia-Sampedro
g,
Shreeya
Parmar
b,
Alison A.
Ritchie
b,
Marian
Meakin
b,
Umut Can
Oz
ah,
Ruman
Rahman
c,
Jennifer C.
Ashworth
ci,
Anna M.
Grabowska
bc,
Cara
Moloney‡
ac and
Cameron
Alexander
*a
aDivision of Molecular Therapeutics and Formulation, School of Pharmacy, University of Nottingham, Nottingham NG7 2RD, UK. E-mail: amr.elsherbeny@nottingham.ac.uk; a.elsherbeny@mesox.co.uk; hulya.bayraktutan@hacettepe.edu.tr; nurcan.gumus@bakircay.edu.tr; cameron.alexander@nottingham.ac.uk
bEx Vivo Cancer Pharmacology Centre, Translational Medical Sciences, Biodiscovery Institute, School of Medicine, University of Nottingham, Nottingham NG7 2UH, UK. E-mail: alison.ritchie@nottingham.ac.uk; shreeya.parmar@nottingham.ac.uk; marian.meakin@nottingham.ac.uk; anna.grabowska@nottingham.ac.uk
cBiodiscovery Institute, School of Medicine, University of Nottingham, Nottingham, NG7 2UH, UK. E-mail: ruman.rahman@nottingham.ac.uk; jennifer.ashworth@nottingham.ac.uk; cara.moloney@nottingham.ac.uk
dDepartment of Pharmaceutical Biotechnology, Faculty of Pharmacy, Hacettepe University, Ankara 06100, Türkiye
eDepartment of Medical Biology, Faculty of Medicine, Izmir Bakircay University, Izmir, Türkiye
fDepartment of Medical Pharmacology, Faculty of Medicine, Izmir Bakircay University, Izmir, Türkiye
gDepartment of Chemical Engineering and Biotechnology, University of Cambridge, Philippa Fawcett Dr, Cambridge CB3 0AS, UK. E-mail: ag2421@cam.ac.uk
hDepartment of Pharmaceutical Technology, Faculty of Pharmacy, Ankara University, Ankara 06560, Türkiye. E-mail: umutcanoz@ankara.edu.tr
iSchool of Veterinary Medicine & Science, University of Nottingham, Sutton Bonington Campus, Leicestershire, LE12 5RD, UK
First published on 10th February 2025
Abstract
The design of biodegradable and thermoresponsive polymeric hydrogels with tuneable properties holds immense promise for localised and sustained drug delivery. In this study, we designed and synthesised a library of novel pentablock copolymers, incorporating poly(D,L-lactide) (PLA) into methoxypoly(ethylene glycol)-poly(ε-caprolactone)-methoxypoly(ethylene glycol) (mPEG-PCL-mPEG, or PECE) hydrogels to enhance the hydrolytic degradation and drug release profiles. A pentablock copolymer, methoxypoly(ethylene glycol)-b-poly(D,L lactide)-b-poly(ε-caprolactone)-b-poly(D,L lactide)-b-methoxypoly(ethylene glycol) (mPEG-PLA-PCL-PLA-mPEG, or PELCLE), was selected based on its thermoresponsive sol–gel transition behaviour at a physiologically relevant temperature (37 °C). Physicochemical characterisation revealed that both PECE and PELCLE hydrogels self-assembled into micellar structures, with PELCLE exhibiting smaller micellar sizes compared to PECE. The incorporation of PLA led to reduced hydrogel stiffness, enhanced degradability, and decreased swelling compared to PECE. In vitro drug release studies demonstrated that both hydrogels exhibited sustained release of various anti-cancer drugs, with PELCLE generally showing slower release kinetics, highlighting its potential for prolonged drug delivery. For potential pancreatic cancer applications, we evaluated the biocompatibility and therapeutic efficacy of PELCLE hydrogels loaded with gemcitabine and oxaliplatin (GEMOX). In vitro and in vivo studies demonstrated safety and some anti-tumour efficacy of GEMOX-loaded PELCLE compared to free drug administration, attributed to enhanced tumour retention and sustained drug release. These findings highlight the potential of the PELCLE hydrogel as a versatile and effective local drug delivery platform for the treatment of pancreatic cancer and other solid tumours, warranting further investigation towards its clinical translation.
Introduction
Systemic chemotherapy, while effective, often results in significant toxicity to healthy tissues, limiting clinically administered doses.1–3 This has spurred the development of local drug delivery systems, aiming to achieve sustained and targeted release, thereby minimizing systemic toxicity while maintaining therapeutic efficacy at the target site.4,5 Biodegradable, thermoresponsive hydrogels have emerged as a promising approach due to their ability to deliver a variety of therapeutic agents, including drugs, proteins, and cell carriers, and their potential for use in tissue engineering.6–10 These injectable hydrogels offer several advantages, including minimally invasive administration and localised drug delivery.11–13 Furthermore, their ability to undergo gelation at body temperature enables depot drug release, reducing administration frequency and improving patient quality of life.14–16 This sustained release also protects the therapeutic agent from rapid metabolism and elimination, potentially extending its efficacy.17Amphiphilic thermosensitive polymers are particularly attractive due to their ability to undergo sol–gel transitions via physical crosslinking and micellar aggregation above a critical concentration, under mild conditions.3,18–20 This eliminates the need for potentially toxic chemical crosslinkers or pH adjustments, offering a safer alternative to chemical gelation. Among these, methoxypoly(ethylene glycol)-poly(ε-caprolactone)-methoxypoly(ethylene glycol) (mPEG-PCL-mPEG, or PECE) has been considered as a promising candidate for various healthcare applications as it is composed of polymers that have been used clinically.21–23 PECE-based hydrogels have been explored as drug delivery systems for diverse therapeutic agents in conditions such as ocular inflammation, bacterial infections, and bone regeneration.22–24
The gelation behaviour and rheological properties of PECE polymers are intrinsically linked to the balance between hydrophilic and hydrophobic components, with factors like chemical composition, block length, and molecular weight of PCL and PEG influencing the sol–gel transition.23,25 Moreover, the hydrophobic nature of PCL units significantly impacts hydrogel degradation rates, typically leading to prolonged in vivo retention (2–3 years).26 To address this, we introduced PLA blocks into PECE using a systematic approach, creating an ABCBA pentablock polymer methoxypoly(ethylene glycol)-b-poly(ε-caprolactone) poly(D,L lactide)-b-poly(ε-caprolactone)-b-methoxypoly(ethylene glycol) (mPEG-PCL-PLA-PCL-mPEG). We also synthesised an analogue methoxypoly(ethylene glycol)-b-poly(D,L lactide)-b-poly(ε-caprolactone)-b-poly(D,L lactide)-b-methoxypoly(ethylene glycol) (mPEG-PLA-PCL-PLA-mPEG), with the aim of enhancing its hydrolytic degradation as a result of the faster degradation times of PLA as compared to PCL blocks.27 Additionally, we hypothesised that the inclusion of PLA blocks would modulate hydrogel swelling and water uptake, potentially leading to modified drug release profiles.28
We made a series of pentablock copolymers through the incorporation of PLA blocks into PECE polymers, and from these selected a polymer (designated as PELCLE) which was freely soluble in aqueous buffer below 20 °C but which formed a viscous hydrogel-like material at 37 °C. DLS and TEM were used to characterize material structures and SEM to examine macro-structural changes. Additionally, we evaluated in vitro swelling behaviour, hydrogel degradation, and in vitro drug release profiles of various chemotherapeutic agents with diverse cLogP values (oxaliplatin, gemcitabine, doxorubicin and olaparib), correlating release mechanisms with mathematical models.
We then explored PELCLEs for localised and sustained GEMOX delivery in pancreatic cancer models. Tolerability of gel formulations was assessed in vitro using pancreatic cancer cell lines and in vivo using PANC1-FLuc xenograft mouse models. We confirmed the synergistic cytotoxic effects of GEMOX at various molar ratios and investigated the impact of GEMOX loading on PELCLE properties and drug release profile. To evaluate tumour retention, we compared the release profile of the hydrophilic fluorescent dye Cy5.5 from PELCLE hydrogels to free Cy5.5 in subcutaneous xenograft models. Finally, the anti-tumour efficacy of GEMOX-loaded PELCLE hydrogels was compared to free GEMOX, both in vitro using PANC-FLuc cells and in vivo in subcutaneous xenograft PANC-FLuc mouse models.
Materials and methods
Materials
Mono-methoxy poly(ethylene glycol) (mPEG, 500 g mol−1), ε-caprolactone (εCL, 97%), tin(II) 2-ethylhexanoate (SnOct2, 92.5–100%), hexamethylene diisocyanate (HMDI, ≥99%), dulbecco's modified eagle media (DMEM), trypsin-EDTA solution, fetal bovine serum (FBS), L-glutamine, and all deuterated solvents were purchased form Sigma-Aldrich. Gemcitabine hydrochloride (≥98%) and oxaliplatin (≥98%) were obtained from Biosynth. Cyanine5.5 (Cy5.5) alkyne was purchased from Lumiprobe. PrestoBlue™ was purchased from Thermo Fisher Scientific. Human pancreatic adenocarcinoma cells (PANC-1) were obtained from the European Collection of Authenticated Cell Cultures (ECACC) and lentivirally transduced in house with the Firefly Luciferase gene (FLuc). All solvents were purchased from Fisher Scientific. Ki67 (ab16667) primary antibody was purchased from Abcam. Normal Goat Serum (10098792) was purchased from Gibco. Dako REAL EnVision detection system (peroxidase/DAB), wash buffer and Dako REAL peroxidase-blocking solution were purchased from Agilent. All chemicals were used as received unless otherwise stated.Polymer synthesis and characterisation
| Polymer | Block orientation | Total LA units | Total εCL units | LA (mol) | εCL (mol) | Đ | GPC Mw (g mol−1) | NMR Mn (g mol−1) | Hydrophilic![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hydrophobic ratio :hydrophobic ratio |
|---|---|---|---|---|---|---|---|---|---|
| PECE | mPEG-PCL-mPEG | 0 | 8 | 0 | 0.111 | 1.1 | 11688 |
3220 | 1:2 |
| P1 | mPEG-PCL-PLA-PCL-mPEG | 4 | 4 | 0.04 | 0.038 | 1.1 | 11069 |
3230 | 1:2 |
| P2 | mPEG-PLA-PCL-PL-mPEG | 4 | 4 | 0.04 | 0.038 | 1.1 | 13240 |
3230 | 1:2 |
| P3 | mPEG-PCL-PLA-PCL-mPEG | 1 | 7 | 0.014 | 0.07 | 1.2 | 13014 |
3230 | 1:2 |
| P4 | mPEG-PLA-PCL-PLA-mPEG | 1 | 7 | 0.014 | 0.07 | 1.2 | 15492 |
3230 | 1:2 |
| P5 | mPEG-PCL-PLA-PCL-mPEG | 6 | 1 | 0.055 | 0.018 | 1.1 | 12896 |
3240 | 1:2 |
| P6 | mPEG-PLA-PCL-PLA-mPEG | 6 | 1 | 0.055 | 0.018 | 1.3 | 21459 |
3120 | 1:2 |
The synthesis began with the azeotropic distillation of mPEG (5 g, 0.010 mmol, 500 g mol−1) in anhydrous toluene at 50 °C. Subsequently, predetermined amounts of D,L-lactide or ε-caprolactone were added to the reaction mixture, which was then stirred at 120 °C under a nitrogen atmosphere until a homogeneous solution was obtained. Polymerisation was initiated by the addition of Sn(Oct)2 (52.2 mg, 0.0004 mmol) dissolved in CH2Cl2. The reaction was allowed to proceed overnight, and complete monomer conversion to the corresponding mPEG-PLA or mPEG-PCL diblock copolymers was confirmed by 1H NMR analysis.
