
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Injectable and self-healable supramolecular hydrogels assembled by quaternised chitosan/alginate polyelectrolyte complexation for sustained drug delivery and cell encapsulation†
Cristiana F. V. Sousa ,
João Borges
* and
João F. Mano*
,
João Borges
* and
João F. Mano*
CICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, Campus Universitário de Santiago, 3810-193 Aveiro, Portugal. E-mail: joaoborges@ua.pt; jmano@ua.pt
First published on 14th May 2025
Abstract
Hydrogels formed through phase separation during the complexation of oppositely charged polymers have unique properties, including fast self-assembly, hierarchical microstructures, and tunable properties. These features make them highly attractive materials for various biomedical applications, such as drug delivery, protective coatings, and surface adhesives. Notably, injectable polyelectrolyte complex (PEC) supramolecular hydrogels stand out for their minimally invasive administration and reduced trauma and side effects, providing attractive alternatives to covalent hydrogels, which are constrained by the irreversibility of their crosslinks, limiting their versatility and broader applicability. Sustainable marine-origin polysaccharides have been used for developing hydrogels due to their proven biocompatibility, non-cytotoxicity and wide bioavailability from renewable resources. In particular, chitosan (CHT) and alginate (ALG) have been widely employed to develop hydrogels, taking advantage of their opposite charge nature. However, the limited solubility of CHT under physiological conditions limits the range of bioapplications. Herein, we report the development of size- and shape-tunable PEC supramolecular hydrogels encompassing water-soluble quaternised CHT and ALG biopolymers, under physiological conditions, by polyelectrolyte complexation. The rheological and mechanical properties of the PECs are studied, demonstrating their injectability, self-healing behaviour, and cytocompatibility towards human adipose-derived stem cells. A sustained and controlled release of encapsulated fluorescein isothiocyanate-labelled bovine serum albumin is observed over fourteen days. This work paves the way for the design and development of advanced CHT-based injectable biomaterial platforms for a wide array of biomedical and biotechnological applications.
 João Borges | Dr João Borges is a Senior Researcher at the Department of Chemistry and CICECO – Aveiro Institute of Materials at the University of Aveiro, Portugal. He received his PhD in chemistry from the University of Porto in 2013. His research focuses on the molecular design, synthesis and development of supramolecular multicomponent biomaterials to be used as bioinstructive matrices to control cell functions and as platforms for controlled drug/therapeutics/cell delivery. He is an Editorial Advisory Board Member of J. Mater. Chem. B and he has been elected Chemistry Europe Fellow (Class of 2022/2023), Fellow of the Global Young Academy, Young Academy of Europe, and Young Academy of Portugal. |
1. Introduction
Over the last century, it has been discovered that mixing dilute solutions of oppositely charged polyelectrolytes leads to the spontaneous formation of a dense phase through the complexation of the two polymers.1,2 Complexation occurs mainly through supramolecular electrostatic interactions, endowing polyelectrolyte complex (PEC) hydrogels with unique properties when compared to the conventional hydrogels, which are typically prepared through covalent bonds, thereby restricting their versatility and potential applications.3–5 In contrast, PEC hydrogels denote a 3D reversible and dynamic nature as they are assembled via reversible interactions, including electrostatic interactions, hydrophobic interactions, host–guest interactions, hydrogen bonding, metal–ligand coordination, π–π stacking, and van der Waals forces.6,7 Furthermore, PEC hydrogels exhibit viscoelastic behaviour, tunable mechanical properties, hierarchical microstructures, injectability, self-healing behaviour, responsiveness to external stimuli, and protective encapsulation, which underscore their range of biomedical applications.3,7–9 The self-assembly, formation and stability of the PECs depend on numerous intrinsic factors, including the degree of ionization of the polyelectrolyte, the distribution of charges, the concentration and mixing ratio of the polyelectrolytes, the molecular architecture and the molecular weight of the polyelectrolytes, the flexibility of the polymer chains, as well as external conditions, such as ionic strength, type of salt, pH and temperature.2,7 PECs are ubiquitous in nature and play important roles in various biological processes, including as membraneless organelles,10 or natural adhesives,11 and in functions like nucleotide transportation,12,13 and protein folding.14,15 These materials have been widely used in various forms, including as membranes,16,17 coatings,18 hydrogels,19 cryogels,20 microcapsules,21 and beads22 for protein/drug/gene/cell delivery, as well as wound healing and tissue engineering strategies.The use of natural polymers, in particular those derived from marine-origin resources has emerged as a promising source for engineering PEC hydrogels, offering a sustainable alternative by repurposing waste from the food industry.23–26 The renewable natural biopolymers are particularly attractive to produce high added-value sustainable multifunctional hydrogel biomaterials for bioapplications owing to their wide and ready bioavailability, biocompatibility, physicochemical and biological properties.27–30 Chitosan (CHT), derived from the deacetylation of chitin extracted mainly from the crustaceans’ shells,31 and alginate (ALG), extracted from brown algae,32 form a polyelectrolyte pair widely used in the development of electrostatically-driven 2D and 3D materials for bioapplications owing to their opposite