To synthesize the triblock copolymers, ε-caprolactone or D,L-lactide was added to the respective mPEG-PLA or mPEG-PCL diblock solutions, and the polymerisation process was repeated as described above. For the final pentablock copolymer formation, the reaction temperature was lowered to 80 °C, HMDI (1.68 g, 0.01 mmol) was introduced and the reaction was maintained for an additional 24 hours. The resulting pale-yellow viscous product was dissolved in CH2Cl2, precipitated in cold hexane, and subsequently centrifuged (5 min, 4200g). The isolated copolymer was then dried under vacuum at 30 °C overnight and analysed by FT-IR, GPC, and 1H NMR.
550 g mol−1. The calibration was achieved using Agilent EasyVial calibrants, incorporating a cubic function to establish a correlation between retention time and molar mass. The Cirrus GPC software was employed for this purpose. Polymer samples were made by dissolving 2 mg mL−1 pure polymer in 1 mL DMF + 0.1% LiBr. 100 μL samples were injected and eluted at 1 ml min−1 for 30 min.
Evaluation of hydrogel formation in synthesized polymers and selection of an optimum polymer
Physicochemical characterisation and comparison of PECE and PELCLE hydrogels
The swelling ratio (%) of the hydrogels was calculated using the following equation:

Drug-loaded hydrogels (100 μL) were injected into Eppendorf tubes and allowed to gel at 37 °C for 30 minutes. Pre-warmed PBS (pH 7.4, 1 mL) was added, and the tubes were incubated at 37 °C. At predetermined time points, the release medium was collected and replaced with fresh PBS. Drug concentrations were quantified using appropriate techniques.
For OXA and GEM, the same high performance liquid chromatography (HPLC) technique was utilised using a Shimadzu UFLC system (Shimadzu Corporation, Kyoto, Japan). A Hichrom 5 C18 (250 × 4.6 mm) column at room temperature was used and the mobile phase consisted of water:acetonitrile (97:3) at a flow rate of 0.8 mL min−1. The injection volume was 40 μL and the drug was monitored at 254 nm for both agents. The retention times of OXA and GEM were found to be 8.5 and 11 min, respectively. The method produced linear responses in the concentration range of 1–2500 μg ml−1 for GEM and 1–250 μg ml−1 for OXA.
The drug release samples of OLA were also analysed using HPLC quantification method (Agilent technologies, USA). A Hichrom 5 C18 column at room temperature (250 × 4.6 mm) was used and the mobile phase consisted of acetonitrile:ammonium acetate (10 mM, pH 4) (45:55) at a flow rate of 1.0 mL min−1. The injection volume was 20 μL and the drug was monitored at 254 nm. The retention time for OLA was found to be at 3.8 min. LC solution software was used to analyse the chromatograms. The method produced linear responses in the concentration range of 0.05–50 μg ml−1.
A fluorescence spectrophotometric method (TECAN Spark 10 M Multimode Microplate Reader (TECAN, Männedorf, Switzerland)) was used to assess the amounts of released DOX at an excitation/emission of 470/595 nm. The method produced linear responses in the concentration range of 0.01–5 μg ml−1.
The drug release mechanism for the initial 60% release was evaluated by fitting the data to zero-order, first-order, Hixson-Crowell (HC), Higuchi, and Korsmeyer-Peppas models. All mathematical equations are described in the ESI.†
Evaluation of gemcitabine and oxaliplatin (GEMOX)-loaded PELCLE hydrogels for pancreatic cancer treatment in a model system
Cell culture. Human pancreatic ductal adenocarcinoma PANC-FLuc cells were cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 2 mM L-glutamine. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. To ensure stable firefly luciferase (FLuc) expression, PANC-FLuc cells were cultured in the presence of 1 μg mL−1 puromycin every 5 passages.
Assessment of in vitro metabolic activity and synergistic effects of drug combinations. The cytotoxic effects of GEM, OXA, and GEMOX on PANC-FLuc cells were assessed using a PrestoBlue assay. Briefly, cells were seeded in 96-well plates (1.2 × 104 cells per well) and incubated for 24 hours. The medium was replaced with fresh medium containing GEM, OXA, or GEMOX at various molar ratios (GEM
:OXA = 6.5:1, 13:1, 26:1) and concentrations. After 72 hours of exposure, the medium was replaced with 10% PrestoBlue™ HS Cell Viability Reagent in DMEM, and fluorescence intensity (Ex/Em = 544/590 nm) was measured using a FLUOstar Omega plate reader (BMG LABTECH, UK). Relative metabolic activity was calculated, normalizing the negative control to 100%. Experiments were performed in triplicate, and results are presented as mean ± SD.
To evaluate the synergistic cytotoxicity of GEM and OXA on PANC-FLuc cells, the combination index (CI) analysis was utilised using the equation:29

In vitro metabolic activity of blank and GEMOX-loaded hydrogels. The biocompatibility of blank P2 hydrogels was assessed using a direct contact method. PANC-FLuc cells were seeded in 96-well plates (1.2 × 104 cells per well) and incubated for 24 hours. P2 hydrogels were prepared as described previously and diluted with cell culture media (DMEM) to create a range of concentrations for testing. Specifically, 86.6 μL of the hydrogel in its sol state was diluted with 913.4 μL of DMEM to form a 2 mg mL−1 P2 solution. Serial dilutions were then performed to achieve final hydrogel concentrations of 0.125 mg mL−1. These diluted hydrogel solutions were then added to the wells containing the cells. After 72 hours of exposure, the medium was replaced with 10% PrestoBlue™ HS Cell Viability Reagent in DMEM, and fluorescence intensity (Ex/Em = 544/590 nm) was measured after 20 minutes using a FLUOstar Omega plate reader. Relative metabolic activity was calculated, with the negative control normalised to 100%. Experiments were performed in triplicate, and results are presented as mean ± SD.
To evaluate the cytotoxicity of GEMOX-loaded PELCLE hydrogels, PANC-FLuc cells were seeded in 24-well plates (7.0 × 104 cells per well) and incubated for 24 hours. Free GEMOX solution (50 μL of DI water containing 2.5 mg GEM and 0.25 mg OXA, molar ratio of 13:1, respectively) or GEMOX-loaded PELCLE hydrogels (50 μL containing 30% w/v of hydrogel loaded with 2.5 mg GEM and 0.25 mg OXA, molar ratio of 13:1, respectively) were added to Greiner Transwell inserts (0.4 μm pore size) and pre-set at 37 °C for 30 minutes before being placed onto the cell-containing wells. Blank hydrogels and cell culture-grade water served as controls. After 72 hours of incubation, the inserts were removed, and cell viability was assessed using the PrestoBlue assay as described above. Experiments were performed in triplicate, and results are presented as mean ± SD.
Establishing subcutaneous in vivo PANC-FLuc models. All animal experiments were conducted in accordance with UK Home Office Project Licence numbers PPL P435A9CF8 and PP5089113, and with the approval of the University of Nottingham Animal Welfare and Ethical Review Body (AWERB). Experimental procedures adhered to the NCRI Guidelines on Experimental Neoplasia, the BVA/FRAME/RSPCA/UFAW Refining Procedures for the Administration of Substances Working Group report, and the NC3Rs Guidance for in vivo techniques, as well as the ARRIVE reporting guidelines.
Forty-two CD-1 NuNu mice (5–7 weeks old, 21 males, 21 females) were obtained from Charles River UK. Animals were housed in groups of three within individually ventilated cages, under a 12-hour light–dark cycle in a controlled environment (21 ± 2 °C, 55% ± 10% humidity). Food and water were provided ad libitum, and animal welfare was monitored throughout the study.
Following a one-week acclimatisation period, pancreatic tumours were initiated by subcutaneous injection of 100 μL of 1 × 107 mycoplasma-free PANC-FLuc cells suspended in Matrigel into the left flank. Tumour growth was monitored weekly via caliper measurements and bioluminescent imaging using the IVIS® Spectrum system. Animals were anesthetised with a ketamine/medetomidine cocktail (75 mg kg−1 + 1 mg kg−1, s.c.) prior to imaging and reversed with atipamezole (1 mg kg−1, s.c.) post imaging.
Biodistribution and peritumoural retention of Cy5.5/PELCLE hydrogels. To evaluate the peritumoural distribution and sustained release of drug-loaded PELCLE hydrogels, the hydrophilic fluorescent dye Cy5.5 was employed as a model drug. Established pancreatic cancer xenograft models (PANC-FLuc) were utilised. After 4 weeks of tumour growth, 12 mice were randomly divided into two groups (n = 6 per group, 3 males, 3 females). One group received intratumoural injections of 50 μL Cy5.5 solution (1 μg mL−1) in cell-culture grade water, while the other received 50 μL Cy5.5-loaded PELCLE hydrogel (1 μg mL−1 Cy5.5, 30% w/v PELCLE). At predetermined time points (4, 24, 72, 192, 264, and 336 hours post-administration), mice were anesthetised and imaged using the IVIS® Spectrum system (excitation/emission = 675/694 nm) to track Cy5.5 fluorescence. At each time point, one mouse from each group was euthanised, and tumours were excised for ex vivo imaging.
In vivo cytotoxicity of different treatment groups. Following four weeks of tumour establishment, 30 mice were randomised into five groups (n = 6 per group, 3 males, 3 females) and received peritumoural injections of 50 μL of either: normal saline, blank PELCLE hydrogel, free GEMOX (intravenously), free GEMOX (peritumourally), or GEMOX-loaded PELCLE hydrogel (peritumourally). All GEMOX formulations were administered at a concentration of 83.3 mg kg−1 GEM and 8.3 mg kg−1 OXA.
Animals were monitored daily for clinical signs and weighed twice weekly for 28 days post-treatment to assess the biocompatibility of the formulations. Tumour growth was also monitored as described above. At the study's conclusion, mice were euthanised, and tumours were excised for ex vivo imaging, weighing, and fixation in neutral buffered formalin for further analysis.
Histology, immunohistochemistry analysis of Ki-67 and TUNEL. Fixed tumour and organ samples were dehydrated and embedded in paraffin using a Leica TP1020 tissue processor. Sections (10 μm) were cut on a Leica RM2245 microtome.
For haematoxylin and eosin (H&E) staining, slides were deparaffinised in xylene, rehydrated through a graded series of industrial methylated spirit (IMS) solutions, and stained with Harris haematoxylin and eosin. Stained sections were dehydrated, mounted with DPX mounting medium, and imaged using a NanoZoomer®-SQ (Hamamatsu).