charge nature.33–43 However, the insolubility of CHT at physiological pH (pH < pKa ∼6–6.5)43,44 hinders the incorporation of therapeutic molecules and living cells into CHT-based materials, thereby restricting its potential for being used in bioapplications. To address this challenge, the functionalization of CHT with quaternary amine groups has been recently reported as a strategy to enhance the solubility of native CHT at physiological pH, thus enabling the incorporation of bioactive molecules and cells under nondenaturing conditions.45 In particular, N-(2-hydroxypropyl)-3-trimethylammonium chitosan chloride (HTCC) has emerged as a promising alternative to native CHT biopolymer.46–49 Despite these advantages, few studies have focused on the development of PEC hydrogels based on HTCC for biomedical purposes.50,51 In this regard, PEC hydrogels prepared through in situ polymerization of poly(acrylic acid) (PAA) monomers in quaternised CHT solution revealed improved mechanical strength and self-recovery properties when compared to PEC hydrogels, developed with unmodified CHT, owing to the higher charge density of the HTCC.51 HTCC has been also combined with divinyl ether and maleic anhydride to produce PEC hydrogels suitable for controlled drug delivery targeting cancer therapy.50 However, these works focused on the preparation of hybrid synthetic/natural polymer-based PEC hydrogels for biomedical applications. In contrast, the development of PEC hydrogels that combine native CHT exclusively with other natural polymers has been extensively explored.52–55 For instance, CHT/ALG PECs have been reported as promising platforms for cell encapsulation. However, these PECs have been developed at pH 4 due to the insolubility of CHT at physiological pH and, as such, cells need to be encapsulated after PECs preparation, followed by an additional centrifugation step.53 We hypothesise that the replacement of CHT with HTCC would not only enable the dissolution of the modified CHT in physiological media, but also the encapsulation of cells within the PEC hydrogels to be performed in a single, one-pot process. In addition, although CHT/ALG PECs have been reported as platforms for drug release, the majority of the drug is released within the first 24 h, thus do not enabling a sustained release over time.56
Herein, we report for the first time the development of fully marine-origin polysaccharides-based injectable PEC hydrogels, at physiological pH, as advanced platforms for drug and cell encapsulation and for the sustained release of model drugs (Scheme 1). The hydrogels are formed in a straightforward manner through electrostatic interactions between HTCC and ALG biopolymers, thus imparting them with reversible structural properties. The physicochemical, morphological, rheological and mechanical properties of the as-produced PEC supramolecular hydrogels were evaluated by attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy, scanning electron microscopy (SEM), rheology, and universal mechanical testing machine, respectively. Moreover, the release profile of the model drug bovine serum albumin, the injectable properties, and the in vitro cytocompatibility of the HTCC/ALG PEC hydrogels towards human adipose-derived stem cells (hASCs) were also studied, revealing the biocompatible, self-healing and injectable nature of the hydrogels. These dynamic HTCC/ALG PEC hydrogels hold great promise as advanced and sustainable nanocarriers for the encapsulation and sustained release of therapeutics and even cells, paving the way for pursuing innovative approaches in controlled therapeutics delivery, customizable tissue engineering and regenerative medicine strategies.
 | ||
| Scheme 1 Schematic illustration of the process for the self-assembly of HTCC/ALG PEC supramolecular hydrogels. After mixing the HTCC and ALG biopolymeric solutions, a dense phase spontaneously forms upon sedimentation. Following a centrifugation step, the PECs can be collected. | ||
2. Materials and methods
2.1 Materials
CHT with a molecular weight (Mw) of 184.84 kDa, viscosity ∼122 cP, and a degree of deacetylation of 94% was kindly provided by Primex ehf (Siglufjordur, Iceland) and used without further purification. Low viscosity sodium ALG derived from brown algae (Mw = 538 kDa, viscosity ∼250 cP), glycidyltrimethylammonium chloride (GTMAC), phosphate buffered saline (PBS), fluorescein isothiocyanate-labelled bovine serum albumin (FITC-BSA), Triton X-100, gelatin and paraformaldehyde were purchased from Sigma-Aldrich (St Louis, MO, USA) and used as received. Glacial acetic acid (CH3COOH) and sodium acetate trihydrate (CH3COONa·3H2O) were purchased from JMGS (Odivelas, Lisboa, Portugal). Sodium alginate NOVATACHTM MVG GRGDSP (GRGDSP-coupled alginate) was purchased from NovaMatrix (Sandvika, Norway). Lithium phenyl(2,4,6-trimethylbenzoyl)phosphinate (LAP), and amicon ultra 50 mL centrifuge tubes with ultracell membrane cut-off of 100 kDa and cellulose filters were purchased from Biosynth (Staad, Switzerland) and Merck Milipore (Darmstadt, Germany), respectively. All other reagents, namely alpha MEM medium, fetal bovine serum (FBS), penicillin, streptomycin, live/dead kit, phalloidin tetramethylrhodamine B isothiocyanate and 4,6-diaminidino-2-phenylindole-dilactate (DAPI) were purchased from Thermo Fischer Scientific (Fair Lawn, NJ, USA). All the aqueous solutions were prepared using ultrapure water from a Milli-Q Plus water purification system (resistivity >18.2 MΩ cm) from Merck Millipore (Burlington, MA, USA).2.2 Preparation of the PECs
Marine-origin polysaccharides-based PECs were prepared as described elsewhere.53 Briefly, 0.5 mg mL−1 freshly aqueous solutions of CHT, HTCC and ALG in 0.1 M acetate buffer (pH 5.5), and HTCC and ALG in 10 mM PBS (pH 7.4) were prepared. The HTCC biopolymer was previously synthesised by our group by reacting CHT with GTMAC, as described elsewhere,42 denoting a degree of quaternisation of 75%. To produce the PECs, the HTCC aqueous solution in PBS at pH 7.4 was heated to 37 °C and the ALG solution was added to it. The mixed solution was stirred at 600 rpm for 10 min and left to sediment at the bottom of the beaker for ∼1 h. After sedimentation, ∼75% of the total solution volume was discarded and the concentrated HTCC/ALG PECs were poured into ultra-centrifugal filter units with a filter membrane cut-off of 100 kDa and centrifuged at 500g for 10 min. The supernatant was removed from the upper part of the tube and the PECs were collected. For comparison, HTCC/ALG and CHT/ALG PECs were prepared in a reminiscent manner in acetate buffer at pH 5.5.2.3 Zeta (ζ)-potential and dynamic light scattering measurements
The net electrical charge of 0.5 mg mL−1 CHT, HTCC and ALG aqueous solutions in 0.1 M acetate buffer (pH 5.5), and HTCC and ALG aqueous solutions in 10 mM PBS (pH 7.4) was determined at 25 °C using a Zetasizer Nano-ZS (Malvern Instruments Ltd, Royston, Hertfordshire, UK) by measuring their ζ-potentials. Additionally, aliquots of CHT/ALG and HTCC/ALG PECs in acetate buffer and HTCC/ALG PEC in PBS were collected to measure their ζ-potentials. The electrophoretic mobility (u) was converted into a ζ-potential value following the Smoluchowski equation (ζ = uη/ε; η and ε refer to the viscosity and permittivity of the solution, respectively).57 The same apparatus was used to measure the average particle size, expressed as the average hydrodynamic diameter (Z-average) of the 0.5 mg mL−1 CHT, HTCC and ALG aqueous solutions at pH 5.5 and 7.4 by means of dynamic light scattering (DLS) at a scattering angle of 173° (backscatter). The ζ-potential and DLS measurements were performed in triplicate and the results averaged for each sample.2.4 Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy
The ATR-FTIR spectra of the individual CHT, HTCC and ALG biopolymers in powder form, as well as CHT/ALG and HTCC/ALG PECs were acquired in the absorbance mode using a Bruker TENSOR 27 FTIR spectrometer (Thermo Scientific, USA) fitted with a “Golden Gate” ATR module equipped with a diamond crystal. The ATR-FTIR spectra were measured in the spectral range of 4000–400 cm−1 by averaging 256 individual scans per sample at a resolution of 4 cm−1. All data were linear baseline corrected and normalized using the OPUS software supplied with the instrument.2.5 Scanning electron microscopy (SEM)
The microstructure and pore morphology of the PECs were analysed in a scanning electron microscope (SEM Hitachi SU-3800, Hitachi, Japan) coupled with a standard cooling stage (−25 to 50 °C, Deben, UK). The microscope operated in high vacuum in the secondary electrons mode at an acceleration voltage of 5 kV and working distances between 5.9 and 7.8 mm. SEM images were analysed by ImageJ (v. 1.52, NIH, USA) software for pore size measurements.2.6 Rheology measurements
The rheological properties of the CHT/ALG and HTCC/ALG PECs in acetate buffer at pH 5.5 and HTCC/ALG PEC in PBS at pH 7.4 were assessed using a rheometer (Kinexus Lab+, Malvern Panalytical, UK) equipped with a stainless-steel parallel plate geometry (8.0 mm diameter) and a solvent trap. The gap setting was fixed to 0.5 mm, and the assays were performed at 25 °C with a frequency of 1 Hz and strain sweep from 0.1 to 500% to determine the linear viscoelastic region (LVR). Oscillatory frequency sweep assays were performed across a frequency range of 0.01 to 100 Hz, with a constant strain of 1%. The shear-thinning profile of the PECs was evaluated by measuring their shear viscosity while increasing the shear rate from 0.1 to 100 s−1. All assays were performed with three independent experiments.2.7 Self-healing behaviour
The self-healing behaviour of HTCC/ALG PEC hydrogels prepared in PBS at pH 7.4 was assessed by dynamic rheology using step-strain experiments. In brief, the hydrogel was subjected to cyclic strain at a frequency of 1 Hz, alternating between low (1%) and high (200%) strains over five cycles, and the self-healing recovery from the first cycle was determined. Three independent experiments were performed. The self-healing properties of the hydrogels were also qualitatively studied by cutting them into six segments and staining them with three different dyes (red, blue and green dyes). The stained pieces were then reassembled at room temperature (RT), forming a continuous filament and a closed circle. Microscopic images of the cross-sections of the HTCC/ALG PEC hydrogels were captured before and after rejoining the pieces, using an SEM (Hitachi SU-3800, Hitachi, Japan) equipped with a standard cooling stage (−25 to 50 °C, Deben, UK). The microscope operated in high vacuum in the secondary electrons mode at an acceleration voltage of 5 kV and working distances between 6.1 and 6.9 nm.2.8 Water content, swelling degree and weight loss
After the preparation of the HTCC/ALG PEC hydrogels in PBS at pH 7.4, the excess of water was removed using a cellulose filter and the wet weight (Ww) was measured. Then, the hydrogels (n = 3) were frozen at −80 °C, freeze-dried for 24 h, and the dry weight (Dw) was measured. The water content (%) was determined using the equation (eqn (1)), as follows:
 | (1) |
The swelling degree of the PEC hydrogels after 1, 3, 7, 14 and 30 days in PBS at pH 7.4, and adjusted to the pH values of 4, 5.5, 9 and 12 using either 0.1 M HCl or 0.1 M NaOH, was determined by measuring the difference in the wet weight (Wt) at each time-point relative to day zero, using the equation (eqn (2)), as follows:
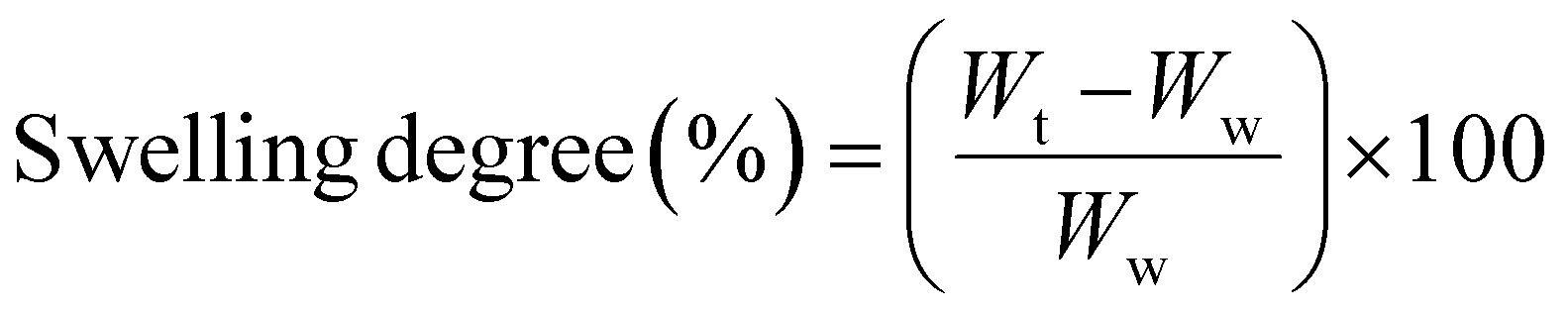 | (2) |
The weight loss was quantified by calculating the differences in the dry weight (Dt) at each time-point relative to day zero, following the equation (eqn (3)), as follows:
 | (3) |
Microscopy images of the PEC hydrogels over time, under different pH conditions, were acquired using a microscope with magnifying glass (Stemi 508 Stereo Microscope, Carl Zeiss, Jena, Germany).