Ki67 immunohistochemistry (IHC) was performed according to the manufacturer's instructions. Briefly, deparaffinised and rehydrated slides underwent antigen retrieval in sodium citrate buffer (pH 6.0) using a microwave. Following incubation with normal goat serum (NGS) and hydrogen peroxide, a peroxidase blocking solution was applied. Slides were then incubated with the primary antibody (Ki67, 1:50 dilution in 30% NGS) overnight at 4 °C, followed by the secondary antibody and DAB solution. After counterstaining with haematoxylin, slides were dehydrated, mounted, and imaged. Positive Ki67 staining was quantified using QuPath-0.4.3.
Apoptosis was assessed using a modified TUNEL assay with an antidigoxigenin-peroxidase system. Deparaffinised and rehydrated slides were permeabilised with proteinase K, and endogenous peroxidases were quenched with H2O2. Following incubation with the TUNEL labelling mixture, slides were counterstained with methyl green, dehydrated, mounted, and imaged. TUNEL-positive cells were quantified using QuPath-0.4.3.
Statistical analyses
Statistical analyses of experimental data were conducted using t-test or analysis of variance (two-way ANOVA with Tukey's post-analysis test, with a single pooled variance). Values of p < 0.05 were considered statistically significant. (****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05).Results and discussion
Polymer synthesis, hydrogel formation, and rheological characterisation
A series of pentablock copolymers, including mPEG-b-PLA-b-PCL-HMDI-PCL-b-PLA-mPEG and mPEG-b-PCL-b-PLA-HMDI-PLA-b-PCL-mPEG, were synthesised via ring-opening copolymerisation, employing varying ratios of poly(ε-caprolactone) (PCL) to poly(D,L-lactide) (PLA) as detailed in Table 1. Methoxypoly(ethylene glycol)-poly(ε-caprolactone)-methoxypoly(ethylene glycol) (mPEG-PCL-mPEG, or PECE), synthesised as previously reported21,22 (Scheme 1A), served as a control to evaluate the impact of PLA incorporation on the dynamic rheology of the final product.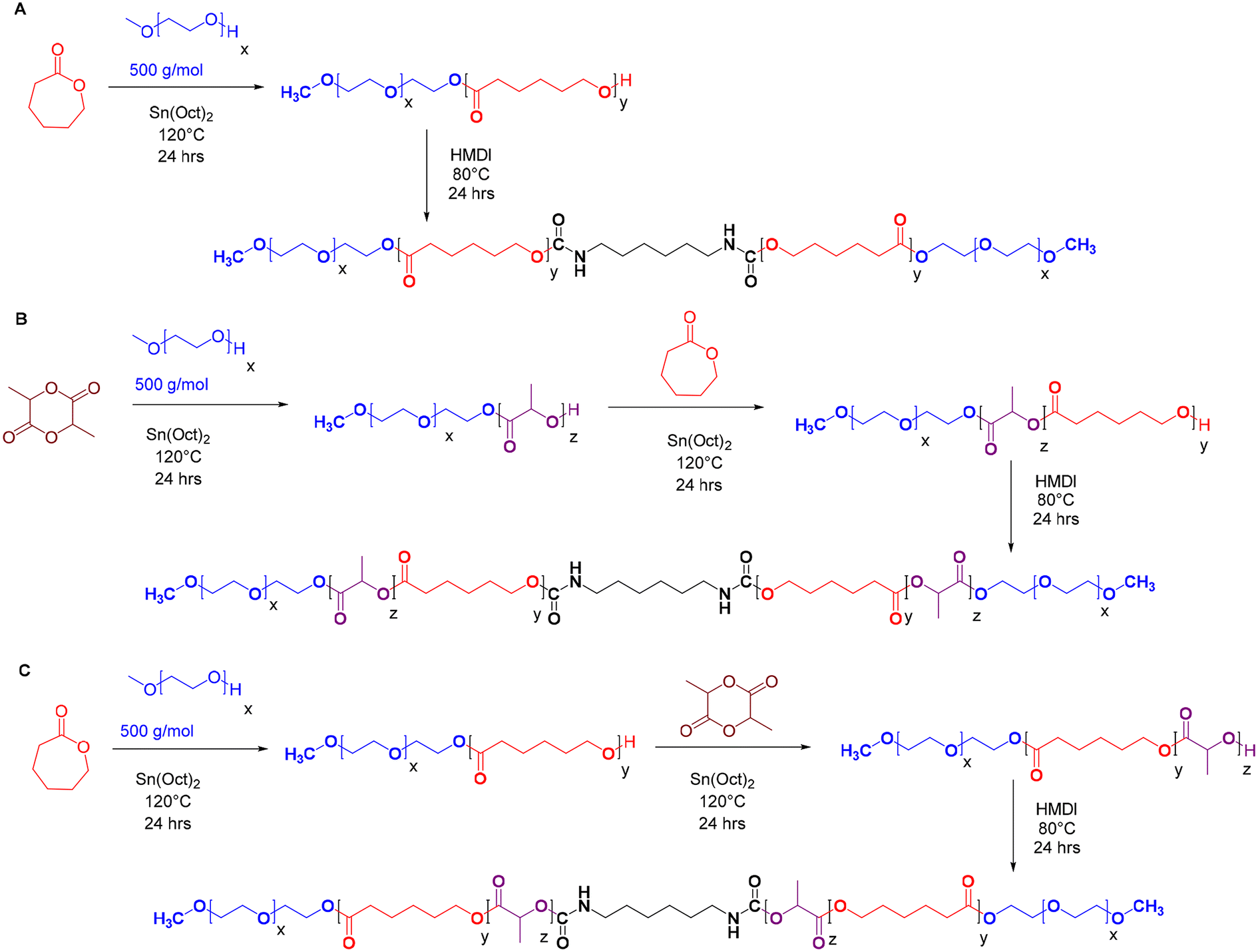 | ||
| Scheme 1 Polymer synthesis. Schematic representation of the synthetic procedures followed to prepare: (A) PECE triblock co-polymer; (B) mPEG-b-PLA-b-PCL-HMDI-PCL-b-PLA-mPEG pentablock co-polymer (PELCLE); and (C) mPEG-b-PCL-b-PLA-HMDI-PLA-b-PCL-mPEG pentablock co-polymer (PECLCE). | ||
The copolymers were synthesised using mPEG500 as a macroinitiator and Sn(Oct)2 as a catalyst. Sequential addition of D,L-lactide or ε-caprolactone followed by coupling with HMDI yielded the pentablock structures (Scheme 1B and C). The successful synthesis and coupling were confirmed by 1H NMR, GPC, and FT-IR analyses. The average molar masses (Mwt) of the prepared copolymers (PECE and P1–P6) were approximately 3200 g mol−1, with a hydrophilic:hydrophobic ratio of 1:2. This was calculated from the 1H-NMR spectra (Fig. S1–S7†), based on the peaks at 3.38 ppm (A,A′: CH3 of mPEG), 2.3 ppm (J,J′: CH2 of εCL), and 5.10 ppm (K,K′: CH of D,L-LA), and the known Mn of mPEG (500 g mol−1). GPC analysis showed the expected 2-fold increase in molar mass after HMDI addition, suggesting success of the coupling reaction (Fig. S8†). All polymers exhibited a unimodal molecular weight distribution with polydispersity indices (Đ < 1.4) (Table 1), however, Mw and Mw values from GPC were higher than those from NMR, which we attribute to the different solution conformations of these amphiphilic polymers in DMF compared to the PMMA standards. FT-IR analysis further validated the successful synthesis (Fig. S9†). A strong C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band at 1750 cm−1 indicated the presence of ester bonds, while the disappearance of –NCO absorption peaks (2250–2270 cm−1) and the appearance of N–H bending vibrations at 1550 cm−1 confirmed the completion of the urethane coupling reaction. The incorporation of a short urethane block into the polymer backbone was expected to enhance mechanical properties and impart self-healing properties owing to the dynamic nature of the N–CO bond and the reversible hydrogen bonding interactions across carbamate linkages.30–32
O stretching band at 1750 cm−1 indicated the presence of ester bonds, while the disappearance of –NCO absorption peaks (2250–2270 cm−1) and the appearance of N–H bending vibrations at 1550 cm−1 confirmed the completion of the urethane coupling reaction. The incorporation of a short urethane block into the polymer backbone was expected to enhance mechanical properties and impart self-healing properties owing to the dynamic nature of the N–CO bond and the reversible hydrogen bonding interactions across carbamate linkages.30–32
The thermoresponsive behaviour of all synthesised hydrogels (30% w/v) was initially assessed using the tube inversion method (Fig. 1A). At 20 °C, all polymers, except P1, exhibited a free-flowing sol state. Upon heating to 37 °C, PECE and P2 underwent a sol–gel transition, while P3–P6 remained viscous liquids.