2.9 Mechanical properties
The mechanical properties of the HTCC/ALG PEC hydrogels prepared in PBS at pH 7.4 were evaluated using a universal mechanical testing machine (Shimadzu MMT-101N, Shimadzu Scientific Instruments, Kyoto, Japan) equipped with a load cell of 50 N. Unidirectional compression tests were performed on hydrogels at a constant rate of 1 mm min−1. Young's modulus was determined as the slope of the linear region of strain-stress curves corresponding to 5–10% strain. The toughness and ultimate strength correspond to the maximum peak determined for each individual stress–strain curve.2.10 Extrudability and injectability
HTCC/ALG PEC hydrogels prepared in PBS at pH 7.4 were loaded into a 1 mL syringe and extruded through needles with gauges of 27, 25 and 22 G using a microfluidic pump. To assess the fluidity and structural integrity of the injectable HTCC/ALG PECs, 10% (w/v) methacrylated gelatin (GelMA) hydrogel constructs embedding a needle were prepared aiming to form a hollow microchannel. After photo-crosslinking for 20 s on each side of the hydrogel using 0.5% LAP as photo-initiator, the needle was removed, allowing the HTCC/ALG hydrogel to be injected through the hollow microchannel created by the needle. The GelMA biopolymer was synthesised as described elsewhere.582.11 In vitro release profile
The ability of the HTCC/ALG PEC hydrogels prepared in PBS (pH 7.4) to encapsulate FITC-BSA was assessed by mixing a 0.25 mg mL−1 FITC-BSA aqueous solution in PBS (pH 7.4) with the ALG solution used for PECs formation. Then, the in vitro release profile of the hydrogels was determined by their incubation, in triplicate, in 5 mL of 10 mM PBS aqueous solution at pH 7.4 at 50 rpm and 37 °C for 14 days. At each predetermined time-points (0.25, 0.5, 1, 2, 4, 6, 8, 24, 48, 72, 96, 120, 168, 216, 264 and 336 h), 0.2 mL of the release medium was collected, and an equal volume of fresh PBS was added to the sample. The fluorescence of the sample solutions collected from both the PEC hydrogel development (loading capacity) and the release assays were measured using a microplate reader (Synergy HTX Biotek, Izasa Scientific, Carnaxide, Portugal) at excitation (λexc) and emission (λem) wavelengths of 495 and 520 nm, respectively. A standard calibration curve of FITC-BSA (0–150 μg mL−1) in PBS (pH 7.4) was used to quantify the cumulative released mass over time. Fluorescence microscopy micrographs of the PEC hydrogels over time were acquired in an upright motorized widefield fluorescence microscope (Axio Imager M2, Carl Zeiss, Jena, Germany) equipped with a 200 W HXP lamp, a 3.0 Mpix monochromatic camera (Axiocam 105 mono; Carl Zeiss, Jena, Germany), and a 5× objective (Carl Zeiss, Jena, Germany).2.12 Metabolic activity and viability assays
The cell viability assays were conducted in a HTCC/ALG-RGD PEC hydrogel. ALG-RGD was used instead of native ALG biopolymer owing to the intrinsic capacity of the RGD peptide motif to bind to the integrin receptors on the cell's surface triggering cell adhesion and proliferation. The cytotoxicity of the hydrogels was evaluated using a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) cell proliferation assay and a live/dead fluorescence study, following the manufacturer's instructions and using hASCs as a model of adherent cells. Briefly, hASCs (25 × 103 cells per mL) were mixed with the ALG-RGD solution and then added to the HTCC solution to form the PEC with hASCs encapsulated. The PECs were centrifuged at 300g for 5 min (a standard condition in cell culture). Afterwards, the HTCC/ALG PEC hydrogel was injected through the hollow microchannel of the GelMA hydrogels, and then incubated in alpha MEM medium with 10% (v/v) FBS and 1% (v/v) penicillin–streptomycin in 48-well plates at 37 °C, 5% CO2 and fully humified for 1, 3 and 7 days. For the metabolic activity assays, the medium of the hydrogels was replaced by dPBS containing MTS (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and incubated for 4 h to determine the metabolic activity of hASCs in the hydrogel channel after 1, 3 and 7 days. The absorbance was measured at 490 nm, using a microplate reader. The metabolic activity assays were conducted in three independent experiments with triplicates.
:1) and incubated for 4 h to determine the metabolic activity of hASCs in the hydrogel channel after 1, 3 and 7 days. The absorbance was measured at 490 nm, using a microplate reader. The metabolic activity assays were conducted in three independent experiments with triplicates.
For the viability assays, hASCs encapsulated within the injected hydrogel were stained with calcein-AM (1:500 in dPBS) and propidium iodide (1:1000 in dPBS) for 30 min at 37 °C, protected from the light. Afterwards, the cells were washed twice with dPBS and immediately visualized by confocal microscopy (Zeiss LSM 900 confocal laser scanning microscope equipped with a GaSP detector, Carl Zeiss, Jena, Germany).
2.13 Cell morphology
The cell morphology of hASCs encapsulated in the HTCC/ALG PEC hydrogel injected into the microchannel of GelMA hydrogels was evaluated after culturing for 1, 3, and 7 days in 48-well plates at 37 °C with 5% CO2 and full humidity. At each time point, the culture medium was removed, and the cells were washed with dPBS. The cells were then fixed with 4% (v/v) paraformaldehyde in PBS for 1 h at RT. After fixation, the samples were rinsed with PBS and incubated with 0.5% Triton X-100 for 5 min at RT to permeabilize the cells. Following permeabilization, the cells were washed with dPBS, and solutions of phalloidin tetramethylrhodamine B isothiocyanate (1:1000 in dPBS) and DAPI (1:1000 in dPBS) were added for 30 min and 5 min, respectively, to stain the F-actin cytoskeleton and nuclei. The samples were then washed with dPBS and immediately visualized using confocal microscopy.