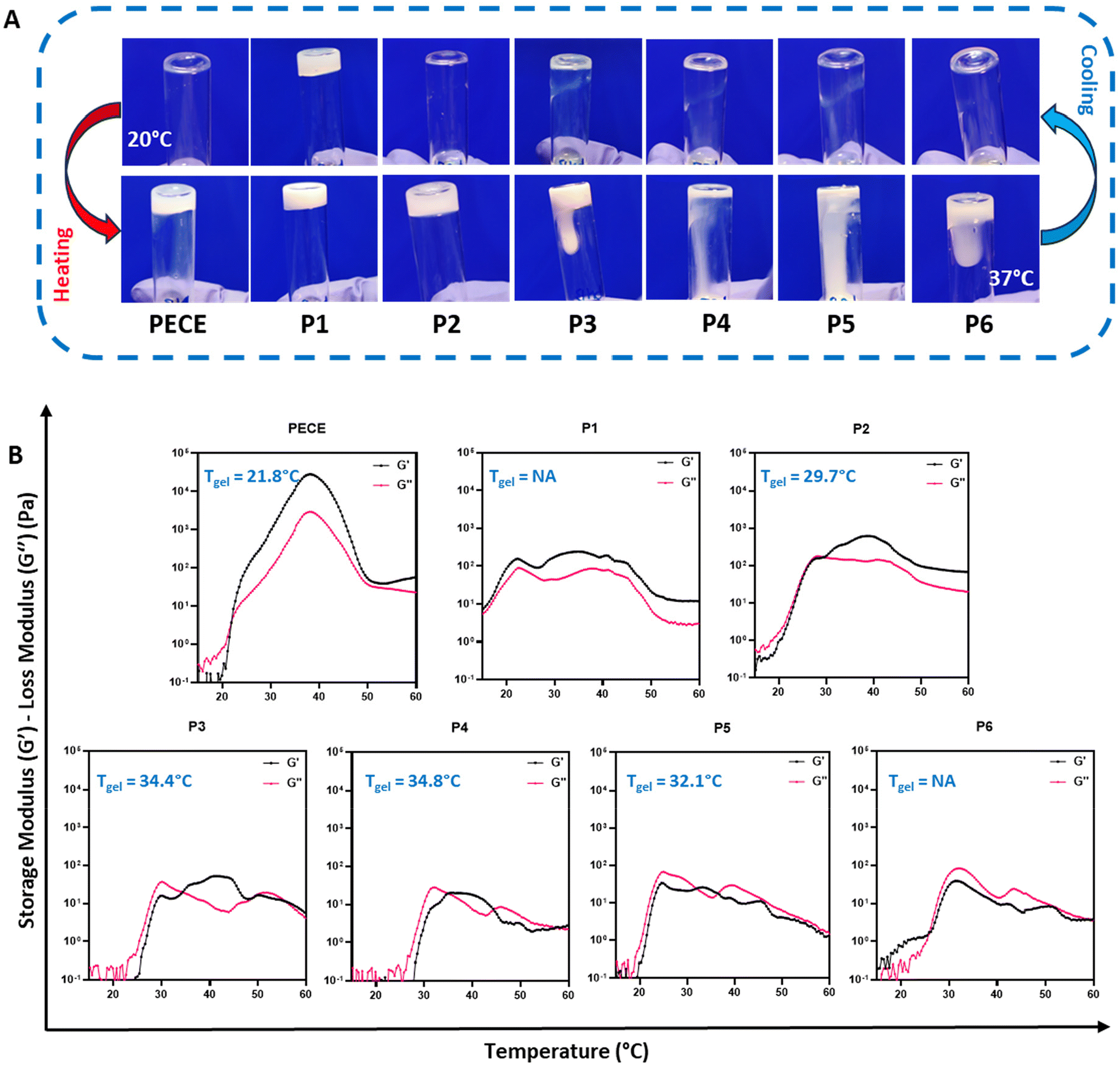 | ||
| Fig. 1 Thermoresponsive behaviour and rheological properties of polymeric materials. (A) Tube inversion test as a proxy for rheological behaviour of the thermoresponsive polymers (30% w/v) at 20 °C and 37 °C. (B) Temperature-dependent changes in storage modulus (G′) and loss modulus (G′′) for polymeric materials (30% w/v), illustrating their rheological behaviour during the gelation process. (Tgel = gelation temperature, NA: not applicable). | ||
Dynamic rheological measurements provided further insights into the gelation process, specifically by assessing their storage (G′) and loss (G′′) moduli. At temperatures below 20 °C, all polymers except P1 displayed low G′ and G′′ values (<1 Pa), indicating good injectability due to their predominantly liquid-like behaviour. Upon heating, distinct differences in gelation behaviour emerged. PECE exhibited a gelation temperature (Tgel) of 21 °C, marked by the crossover of G′ surpassing G′′, and showed a substantial increase in both moduli at 37 °C, indicative of a relatively stiff hydrogel (Fig. 1B). In contrast, P1, with G′ consistently exceeding G′′, exhibited a predominantly solid-like behaviour throughout the temperature range, suggesting poor injectability. P2 demonstrated a Tgel at 29 °C, implying a wider injection window compared to PECE. Furthermore, at 37 °C, P2 formed a softer hydrogel compared to PECE, as evidenced by its lower storage modulus (G′). It is important to note that only PECE and P2 exhibited a clear sol–gel transition state where G′ consistently exceeded G′′ upon heating.
On the other hand, P3 and P4 displayed similar gelation behaviour, with Tgel at 34 °C. P5 exhibited an atypical pattern, with G′ briefly exceeding G′′ at 31 °C before G′′ dominated again. P6, with G′′ consistently higher than G′, remained a viscous liquid throughout the tested temperature range (Fig. 1B). This observation highlights that P3–P6 do not behave as classical hydrogels, but rather exhibit complex temperature-dependent viscoelastic behaviour.
These rheological observations, along with the observed macroscopic gelation behaviour, suggest that both block orientation and the PLA-PCL ratio significantly influence the gelation and mechanical properties of the resulting hydrogels. While no clear trend correlating block properties and gelation behaviour was discernible, likely due to the inherent disorder associated with PLA and the use of D,L-lactide,33,34 a consistent finding was the decrease in hydrogel stiffness upon PLA incorporation into the PECE backbone. This reduction in stiffness is attributed to the contrasting structural characteristics of PLA and PCL. The low crystallinity and quasi-amorphous nature of PLA, arising from the random incorporation of D,L-lactide, contribute to enhanced chain flexibility and reduced rigidity in PLA-containing hydrogels.28,35 In contrast, PECE hydrogels, lacking the PLA component, exhibit a more ordered and tightly packed crystalline structure due to their PCL segments, resulting in increased resistance to deformation and a stiffer hydrogel network.28,35
Based on our objective of developing an injectable hydrogel that remains liquid below 20 °C and forms a stable gel at physiological temperature (35–40 °C), we selected PECE and P2 (PELCLE) as the candidate materials for further investigation.
Influence of PLA incorporation on nanoparticle formation
The amphiphilic nature of PECE and P2 block copolymers suggested their potential for self-assembly into micellar-like nanoparticles (NPs) in aqueous solutions.36 To investigate the impact of PLA incorporation on NP formation, we employed dynamic light scattering (DLS) and transmission electron microscopy (TEM). DLS analysis of 1% w/w aqueous solutions prepared using the temperature cycling approach revealed that both PECE and P2 (PELCLE) formed NPs at room temperature, with average sizes of ∼76 and 25 nm, respectively. Upon heating to 37 °C, both NP types exhibited increased aggregation and size, reaching ∼150 nm for PECE and ∼75 nm for P2 (Fig. 2A and B). Interestingly, P2 NPs were consistently smaller than PECE NPs across all tested temperatures (Fig. 2C).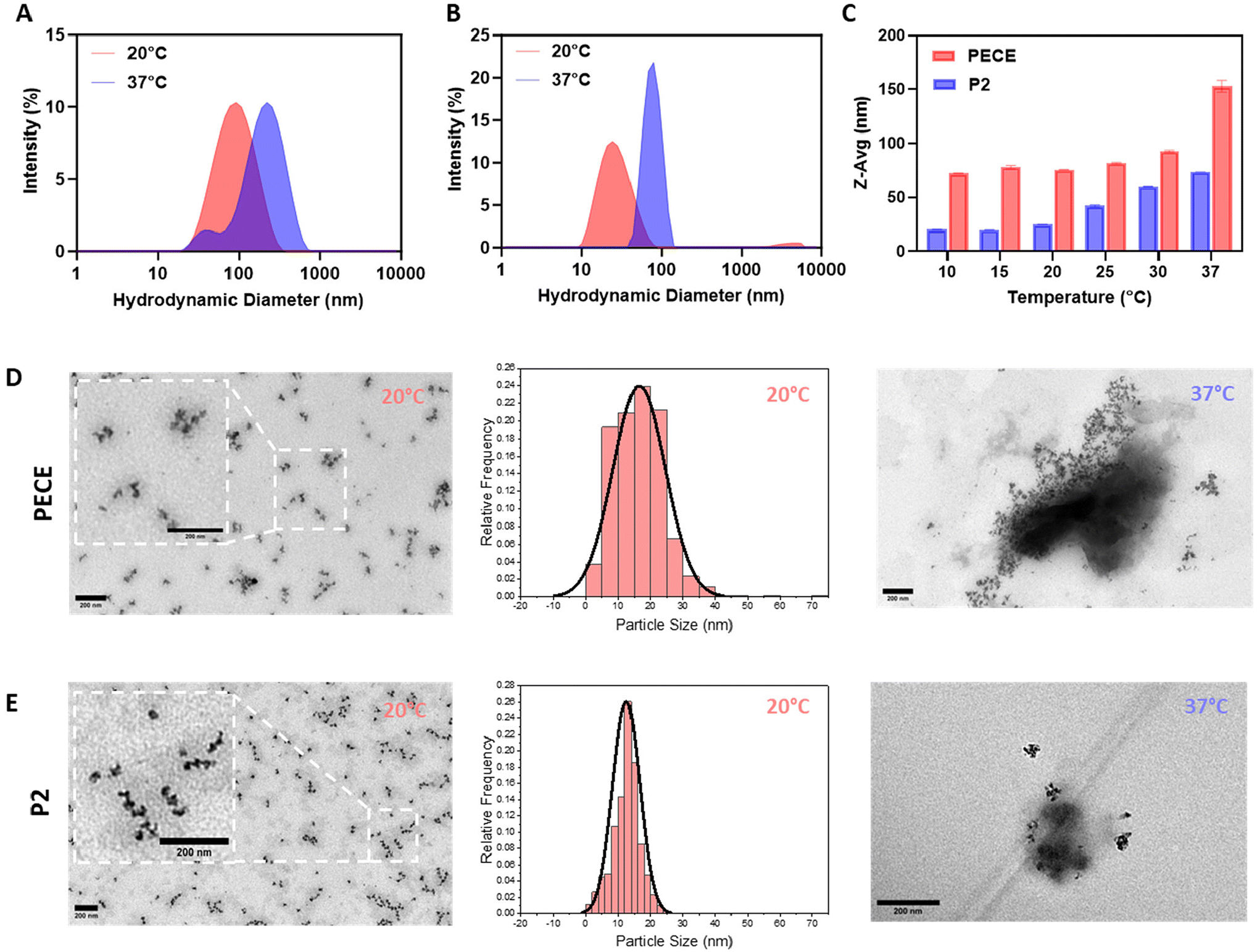 | ||
| Fig. 2 Characterisation of thermo-responsive PECE and P2 NPs. Size distribution analysis of 1% (w/w) of (A) PECE (20 °C: Z-avg = 75.7 nm, PDI = 0.221; 37 °C: Z-avg = 152.9 nm, PDI = 0.296) and (B) P2 NPs (20 °C: Z-avg = 24.9 nm, PDI = 0.207; 37 °C: Z-avg = 73.7 nm, PDI = 0.043). (C) Z-avg (nm) differences between PECE and P2 NPs at different temperatures. Transmission electron microscopy (TEM) imaging and TEM size distribution analysis of (D) PECE and (E) P2 NPs at 20 °C (PECE: mean = 16.5 nm, PDI = 0.239; P2: mean = 12.5 nm, PDI = 0.112) and 37 °C (scale bar = 200 nm). | ||
TEM imaging supported these findings, revealing spherical morphologies for both polymers at 20 °C (Fig. 2D and E). However, some aggregation was observed, being less pronounced in P2 micelles. TEM analysis showed that P2 micelles had a smaller average particle size (mean = 12.5 nm), and a narrower size distribution (PDI = 0.112) compared to PECE (mean = 16.5 nm, PDI = 0.239). At 37 °C, aggregation increased for both polymers, consistent with the DLS results.
The observed temperature-induced aggregation of PECE and P2 micelles is consistent with their inherent thermoresponsive sol–gel transition. As reported for similar amphiphilic copolymers, micellar properties are often governed by interactions within their hydrophobic core.36 Elevated temperatures weaken polymer–water interactions, promoting chain collapse, aggregation, and the formation of physical crosslinks that culminate in a hydrogel network.37,38 This suggests that the temperature sensitivity and propensity for physical crosslinking inherent to PECE and P2 micelles drive their gelation upon heating. This size difference is likely due to the PLA block, which has been shown to enhance chain packing within the hydrophobic core through alkyl chain–chain interactions.39 This observation suggested in turn that micellar size might influence the macroscopic mechanical properties of the resulting hydrogels. Smaller micelles, as seen in P2, were expected to alter chain entanglement and cross-linking density within the hydrogel network, potentially impacting its stiffness, elasticity, and overall mechanical performance.
PLA incorporation impacts PECE hydrogel degradation and swelling
To investigate further the influence of PLA incorporation on hydrogel properties, we evaluated the degradation and swelling behaviour of PECE and P2 hydrogels at 37 °C in PBS pH 7.4. Visual observation revealed that PECE hydrogels maintained their volume over 90 days (Fig. 3A), while P2 hydrogels exhibited a noticeable decrease (Fig. 3B). However, measurements of polymer mass after 90 days showed an increase for PECE hydrogels (from 104.7 ± 7.6 mg to 174.8 ± 8.6 mg) and a decrease for P2 hydrogels (from 104.2 ± 7.5 mg to 70.3 ± 6.3 mg). The increase in PECE mass suggests swelling, which could mask degradation if assessed by mass alone. Therefore, Gel Permeation Chromatography (GPC) was employed to assess degradation in both hydrogels. GPC analysis revealed a shift to lower molecular weights for both PECE and P2, confirming degradation. Specifically, PECE exhibited a 1.7-fold decrease in molecular weight over 90 days, changing from ∼11.7 kg mol−1 to 7.3 kg mol−1. P2 underwent a more substantial reduction, with a 3.2-fold decrease from 13.2 kg mol−1 to 4.3 kg mol−1 over the same period (Fig. 3C and D). The accelerated degradation of P2 is attributable to the inherent higher hydrolytic degradation of PLA, which is known to degrade faster than PCL.27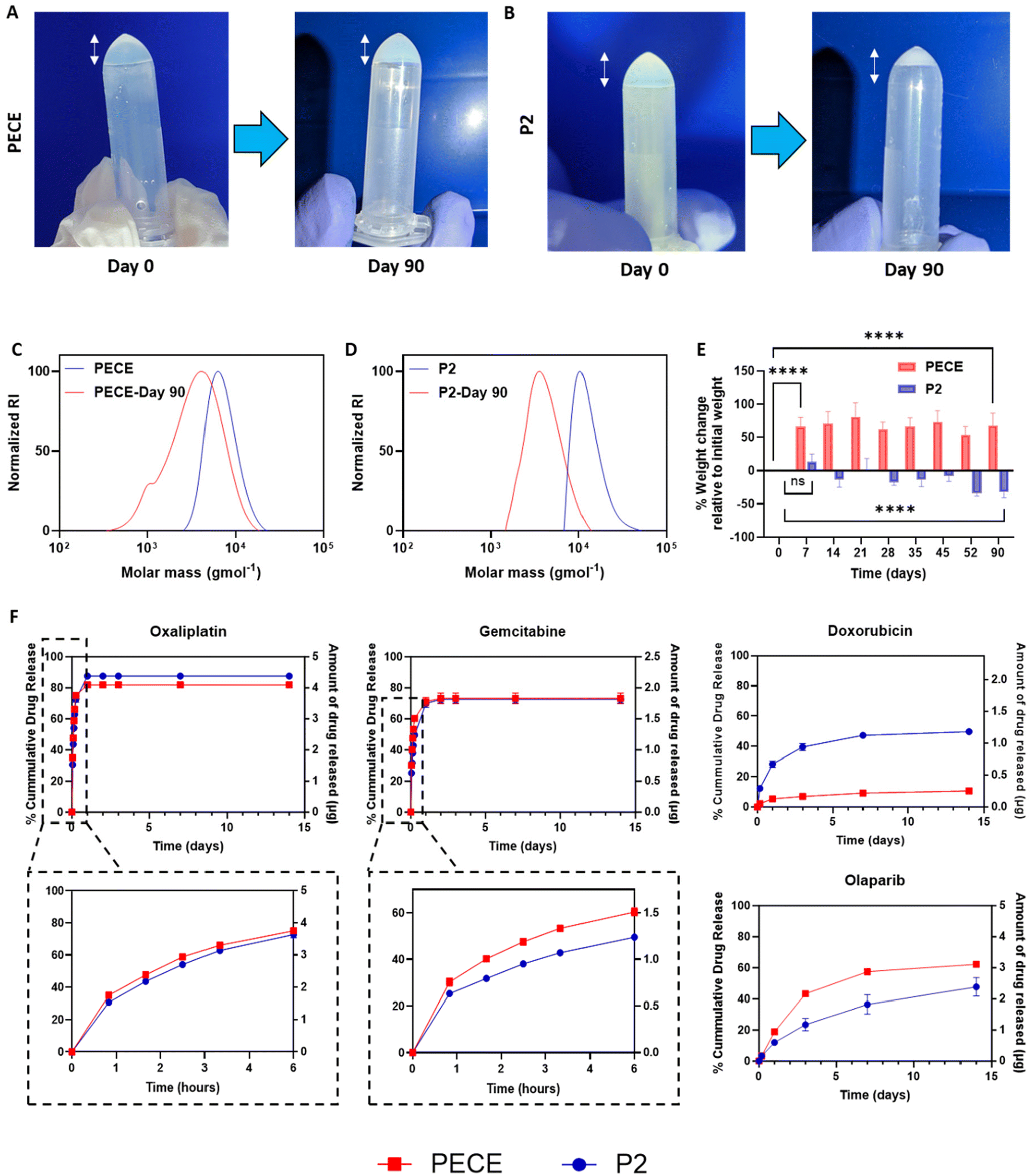 | ||
| Fig. 3 Comparative characterisation of PECE and P2 hydrogels: degradation, swelling, and drug release. Visual observation changes of (A) PECE and (B) P2 hydrogel over 90 days. Changes in the molecular weight following 90 days incubation in PBS pH 7.4 for (C) PECE and (D) P2 hydrogel, assessed by GPC. (E) % weight change relative to initial weight of PECE and P2 hydrogels measured at predetermined times over 90 days period (n = 8) (****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05 ordinary one-way ANOVA with Tukey's post-analysis test). (F) In vitro drug release studies conducted in PBS at pH 7.4, showcasing diverse agents employed in cancer treatments for both PECE and P2 hydrogels. | ||
In terms of swelling behaviour, PECE hydrogels demonstrated a significant mass increase of 73 ± 6% within the first 7 days (p < 0.0001), highlighting their acute high water absorption capacity. No further significant change in hydrogel mass was seen in the remainder study period (p > 0.05) (Fig. 3E). Conversely, P2 hydrogels exhibited non-significant increase in mass (p-value = 0.452) during the first 7 days, potentially due to the PLA blocks, which can decrease hydrogel swelling ratio through increased cross-link density and reduced viscoelastic restoring forces.40 Instead, P2 showed a gradual mass loss, reaching a significant reduction of 30 ± 6% by 90 days (p < 0.0001) further emphasizing its propensity for degradation rather than swelling (Fig. 3E).27
In vitro assessment of injectability, gelation, drug retention, drug release
To evaluate injectability, gelation kinetics, and immediate hydrophilic drug retention of the materials, PECE and P2 solutions (30% w/v) were loaded with Cyanine5.5.alkyne (Cy5.5, 10 μg mL−1) and injected through a 29-gauge needle at room temperature (Videos 1A and B, ESI†). The materials were then injected dropwise into pre-heated deionised water (37 °C, 25 mL). The solutions were easy to inject and gelled rapidly upon contact with the warm water, with Cy5.5 clearly entrapped within the formed hydrogel network (Videos 1A and B, ESI†). Subsequent cooling of the hydrogels on ice resulted in complete dissolution and release of the dye into the surrounding solution (Fig. S10A and B†), demonstrating the reversibility of the sol–gel transition. This experiment provides a visual assessment of the immediate behaviour of the hydrogels upon injection at physiological temperature.Following the initial characterisation, various anti-cancer agents with diverse cLogP values were loaded into PECE and P2 hydrogels at comparable concentrations to assess their in vitro release profiles over a 2-week period (Fig. 3F). The release data, up to a maximum of 60%, or the total amount released within the experimental timeframe if less than 60%, were tested for fit to zero-order, first-order, Hixson-Crowell, Higuchi, and Korsmeyer-Peppas models to elucidate the release mechanisms and investigate any potential correlation between drug lipophilicity (cLogP) and release kinetics (Table 2).
| cLogP | Zero | First | Hixson | Higuchi | K-P | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R 2 | K | R 2 | K | R 2 | K | R 2 | K | R 2 | K | n | ||
| OXA-PECE | −1.39 | 0.99 | 10.4 | 0.97 | 0.3 | 0.95 | 0.3 | 1.00 | 33.2 | 1.00 | 35.0 | 0.5 |
| OXA-P2 | 0.99 | 10.7 | 0.98 | 0.2 | 0.97 | 0.3 | 1.00 | 31.3 | 1.00 | 30.6 | 0.5 | |
| GEM-PECE | −1.06 | 0.68 | 1.4 | 0.67 | 0.0 | 0.61 | 0.1 | 0.79 | 13.5 | 0.91 | 33.8 | 0.3 |
| GEM-P2 | 0.87 | 1.7 | 0.85 | 0.0 | 0.79 | 0.1 | 0.91 | 13.5 | 0.98 | 26.2 | 0.3 | |
| DOX-PECE | 0.317 | 0.80 | 0.0 | 0.75 | 0.0 | 0.74 | 0.0 | 0.93 | 0.6 | 0.97 | 1.6 | 0.3 |
| DOX-P2 | 0.66 | 0.1 | 0.70 | 0.0 | 0.67 | 0.0 | 0.86 | 2.7 | 0.95 | 9.0 | 0.3 | |
| OLA-PECE | 1.24 | 0.73 | 0.2 | 0.83 | 0.0 | 0.80 | 0.0 | 0.93 | 3.8 | 0.94 | 1.7 | 0.7 |
| OLA-P2 | 0.91 | 0.1 | 0.94 | 0.0 | 0.93 | 0.0 | 0.99 | 2.7 | 0.99 | 1.6 | 0.6 | |
For all therapeutic agents, the release profiles were best described by the Higuchi and/or Korsmeyer-Peppas (K-P) models as demonstrated by the R2 values in Table 2. The Higuchi model, commonly used for polymeric matrices, suggests that drug release is primarily governed by diffusion, assuming negligible swelling of the matrix.41,42 The K-P model, an extension of the Higuchi model, is specifically tailored for drug release from swellable polymeric systems like hydrogels and is a valuable tool for discerning the contributions of diffusion and hydrogel swelling.43,44 The exponent ‘n’ characterizes the release mechanism: n ≤ 0.5 indicates diffusion-controlled release, n = 1 signifies swelling-controlled release, 0.5 < n < 1 suggests anomalous transport (a combination of diffusion and swelling), and n > 1 implies polymer erosion or degradation.