2.14 Statistical analysis
Unless otherwise noted, all experiments were performed in triplicates (n = 3) and the results presented as mean ± standard deviation (SD). The statistical analysis was performed by one-way ANOVA followed by Tukey's post-hoc multiple comparison test using the GraphPad Prism 9.4.0 (GraphPad Inc.) software. Statistically significant differences were considered for * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, and **** p ≤ 0.0001.3. Results and discussion
3.1 Physicochemical and morphological characterisation of the PECs
Prior to the production of the PECs, the net electrical charge of the freshly prepared 0.5 mg mL−1 CHT, HTCC and ALG aqueous solutions in 0.1 M acetate buffer at pH 5.5 and 0.5 mg mL−1 HTCC and ALG aqueous solutions in 10 mM PBS at pH 7.4 were evaluated by measuring their ζ-potentials. Fig. 1a shows the ζ-potentials of the biopolymers proving the positive and negative charge of CHT and HTCC, and ALG, respectively, thus confirming their cationic and anionic nature. Furthermore, the ζ-potentials of CHT, HTCC and ALG biopolymers were similar, in moduli, at both pH 5.5 and 7.4, which could lead to efficient CHT/ALG and HTCC/ALG PECs formation by charge compensation. In this regard, we hypothesised that electrostatically driven PECs could be developed using either CHT or HTCC and ALG. Based on the literature, we decided to work with a 1:1 (w/w) ratio of CHT and ALG biopolymers since PEC hydrogels developed with the same polymer concentration have been reported to exhibit higher drug encapsulation efficiencies. For instance, 1:1 (w/w) CHT/ALG PEC hydrogels were reported to exhibit a diclofenac sodium entrapment efficiency of ca. 89%, whereas similar hydrogels denoting a 1:2 and 2:1 (w/w) CHT/ALG ratios revealed encapsulation efficiencies of ca. 86% and 29%, respectively. In addition to the superior drug loading capacity assured by the CHT/ALG PECs with 1:1 (w/w) ratio, higher amounts of either CHT or ALG also led to reduced release efficiency.59 Thus, PECs encompassing CHT/ALG and HTCC/ALG at pH 5.5 and HTCC/ALG at pH 7.4 were developed and their ζ-potentials measured. While the ζ-potential of the CHT/ALG PEC at pH 5.5 was found to be +7.2 ± 0.6 mV, the HTCC/ALG PEC exhibited a ζ-potential of −10.6 ± 1.2 mV at the same pH. This difference could be assigned to the behaviour of the amine groups in CHT and HTCC, as the HTCC biopolymer contains fewer primary amino groups (∼25% remain unmodified) than CHT due to its quaternisation procedure.42 At pH 5.5, CHT is below its pKa (∼6–6.5),43,44 showing positively charged amino groups that probably did not react with ALG, which could lead to the overall positive charge of the CHT/ALG PEC. On the other hand, the HTCC/ALG complexes exhibited negative ζ-potential values due to the full neutralization of the amino groups in HTCC by ALG. The HTCC/ALG PEC at pH 7.4 also demonstrated a negative ζ-potential value of −5.4 ± 0.6 mV, suggesting that the surface is negatively charged, with the remaining unreacted deprotonated ALG carboxyl groups, i.e. negatively charged at pH 7.4, which is above the pKa of ALG (∼3.38 or 3.65 for mannuronic and guluronic acid residues, respectively).32,60
 | ||
| Fig. 1 (a) Zeta (ζ)-potential measurements (black bars) for CHT, HTCC and ALG biopolymers, and CHT/ALG and HTCC/ALG PECs, and average particle size, expressed as the average hydrodynamic diameter (Z-average) for CHT, HTCC and ALG biopolymers (patterned bars). Data are presented as mean ± SD of three independent experiments (n = 3) performed in triplicates. (b) ATR-FTIR spectra of CHT, HTCC and ALG biopolymers, and CHT/ALG and HTCC/ALG PECs. Representative optical images of (c) CHT/ALG and (d) HTCC/ALG PECs in acetate buffer and (e) HTCC/ALG PECs in PBS. Scale bars represent 1 mm. Representative SEM micrographs of the (f and i) CHT/ALG and (g and j) HTCC/ALG PECs in acetate buffer and (h and k) HTCC/ALG PECs in PBS. Scale bars represent 500 μm in the panels f, g and h, and 100 μm in the panels i, j and k, respectively. (l) Pore size measurements of the structures observed in the SEM images. | ||
The CHT, HTCC and ALG biopolymer powders, and CHT/ALG and HTCC/ALG PECs were also inspected using ATR-FTIR spectroscopy (Fig. 1b). As previously reported, the ATR-FTIR spectra of HTCC powder showed two well-defined absorption peaks at 1477 and 1647 cm−1, assigned to the methyl groups in the quaternary ammonium side chains and GTMAC molecules, respectively.42 Moreover, the absence of the peaks presented by the native CHT biopolymer at 1645 and 1587 cm−1, attributed to amide I and amide II, respectively, confirms that the –NH2 groups in the CHT biopolymer were replaced by N-(2-hydroxypropyl)-3-trimethylammonium chloride groups, confirming the successful modification of the native CHT biopolymer. The ATR-FTIR spectra of ALG biopolymer revealed two absorption peaks at 1593 and 1408 cm−1, corresponding to the asymmetric and symmetric stretching vibrations of –COOH groups in the ALG structure.61 The spectra of the PECs showed a peak at ∼1408 cm−1, confirming the interaction between CHT or HTCC and ALG. The PECs developed with HTCC also reveal a peak at ∼1477 cm−1, being indicative of the presence of HTCC in the complexes.