We first examined the release of oxaliplatin (OXA, logP = −1.39). OXA exhibited a similar biphasic release profile from both hydrogels, with a burst release in the first 6 hours followed by a slower sustained release. Approximately 75 ± 0.5% and 73 ± 2% of the drug were released within the first 6 hours from PECE and P2, respectively (Fig. 3F). The release profiles were well-described by both the Higuchi and Korsmeyer-Peppas (K-P) models, suggesting a diffusion-controlled mechanism, which is consistent with the K-P model ‘n’ value of 0.3. Notably, P2 hydrogels exhibited a slightly slower release rate compared to PECE (Table 2). This difference may be attributed to the denser structure and reduced swelling of P2, hindering the diffusion of OXA through the hydrogel matrix. Gemcitabine (GEM, cLogP = −1.06), another hydrophilic drug, also showed a slower and more sustained release from P2 compared to PECE, particularly in the initial 6 hours (50 ± 0.5% vs. 60 ± 1.5% release, respectively). The K-P model confirmed diffusion-controlled release (n = 0.3 for both), with P2 demonstrating a slower rate (K = 26.2) than PECE (K = 33.8) (Fig. 3F).
In contrast, the release of doxorubicin hydrochloride (DOX.HCl, cLogP = 0.317) was slower from PECE than P2 (40 ± 2% vs. 7 ± 1% release in 72 hours) (Fig. 3F). The K-P model again suggested diffusion-controlled release (n = 0.3 for both), but with PECE exhibiting a slower rate (K = 1.6) than P2 (K = 9.0). These data might be partly explained via specific interactions between the charged pendant aminoglycoside ring of DOX with lactate and caproic acid terminated fragments resulting from polymer degradation. The more rapid degradation of the poly(lactic acid) regions in P2 compared to the poly(caprolactone)-only polymer PECE would lead to more rapid formation of carboxyl end-groups, potentially trapping DOX via electrostatic interactions.
The hydrophobic drug olaparib (OLA, cLogP = 1.24) exhibited a slower and more sustained release profile from P2 compared to PECE (Fig. 3F). Within the first 72 hours, OLA release was 23.0 ± 3.3% from P2 and 43.3 ± 0.7% from PECE. The K-P model indicated anomalous transport for both formulations (n = 0.6 for P2, n = 0.7 for PECE), with P2 demonstrating a slower release rate (K = 1.6 vs. 1.7 for PECE).
These findings reveal several key trends. First, increasing drug hydrophobicity (higher cLogP) generally correlates with decreased release percentage and rate for both hydrogels. This is likely due to the reduced water solubility and slower dissolution of more hydrophobic drugs.45 Second, drug release mechanisms appear to be influenced by cLogP values, with more hydrophilic drugs following diffusion-controlled release and more hydrophobic drugs exhibiting anomalous transport. This aligns with previous reports.43 Third, while P2 hydrogels generally exhibited slower release rates than PECE for most agents, the data for DOX.HCl highlighted the potential influence of specific polymer–drug interactions on release kinetics.
Preclinical assessment of GEMOX-loaded P2 hydrogels for pancreatic cancer therapy
The thermoresponsive behaviour of P2 suggested it might be useful as an in situ gelling depot for drug delivery in the challenging conditions of pancreatic ductal adenocarcinoma (PDAC).44 The poor prognosis and limited treatment options for PDAC, in conjunction with the challenges associated with current chemotherapeutic regimens, highlight the urgent need for improved therapeutic strategies. The combination of injectability, hydrolytic degradation, controlled drug release, and minimal swelling might offer a means to overcome some of these obstacles, hence we investigated the use of P2 hydrogels as a delivery system for the combination therapy of gemcitabine and oxaliplatin in the context of PDAC treatment.:OXA molar ratios (6.5:1, 13:1, and 26:1) centered around the clinically relevant molar ratio of 13:1 (1000 mg m−2 GEM, 100 mg m−2 OXA).11,46 Cytotoxicity was evaluated using the PrestoBlue assay. All treatments exhibited dose-dependent cytotoxicity, with GEMOX combinations consistently demonstrated lower IC50 values compared to the free drugs (Fig. 4A). Specifically, GEMOX 6.5:1, 13:1, and 26:1 showed IC50 values of 0.193 μM, 0.213 μM, and 0.280 μM, respectively, compared to free GEM (IC50 = 0.451 μM) and free OXA (IC50 = 9.994 μM) highlighting the enhanced cytotoxic effect achieved when GEM and OXA are combined.
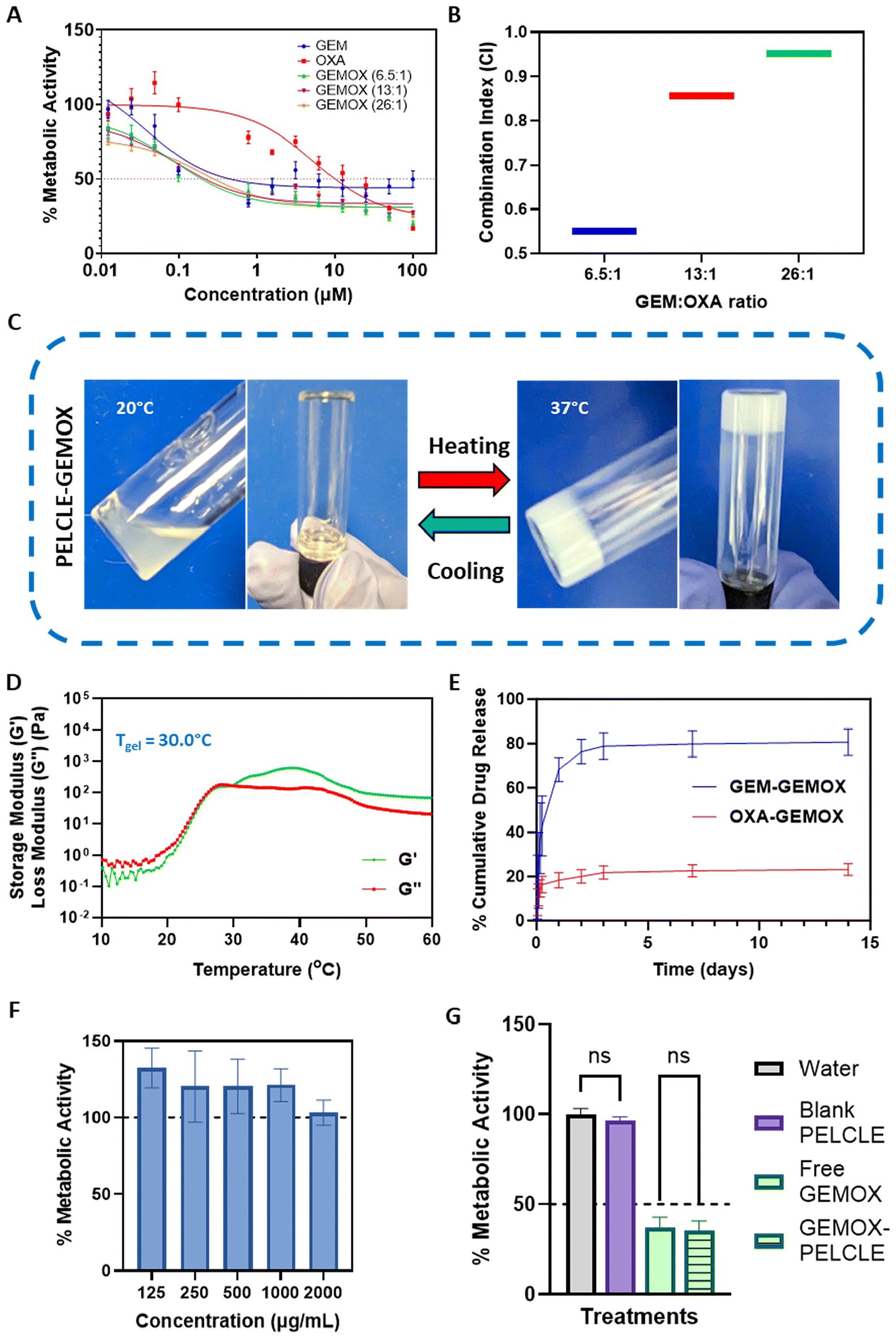 | ||
| Fig. 4 In vitro evaluation of GEMOX-loaded P2 hydrogels. (A) Dose–response curves comparing the efficacy of free GEMOX treatments at various molar ratios to individual free GEM and free OXA treatments. (B) Combination index (CI) analysis demonstrating the synergistic activity of GEMOX at IC50. (C) Tube inversion test visualising the sol–gel transition of P2-GEMOX hydrogels at 20 °C and 37 °C. (D) Temperature-dependent changes in storage modulus (G′) and loss modulus (G′′) for P2-GEMOX hydrogels, illustrating their rheological behaviour. (E) In vitro drug release profiles of GEM (50 mg mL−1) and OXA (5 mg mL−1) from GEMOX-loaded P2 hydrogels in PBS at pH 6.8. (F) Metabolic activity of PANC-1 cells after direct contact with varying concentrations of P2 gels. (G) Comparative analysis of the effects of free GEMOX and GEMOX-P2 on PANC-FLuc cells over a 72-hour period, presented as mean ± SD (n = 3). | ||
Combination index (CI) analysis at the IC50 further confirmed the synergistic effect of GEMOX, with CI values below 1 (Fig. 4B). Considering the established GEMOX dosage regimen and the dose-limiting toxicity of OXA, we selected the clinically relevant 13:1 molar ratio for subsequent studies.11
:1 – GEM:OXA molar ratio) did not significantly affect the gelation properties, causing only a marginal increase in the apparent gelation temperature (Tgel). The observed Tgel was 29.7 °C without drug loading and 30.0 °C with GEMOX loading (Fig. 4D), indicating no significant disruptions in the P2 network.47In vitro drug release studies in PBS (pH 6.8), mimicking the acidic tumour microenvironment,11 showed sustained release of both GEM and OXA from P2-GEMOX HGs over an extended period (Fig. 4E). GEM release from GEMOX-loaded P2 was 78% in 72 hours while OXA release reached 21% at 72 hours.
To assess the cytotoxicity of the blank hydrogel, a direct contact assay was performed. Varying concentrations of the gel (125–2000 μg mL−1) were applied to PANC-1 cells, and no reduction in metabolic activity was observed, confirming the lack of acute toxicity of the polymer (Fig. 4F). These results were further confirmed in Transwell® systems, where no significant difference (p > 0.05) was found between P2 hydrogels loaded in Transwells® and negative controls. This suggests that the products of acute degradation of the polymer do not produce acute toxicity in cells. Similarly, no significant difference (p > 0.05) was observed between GEMOX-P2 and free GEMOX loaded in Transwells® on PANC-FLuc cells, indicating that the hydrogel formulation does not hinder drug release and activity in this more biorelevant environment (Fig. 4G).
 | ||
| Fig. 5 In vivo evaluation of P2 hydrogels in a pancreatic cancer model. (A) In vivo release study of Cy5.5 as a model hydrophilic drug from Cy5.5/P2 hydrogels compared to free Cy5.5 solution over a 2-week duration, shedding light on the sustained release properties of the hydrogel. (B) Visualisation of excised tumours demonstrating the retention of Cy5.5/hydrogel around the tumour over time. (C) Representative immunohistochemistry images depicting H&E, TUNEL, and KI-67 analyses (scale bar = 100 μm). Arrows represent proliferation positive regions. (D) Quantification of the TUNEL assay illustrating results for various treatments. (E) Quantification of the KI-67 assay showcasing results for different treatments. (****p < 0.0001, ***p < 0.001, **p < 0.01, and *p < 0.05 ordinary one-way ANOVA with Tukey's post-analysis test). | ||
Ex vivo imaging of excised tumours confirmed that the dye remained localised at the tumour site throughout the study period, indicating that the hydrogel was not degraded to fragments that could be cleared during this time (Fig. 5B). This enhanced tumour retention suggests that chemotherapeutic agents encapsulated within the hydrogel might persist within the tumour microenvironment for extended durations, potentially leading to a more prolonged therapeutic effect.