Although CHT, HTCC and ALG biopolymers were successfully employed to form PECs, their visual appearance is significantly different. The PECs prepared at pH 5.5 exhibited a viscous liquid consistency (Fig. 1c and d and ESI Movie S1 and S2†), whereas those formed at pH 7.4 displayed a more solid structure, resembling hydrogels (Fig. 1e and Movie S3†). The difference in consistency emphasises the strong impact of the buffer solution and pH on the PECs’ physical properties. The lower pH of the acetate buffer resulted in a more fluid and unstable network, whereas the neutral pH of PBS led to a faster and more cohesive structure and efficient gel formation. To assess the role of salts in the formation of PECs, HTCC/ALG PECs were prepared at pH 7.4 using PBS without sodium chloride and potassium chloride. The resulting PECs denoted an appearance like those prepared with PBS containing the salts, demonstrating that pH is the primary component driving the formation of PEC hydrogels (Movie S4†). Moreover, strong polyelectrolyte pairs, i.e. polymers that remain charged in solution across all pH values, like HTCC,42 are known to form solid complexes, in contrast to weak polyelectrolytes,62 such as CHT, whose charge varies from fully charged to uncharged. This suggests that the combination of a strong polyelectrolyte pair with a neutral pH enables the formation of solid PECs. The PECs also reveal clear differences in the microstructure and pore morphology, as observed by SEM. The HTCC/ALG PECs formed in PBS appear to be more porous than the other complexes (Fig. 1f–k). Additionally, the pore size within these PECs is larger and more uniform than the one in the CHT/ALG or HTCC/ALG PECs assembled at pH 5.5 (Fig. 1l). We hypothesise that this behaviour might be due to the more hydrophobic structure and possible assembly of the HTCC biopolymer into a globular conformation. In fact, as shown in Fig. 1a, DLS measurements performed for individual CHT (pH 5.5 = 732.9 ± 164.6 nm), HTCC (pH 5.5 = 463.6 ± 62.4 nm; pH 7.4 = 352.6 ± 75.4 nm) and ALG (pH 5.5 = 548.5 ± 94.6 nm; pH 7.4 = 615.6 ± 150.9 nm) aqueous solutions revealed the formation of particles denoting an average hydrodynamic diameter (Z-average) smaller for HTCC, which resonate well with our hypothesis. In addition, the lower availability and number of protonated primary amines to form complexes with ALG at pH 7.4 extensively reduce the electrostatic interaction between HTCC and ALG and enable the entry of dissolution media with counterions, which may contribute to the larger micropore size denoted by the HTCC/ALG PEC hydrogels at pH 7.4.
3.2 Rheological characterisation of the PECs
The rheological properties of the PECs were investigated by evaluating their oscillatory amplitude and frequency sweep and viscoelasticity (Fig. 2). Oscillatory amplitude sweep tests were conducted at 25 °C to determine the LVR, where the storage modulus (G′, elastic modulus) and loss modulus (G′′, viscous modulus) remain independent of the shear strain, as well as the values of yield and flow point. As shown in Fig. 2g, HTCC/ALG PECs prepared in PBS exhibited a higher and a more stable LVR (0.1% to 10%) than the remaining conditions (0.1% to 5%) (Fig. 2a and d). For each condition, a strain value within this range was selected for further studies. The yield point, i.e. the shear strain level at which G′ starts to decrease,63 indicating the breakdown of the internal structure,64 was higher (∼10%) in the HTCC/ALG PECs formed in PBS when compared to the other complexes prepared in acetate buffer (∼5%). This suggests that the internal structure of the PECs at pH 7.4 is more robust, whereas the complexes at pH 5.5 have a more fragile network. After the yield point, the G′ is still higher than G′′, but the yield stress has been overcome, suggesting a solid-like behaviour with irreversible deformation. Then, the flow point, i.e. G′ = G′′, for HTCC/ALG PECs prepared in PBS occurred at ∼31.88% of shear strain, indicating the transition from a solid-like to a liquid-like behavior.64 Afterwards, the viscous behaviour dominates over the elastic behaviour (G′′ > G′), leading to the flow of the material.64 The PECs formed at pH 5.5 showed a G′ consistently higher than G′′, reflecting a predominantly solid-like behaviour. However, the G′ and G′′ values for HTCC/ALG PECs formed in PBS were significantly higher than those observed in the other complexes, highlighting the formation of a more robust material suitable for applications requiring enhanced structural integrity. | ||
| Fig. 2 Rheological characterisation of the PECs. (a) Strain sweep at 1 Hz, (b) frequency sweep using a strain of 1% and (c) viscosity measurements of CHT/ALG PECs in acetate buffer (pH 5.5). (d and g) Strain sweep at 1 Hz, (e and h) frequency sweep using a strain of 1% and (f and i) viscosity measurements of HTCC/ALG PECs in acetate buffer (pH 5.5) and PBS (pH 7.4), respectively. | ||
The frequency dependence of both moduli was also assessed over the range of 0.01 to 100 Hz. The PECs exhibited minimal frequency-dependent behaviour, with G′ consistently exceeding G′′, suggesting a solid-like nature across all complexes studied (Fig. 2b, e and h). Nevertheless, a significant increase in both moduli was denoted at frequencies higher than 10 Hz. This behaviour is assigned to the short relaxation time of the physical crosslinks, which prevents the dissociation of the PECs during high frequency oscillations and, in turn, increases the G′ and G′′. On the other hand, at lower frequencies, there is sufficient time for the physical interactions to dissociate, thereby reducing their contribution to the overall moduli.65 The stiffness of each condition can be calculated as the value of G′ at 1 Hz, which for CHT/ALG and HTCC/ALG PECs in acetate buffer, and HTCC/ALG PECs in PBS were found to be 0.3 ± 0.1 kPa, 0.2 ± 0.1 kPa, and 4.2 ± 1.1 kPa, respectively. This suggests that the HTCC/ALG PECs formed in PBS are significantly stiffer than the other PECs. We hypothesise that the combination of physiological pH and a strong polyelectrolyte (HTCC) enhances the electrostatic interactions in comparison to the acidic pH used to prepare the other PECs.
Furthermore, supramolecular self-assembly is associated with shear-thinning properties.66 In this context, the viscosity profile of the PECs was evaluated at increasing shear rates. As shown in Fig. 2c, f and i, the viscosity of all complexes decreased as the shear rate increased, indicating a characteristic non-Newtonian shear-thinning behaviour, which is essential for injectability.67 In addition, the highest shear viscosity of HTCC/ALG PECs in PBS (∼6597 Pa s) was ∼225- and 43-fold higher than that of the HTCC/ALG (∼29.3 Pa s) and CHT/ALG PECs (∼152 Pa s) in acetate buffer, respectively. This marked increase in viscosity highlights their enhanced robustness, further demonstrating their superior ability to maintain structural integrity and withstand mechanical stress, as well as suitability for fulfilling demanding biomedical applications. The PECs prepared at pH 5.5 were used as control owing to the insolubility of CHT at pH 7.4. However, due to the inferior rheological properties of these complexes when compared to those obtained with HTCC/ALG at pH 7.4, all subsequent studies were performed exclusively with the HTCC/ALG PECs prepared in PBS (pH 7.4).
3.3 Self-healing properties of HTCC/ALG PEC hydrogels
HTCC/ALG PEC hydrogels were prepared by supramolecular electrostatic interactions (Fig. 3a), which are known to contribute to self-healing properties.68–70 To confirm such behaviour, five step-strain cycles, alternating between low (1%) and high (200%) strain, were applied to the hydrogel to assess the autonomous structural recovery ability of its mechanical properties (Fig. 3b). The high strain value was chosen to exceed the crossover point (∼31.9% of shear strain) observed in the amplitude sweep experiments, indicating the deformation of the 3D hydrogel network. When a low strain (1%) was applied, the PEC hydrogel retained its solid-like behaviour (G′ > G′′), while under high strain (200%) the PEC hydrogel underwent a temporary transition into a liquid-like material (G′′ > G′). However, upon reapplying a low strain, the HTCC/ALG PEC hydrogels demonstrated a fast recovery of the original G′ values, with G′ once again exceeding G′′ due to the reestablishment of dynamic electrostatic bonds, restoring their original microstructure and mechanical properties.67 In fact, the PECs recovered ∼95% of their original G′ values after one deformation cycle, and this recovery remained consistent throughout all cycles, thus demonstrating its excellent self-healing behaviour, and ability to be repeatedly reshaped. In addition to the rheological assays, the PEC hydrogels’ self-healing capacity was visually assessed by cutting the hydrogel into six pieces and staining them with three different dyes (Fig. 3c). Then, the six pieces were brought into contact for 3 min at RT, allowing them to hold different shapes when handled. As shown in Fig. 3d–h, the fragments perfectly healed into a single, integral piece that could be shaped into virtually any type of geometry and enable the assembly of both rather simple and more convoluted structures, including a fibre-like structure and a continuous closed circle, which can be easily handled and maintain their structural integrity, all without the need for any external stimuli. Therefore, this qualitative, visual assessment corroborates the rheological findings, highlighting the remarkable self-healing properties of the HTCC/ALG PEC hydrogels. We also evaluated the microstructure of the hydrogels cut into two pieces, both before (Fig. 3i) and after (Fig. 3j) they were rejoined. It was found that, after rejoining the two pieces, the hydrogel structure was completely healed into a single piece at the microscopic level, with the pores of the two pieces being interconnected.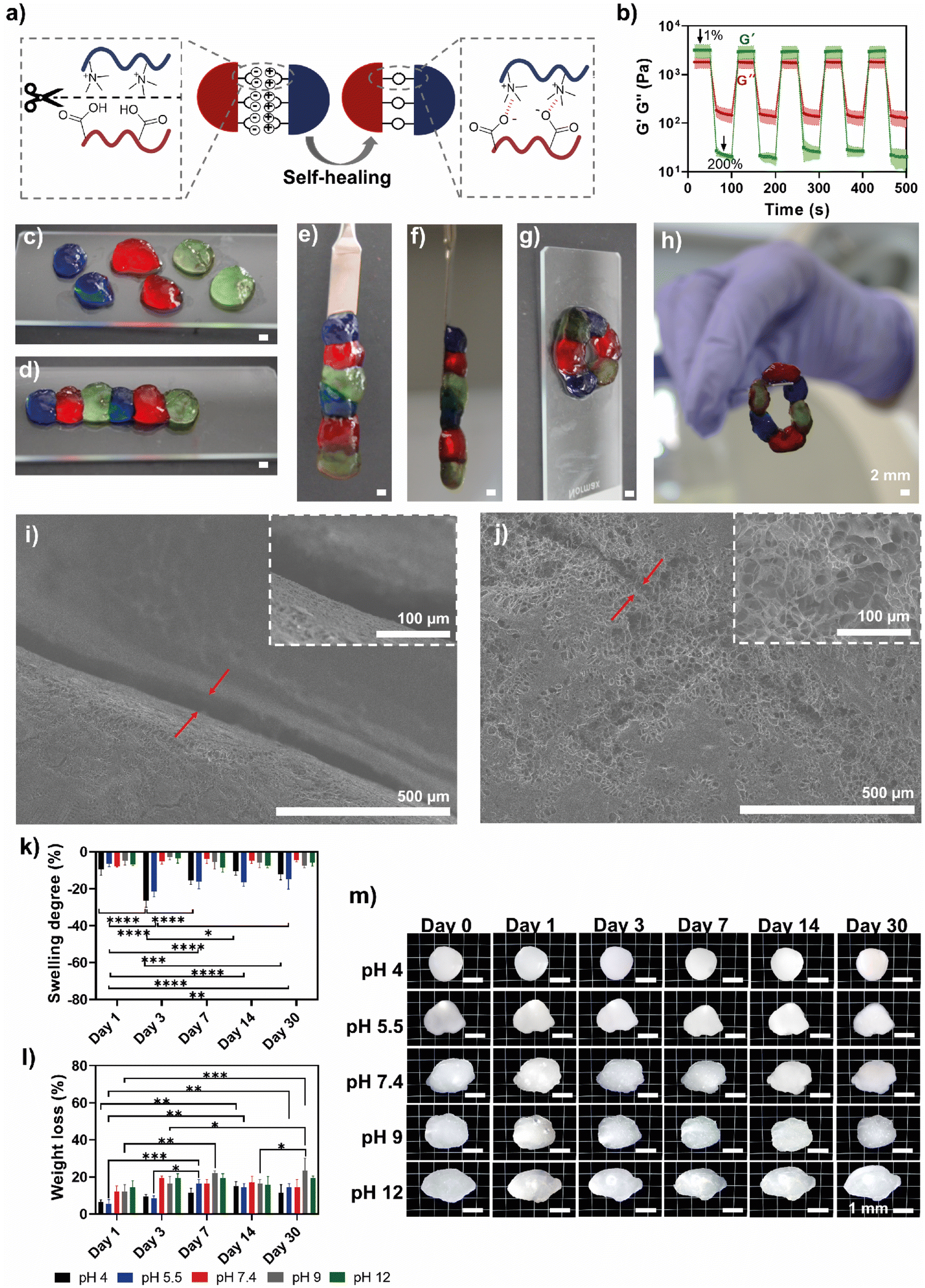 | ||
| Fig. 3 (a) Schematic representation of the self-healing process in HTCC/ALG PEC hydrogels. After mechanical disruption, the hydrogel structure repairs itself through reversible electrostatic interactions between HTCC and ALG biopolymeric chains. This dynamic crosslinking enables the hydrogel to restore its structural integrity, maintaining both mechanical strength and functionality. (b) Dynamic oscillatory test at 1 Hz using low (1%) and high (200%) strain amplitudes. Data are presented as mean ± SD of three independent experiments (n = 3). Optical images of the (c) hydrogel cut into six stained pieces, which are then reassembled to form a single and integral (d–f) fibre-like