:1 molar ratio (83.3 mg kg−1 GEM, 8.3 mg kg−1 OXA).
Throughout the 28-day study period, no significant changes in body weight or behaviour were observed in any treatment group, suggesting good tolerability (Fig. S11A†). However, low and variable tumour growth was observed across all groups, including the negative controls (Fig. S11B†), hindering the assessment of tumour regression. While high variability in xenograft pancreatic tumour growth is not unprecedented,48 we were not able in the time of study to ascertain why this occurred, and thus for ethical reasons were not able to repeat the experiments. Additionally, the subcutaneous model presented challenges in accurately measuring tumour volume due to the hydrogel adherence, leading to variability in tumour weight within different groups (Fig. S11C and D†).
We therefore focused on immunohistochemical analysis to evaluate the effects of different treatments on tumour and major organ tissues. H&E staining was used to assess changes in tissue architecture and cellular organisation.49 No histological alterations were observed in major organs across all treatment groups, suggesting minimal systemic toxicity (Fig. S12†). In tumours treated with blank hydrogel, tissue morphology closely resembled that of the normal saline control, with intact cell structures (Fig. 5C). Both GEMOX IV and GEMOX PT treatments resulted in a slight loss of tissue integrity with occasional empty nuclei (Fig. 5C), indicative of limited therapeutic activity. In contrast, GEMOX-P2 treatment induced detectable changes, with evidence of nuclear shrinkage and loss of cell integrity (Fig. 5C). Subsequently, a TUNEL assay was employed to evaluate the extent of apoptosis within the tumour tissues under different treatment conditions (Fig. 5C). Consistent with the H&E staining, the TUNEL assay revealed a significantly higher number of apoptotic cells per μm2 in the GEMOX-P2 group compared to both GEMOX IV and PT groups (211.7 ± 80.0 vs. 9.3 ± 5.7 and 4.1 ± 1.6, respectively; p = 0.0003 and p = 0.0002, respectively) (Fig. 5D). Similarly, the KI-67 proliferation assay, which detects the expression of the KI-67 proliferation marker,50 demonstrated a significantly lower number of proliferative cells per μm2 in the GEMOX-P2 group compared to GEMOX IV and PT (9.9 ± 2.0 vs. 70.4 ± 5.2 and 130.5 ± 7.8, respectively; p = 0.0065 and p < 0.0001, respectively) (Fig. 5E).
These immunohistochemical assessments collectively suggest that peritumoural injection of GEMOX-P2 hydrogel induced significantly greater apoptosis and inhibited cell proliferation in the tumour tissues more effectively than both intravenous and peritumoural GEMOX. This enhanced anti-tumour activity was achieved while maintaining minimal systemic toxicity, as evidenced by stable body weight throughout the study. At study termination, mice treated with GEMOX-P2 hydrogel had an average body weight of 29.6 ± 2.6 g, a 1.03-fold increase from their pre-treatment baseline of 28.6 ± 1.26 g. We suggest that the sustained drug release from the hydrogel likely contributed to this enhanced therapeutic effect, at least in the regions of the tumours in proximity to the gels, underscoring the potential of this approach as a treatment option for pancreatic cancer.
It is important also to note the limitations of the in vivo studies. Xenograft tumours do not replicate the physiological environment of pancreatic cancers but are often adopted because the establishment of orthotopic pancreatic tumours in mice is complex experimentally and difficult ethically. Tumours grow rapidly in pancreatic sites in mice, leading to limited treatment windows and the need for unacceptably large numbers of animals in a study if survival in control arms is short. We chose a xenograft model to establish the safety of the gels and also to evaluate if local anti-tumour effects could be achieved. However, while these primary goals were achieved, this model did not allow a full efficacy study in terms of tumour regression because of highly variable tumour growth in the animals. In addition, we noted a sex-dependent component in both tumour size and in reduction following treatment, which warrants further investigation (Fig. S13†). A more extensive study now that we have established safety is under consideration, with a likely additional treatment component of an external stimulus, such as ultrasound, to disrupt the gel and pancreatic stromal matrix locally, ensuring site-specific release and optimal therapeutic window for the drug combinations.
Conclusion
This study successfully demonstrated the incorporation of PLA blocks into PECE hydrogels, yielding PELCLE pentablock copolymers with tailored properties. Our primary objective was to investigate the impact of PLA incorporation on hydrogel characteristics, not to replace PECE, but to provide an alternative system with enhanced hydrolytic degradation, reduced swelling, and slower drug release profiles – attributes desirable for certain applications. The introduction of PLA maintained the thermoresponsive behaviour of PECE, allowing both polymers to self-assemble into micelles, as confirmed by DLS and TEM. Notably, PELCLE exhibited smaller micellar sizes compared to PECE, potentially influencing drug delivery and release. Rheological and mechanical analyses revealed that PELCLE hydrogels possessed lower storage modulus, complex viscosity, and stiffness, alongside increased deformability compared to PECE, attributable to the distinct characteristics of PLA blocks. Furthermore, PLA incorporation enhanced hydrolytic biodegradation and reduced swelling in PELCLE hydrogels.Both PECE and PELCLE demonstrated desirable features such as injectability and rapid gelation at body temperature, effectively entrapping both hydrophilic and hydrophobic drugs. Drug release studies revealed that hydrophobicity and cLogP values influence release kinetics, with PELCLE generally exhibiting slower release compared to PECE.
To showcase the potential of PELCLE, we investigated its application in pancreatic cancer treatment using GEMOX as a model drug combination. In vitro studies demonstrated no acute toxicities of PELCLE gels, and in vivo experiments demonstrated sustained GEMOX release and enhanced injection site retention compared to free drug administration. Importantly, PELCLE hydrogels exhibited minimal systemic toxicity and drug-loaded P2 gels were shown to induce apoptosis in regions of tumour tissue to a higher extent than those of the free drugs injected by the same routes.
In conclusion, the pentablock PELCLE hydrogels represent a versatile platform for extended drug delivery, offering tuneable physicochemical properties and drug release profiles. Their potential extends beyond pancreatic cancer, suggesting broad applicability in various biomedical fields where localised and sustained drug delivery is desired.
Author contributions
ElSherbeny Amr: conceptualisation, methodology, validation, formal analysis, investigation, visualisation, writing – original draft. Cara Moloney: methodology, investigation, supervision, writing – review & editing. Hulya Bayraktutan: investigation, visualisation, writing – review & editing. Nurcan Gumus: investigation, formal analysis, writing – review & editing. Andres Garcia Sampedro: investigation, visualisation, writing – review & editing. Oz Umut Can: investigation, supervision, writing – review & editing. Alison Ritchie: methodology, investigation, writing – review & editing. Shreeya Parmar: investigation. Marian Meakin: investigation. Ruman Rahman: supervision, writing – review & editing. Jennifer Ashworth: supervision, writing – review & editing. Anna Grabowska: supervision, writing – review & editing. Cameron Alexander: conceptualisation, supervision, writing – review& editing, funding acquisition, resources.Data availability
The data supporting this article have been included as part of the ESI† or are available from the corresponding authors Amr Elsherbeny and Cameron Alexander.Conflicts of interest
Jennifer C. Ashworth is a co-founder, shareholder and on the Scientific Advisory Board of Peptimatrix Limited.Acknowledgements
We thank UKRI/EPSRC for funding [grants EP/R035563/1, EP/S021434/1, EP/V049291/1], the Little Princess Trust in partnership with CCLG and the Royal Society [Wolfson Research Merit Award WM150086 to CA]. UCO is thankful for a fellowship (2219) from the TUBITAK. We acknowledge Dr Patricia Monteiro for expert assistance in polymer synthesis and characterisation. This work was also supported via Anne McLaren fellowship funding from the University of Nottingham (JCA). We also thank Tom Hyde, Esme Ireson and Paul Cooling for expert technical support. The Nanoscale & Macroscale Research Centre (NMRC) is acknowledged for providing the facilities for TEM, SEM, and related analysis.References
- N. Bhattarai, J. Gunn and M. Zhang, Chitosan-based hydrogels for controlled, localized drug delivery, Adv. Drug Delivery Rev., 2010, 62(1), 83–99 CrossRef CAS PubMed.
- S. Tanga, M. Aucamp and P. Ramburrun, Injectable Thermoresponsive Hydrogels for Cancer Therapy: Challenges and Prospects, Gels, 2023, 9(5), 418–437 CrossRef CAS PubMed.
- R. Fan, Y. Cheng, R. Wang, T. Zhang, H. Zhang and J. Li, et al., Thermosensitive Hydrogels and Advances in Their Application in Disease Therapy, Polymers, 2022, 14(12), 2379–2400 CrossRef CAS PubMed.
- K. Zhang, K. Xue and X. J. Loh, Thermo-Responsive Hydrogels: From Recent Progress to Biomedical Applications, Gels, 2021, 7(3), 77–94 CrossRef CAS PubMed.
- G. Molinaro, J. C. Leroux, J. Damas and A. Adam, Biocompatibility of thermosensitive chitosan-based hydrogels: an in vivo experimental approach to injectable biomaterials, Biomaterials, 2002, 23(13), 2717–2722 CrossRef CAS PubMed.
- L. Klouda and A. G. Mikos, Thermoresponsive hydrogels in biomedical applications, Eur. J. Pharm. Biopharm., 2008, 68(1), 34–45 CrossRef CAS PubMed.
- V. H. G. Phan, E. Lee, J. H. Maeng, T. Thambi, B. S. Kim and D. Lee, et al., Pancreatic cancer therapy using an injectable nanobiohybrid hydrogel, RSC Adv., 2016, 6(47), 41644–41655 RSC.
- B. Jeong, S. W. Kim and Y. H. Bae, Thermosensitive sol-gel reversible hydrogels, Adv. Drug Delivery Rev., 2012, 64, 154–162 CrossRef.
- S. Pardeshi, F. Damiri, M. Zehravi, R. Joshi, H. Kapare and M. K. Prajapati, et al., Functional Thermoresponsive Hydrogel Molecule to Material Design for Biomedical Applications, Polymers, 2022, 14(15), 3126–3170 CrossRef CAS PubMed.
- H. Gholizadeh, E. Landh, D. M. Silva, A. Granata, D. Traini and P. Young, et al., In vitro and in vivo applications of a universal and synthetic thermo-responsive drug delivery hydrogel platform, Int. J. Pharm., 2023, 635, 122777–122789 CrossRef CAS PubMed.
- A. Elsherbeny, H. Bayraktutan, U. Can Oz, C. Moloney, J. C. Ashworth and A. M. Grabowska, et al., Responsive Nanomaterial Delivery Systems for Pancreatic Cancer Management, Adv. Ther., 2023, 2300330–2300362 Search PubMed.
- C. Y. Gong, P. W. Dong, S. Shi, S. Z. Fu, J. L. Yang and G. Guo, et al., Thermosensitive PEG–PCL–PEG Hydrogel Controlled Drug Delivery System: Sol–Gel–Sol Transition and In Vitro Drug Release Study, J. Pharm. Sci., 2009, 98(10), 3707–3717 CrossRef CAS PubMed.
- K. Wu, L. Yu and J. Ding, Synthesis of PCL-PEG-PCL Triblock Copolymer via Organocatalytic Ring-Opening Polymerization and Its Application as an Injectable Hydrogel – An Interdisciplinary Learning Trial, J. Chem. Educ., 2020, 97(11), 4158–4165 CrossRef CAS.
- T. R. Hoare and D. S. Kohane, Hydrogels in drug delivery: Progress and challenges, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- A. C. Marques, P. J. Costa, S. Velho and M. H. Amaral, Stimuli-responsive hydrogels for intratumoral drug delivery, Drug Discovery Today, 2021, 26(10), 2397–2405 CrossRef CAS PubMed.
- J. Luo, X. Zhao, B. Guo and Y. Han, Preparation, thermal response mechanisms and biomedical applications of thermosensitive hydrogels for drug delivery, Expert Opin. Drug Delivery, 2023, 20(5), 641–672 CrossRef CAS PubMed.
- K. Dutta, R. Das, J. Ling, R. M. Monibas, E. Carballo-Jane and A. Kekec, et al., In Situ Forming Injectable Thermoresponsive Hydrogels for Controlled Delivery of Biomacromolecules, ACS Omega, 2020, 5(28), 17531–17542 CrossRef CAS PubMed.
- J. Shi, L. Yu and J. Ding, PEG-based thermosensitive and biodegradable hydrogels, Acta Biomater., 2021, 128, 42–59 CrossRef CAS.
- S. Salehi, S. M. Naghib, H. R. Garshasbi, S. Ghorbanzadeh and W. Zhang, Smart stimuli-responsive injectable gels and hydrogels for drug delivery and tissue engineering applications: A review, Front. Bioeng. Biotechnol., 2023, 11, 1104126–1104146 CrossRef PubMed.
- B. Khan, A. Arbab, S. Khan, H. Fatima, I. Bibi and N. P. Chowdhry, et al., Recent progress in thermosensitive hydrogels and their applications in drug delivery area, MedComm: Biomater. Appl., 2023, 2(3), e55–76 Search PubMed.
- C. Y. Gong, S. Shi, P. W. Dong, B. Kan, M. L. Gou and X. H. Wang, et al., Synthesis and characterization of PEG-PCL-PEG thermosensitive hydrogel, Int. J. Pharm., 2009, 365(1–2), 89–99 CrossRef CAS PubMed.
- S. Singh, M. M. Alrobaian, N. Molugulu, N. Agrawal, A. Numan and P. Kesharwani, Pyramid-Shaped PEG-PCL-PEG Polymeric-Based Model Systems for Site-Specific Drug Delivery of Vancomycin with Enhance Antibacterial Efficacy, ACS Omega, 2020, 5(21), 11935–11945 CrossRef CAS PubMed.
- P. Grossen, D. Witzigmann, S. Sieber and J. Huwyler, PEG-PCL-based nanomedicines: A biodegradable drug delivery system and its application, J. Controlled Release, 2017, 260, 46–60 CrossRef CAS PubMed.
- S. Z. Fu, P. Y. Ni, B. Y. Wang, B. Y. Chu, L. Zheng and F. Luo, et al., Injectable and thermo-sensitive PEG-PCL-PEG copolymer/collagen/n-HA hydrogel composite for guided bone regeneration, Biomaterials, 2012, 33(19), 4801–4809 CrossRef CAS PubMed.
- M. R. Dethe, P. A, H. Ahmed, M. Agrawal, U. Roy and A. Alexander, PCL-PEG copolymer based injectable thermosensitive hydrogels, J. Controlled Release, 2022, 343, 217–236 CrossRef CAS PubMed.
- K. Miller, J. E. Hsu and L. J. Soslowsky, Materials in Tendon and Ligament Repair, Compr. Biomater., 2011, 6, 257–279 Search PubMed.
- F. Ebrahimi and H. R. Dana, Poly lactic acid (PLA) polymers: from properties to biomedical applications, Int. J. Polym. Mater. Polym. Biomater., 2022, 71(15), 1117–1130 CrossRef CAS.
- D. Łysik, J. Mystkowska, G. Markiewicz, P. Deptuła and R. Bucki, The Influence of Mucin-Based Artificial Saliva on Properties of Polycaprolactone and Polylactide, Polymers, 2019, 11(11), 1880–1899 CrossRef PubMed.
- A. Elwakeel, H. Soudan, A. Eldoksh, M. Shalaby, M. Eldemellawy and D. Ghareeb, et al., Implementation of the Chou-Talalay method for studying the in vitro pharmacodynamic interactions of binary and ternary drug combinations on MDA-MB-231 triple negative breast cancer cells, Synergy, 2019, 8, 100047–100057 CrossRef.
- H. Chen, L. R. Hart, W. Hayes and C. R. Siviour, Mechanical characterisation and modelling of a thermoreversible superamolecular polyurethane over a wide range of rates, Polymer, 2021, 221, 123607–123621 CrossRef CAS.
- B. X. Cheng, W. C. Gao, X. M. Ren, X. Y. Ouyang, Y. Zhao and H. Zhao, et al., A review of microphase separation of polyurethane: Characterization and applications, Polym. Test., 2022, 107, 107489–107499 CrossRef CAS.
- J. Xie, L. Fan, D. Yao, F. Su, Z. Mu and Y. Zheng, Ultra-robust, self-healable and recyclable polyurethane elastomer via a combination of hydrogen bonds, dynamic chemistry, and microphase separation, Mater. Today Chem., 2022, 23, 100708–100718 CrossRef CAS.
- S. Farah, D. G. Anderson and R. Langer, Physical and Mechanical Properties of PLA, and Their Functions in Widespread Applications-A Comprehensive Review, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS.
- L. Ranakoti, B. Gangil, S. K. Mishra, T. Singh, S. Sharma and R. A. Ilyas, et al., Critical Review on Polylactic Acid: Properties, Structure, Processing, Biocomposites, and Nanocomposites, Materials, 2022, 15(12), 4312–4331 CrossRef CAS PubMed.
- Y. Lin, E. Bilotti, C. W. M. Bastiaansen and T. Peijs, Transparent semi-crystalline polymeric materials and their nanocomposites: A review, Polym. Eng. Sci., 2020, 60(10), 2351–2376 CrossRef CAS.
- G. Pasparakis and C. Tsitsilianis, LCST polymers: Thermoresponsive nanostructured assemblies towards bioapplications, Polymer, 2020, 211, 123146–123162 CrossRef CAS.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Thermoresponsive polymers with lower critical solution temperature: from fundamental aspects and measuring techniques to recommended turbidimetry conditions, Mater. Horiz., 2017, 4(2), 109–116 RSC.
- M. T. Cook, P. Haddow, S. B. Kirton and W. J. Mcauley, Polymers Exhibiting Lower Critical Solution Temperatures as a Route to Thermoreversible Gelators for Healthcare, Adv. Funct. Mater., 2021, 31(8), 2008123–2008148 CrossRef CAS.
- P. Stipa, S. Marano, R. Galeazzi, C. Minnelli, G. Mobbili and E. Laudadio, Prediction of drug-carrier interactions of PLA and PLGA drug-loaded nanoparticles by molecular dynamics simulations, Eur. Polym. J., 2021, 147, 110292–110301 CrossRef CAS.
- M. Shabani Samghabadi, A. Karkhaneh and A. A. Katbab, Synthesis and characterization of biphasic layered structure composite with simultaneous electroconductive and piezoelectric behavior as a scaffold for bone tissue engineering, Polym. Adv. Technol., 2023, 34(4), 1367–1380 CrossRef CAS.
- M. Haider, A. Elsherbeny, J. Jagal, A. Hubatová-Vacková and I. S. Ahmed, Optimization and evaluation of poly(Lactide-co-glycolide) nanoparticles for enhanced cellular uptake and efficacy of paclitaxel in the treatment of head and neck cancer, Pharmaceutics, 2020, 12(9), 1–22 CrossRef PubMed.
- D. Paolino, A. Tudose, C. Celia, L. Di Marzio, F. Cilurzo and C. Mircioiu, Mathematical Models as Tools to Predict the Release Kinetic of Fluorescein from Lyotropic Colloidal Liquid Crystals, Materials, 2019, 12(5), 693–716 CrossRef CAS PubMed.
- X. Wang, S. S. Venkatraman, F. Y. C. Boey, J. S. C. Loo and L. P. Tan, Controlled release of sirolimus from a multilayered PLGA stent matrix, Biomaterials, 2006, 27(32), 5588–5595 CrossRef CAS PubMed.
- Q. Yu, X. Tang, W. Zhao, Y. Qiu, J. He and D. Wan, et al., Mild hyperthermia promotes immune checkpoint blockade-based immunotherapy against metastatic pancreatic cancer using size-adjustable nanoparticles, Acta Biomater., 2021, 133, 244–256 CrossRef CAS PubMed.
- Y. Fu and W. J. Kao, Drug Release Kinetics and Transport Mechanisms of Non-degradable and Degradable Polymeric Delivery Systems, Expert Opin. Drug Delivery, 2010, 7(4), 429–444 CrossRef CAS PubMed.
- T. C. Chou, Drug combination studies and their synergy quantification using the Chou-Talalay method, Cancer Res., 2010, 70(2), 440–446 CrossRef CAS PubMed . Available from: https://cancerres.aacrjournals.org/cgi/content/full/70/2/440.
- W. Wang, H. Song, J. Zhang, P. Li, C. Li and C. Wang, et al., An injectable, thermosensitive and multicompartment hydrogel for simultaneous encapsulation and independent release of a drug cocktail as an effective combination therapy platform, J. Controlled Release, 2015, 203, 57–66 CrossRef CAS PubMed.
- N. A. Pham, N. Radulovich, E. Ibrahimov, S. N. Martins-Filho, Q. Li and M. Pintilie, et al., Patient-derived tumor xenograft and organoid models established from resected pancreatic, duodenal and biliary cancers, Sci. Rep., 2021, 11(1), 1–12 CrossRef PubMed.
- A. H. Fischer, K. A. Jacobson, J. Rose and R. Zeller, Hematoxylin and eosin staining of tissue and cell sections, CSH Protoc., 2008,(5), 4986–4989 Search PubMed.
- I. Miller, M. Min, C. Yang, C. Tian, S. Gookin and D. Carter, et al., Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence, Cell Rep., 2018, 24(5), 1105–1112 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4bm01629g |
| ‡ AE and CM contributed equally in this work. |
| This journal is © The Royal Society of Chemistry 2025 |