structure or (g and h) a continuous closed circle, demonstrating the self-healing properties of the HTCC/ALG hydrogels. Scale bars represent 2 mm in the panels c, d, e, f, g and h. SEM micrographs of the cross-sections of the hydrogel (i) before and (j) after the pieces were rejoined. The red arrows indicate the gap (i) between the two pieces and (j) the rejoined region, respectively. The magnification factor is 100× (main images) and 500× (insets). (k) Swelling degree (%) and (l) weight loss (%) of HTCC/ALG hydrogels over 30 days at different pH values. (m) Microscopy images of the hydrogels immersed in PBS solutions with the pH adjusted to 4, 5.5, 9 and 12 over 30 days. Scale bars represent 1 mm. | ||
Furthermore, having in mind that the water content of the hydrogels can significantly enhance their biocompatibility and mimic the highly hydrated native ECM microenvironment,71 the water content (%) of the HTCC/ALG PEC hydrogels was measured by weighing them immediately after their preparation and again after freeze-drying, resulting in a water content of 81.7 ± 0.5%. We also evaluated the swelling degree (i.e. water uptake, Fig. 3k) and weight loss (Fig. 3l) of the HTCC/ALG PEC hydrogels over time at different pH values. The moulded hydrogels did not lose their structural integrity and maintained a stable morphology after immersion in acidic, neutral or basic pH (Fig. 3m) and demonstrated a consistent ability to maintain most of their water content, with some weight loss observed over time. The PECs exposed to acidic environments (pH 4 and 5.5) shrank more in comparison to those at neutral or basic pH. We hypothesise that this behaviour might be due to the pH responsiveness of the HTCC biopolymer to acidic media. In fact, the HTCC/ALG PECs subjected to acidic pH experienced a phenomenon of shrinkage followed by swelling due to the weight loss (dry mass) shown by the PECs, which translated into a higher capacity of the PEC hydrogel to absorb water molecules.
3.4 Mechanical properties of HTCC/ALG PEC hydrogels
The mechanical properties of the HTCC/ALG PEC hydrogels were evaluated upon compressive tests. The HTCC/ALG hydrogels revealed a Young's modulus of 6.9 ± 2.1 kPa, an ultimate strength of 18.0 ± 3.3 kPa, and toughness of 2.3 ± 0.7 kJ m−3. The softer mechanical properties imparted by the HTCC/ALG hydrogels make them particularly suitable for applications requiring injectable properties, surpassing significant challenges in injectability and the high risk of needle clogging denoted by stiff hydrogels. In fact, injectable hydrogels require a balance between fluidity for ease of injection and sufficient mechanical stability post-extrusion to maintain their structural integrity.72,73 In this regard, the Young's modulus represents the elasticity or stiffness of a material by describing the relationship between the applied force and the resulting deformation of the hydrogel. Hydrogels with a higher Young's modulus require more stress to achieve the same deformation when compared to those with a lower modulus, remaining stiffer and more resistant to deformation under applied forces.74 Therefore, a Young's modulus of 6.9 ± 2.1 kPa ensures adequate flexibility, allowing the hydrogel to deform easily under stress,75 while also offering enough support in situ, allowing the hydrogel to mimic the mechanical environment of soft tissues, which typically present similar elastic properties.76,77 CHT/hyaluronic acid (HA) PEC hydrogels exhibited a Young's modulus of ∼3–4 kPa at pH 7.0, 7.5 and 8.0, when the HA concentration was 1.5%.78 In comparison, the Young's modulus of our PEC hydrogels was 2-times higher, even with a polymer concentration of only 0.05%, indicating a significantly greater number of physical crosslinks between HTCC and ALG biopolymers. We hypothesise that this behaviour might be due to the different polymer combinations, Mw and stoichiometric ratio of the oppositely charged polymeric materials. Moreover, the ultimate strength of 18.0 ± 3.3 kPa enables the hydrogel to deform during injection without compromising its structure, allowing it to recover the shape afterwards.70 Additionally, the toughness of 2.3 ± 0.7 kJ m−3 supports the material's ability to absorb energy without fracturing, which is crucial for maintaining structural integrity during and after the injection process. On the other hand, PEC hydrogels composed of quaternised CHT and PAA were prepared using various polymer ratios, and the Young's modulus, ultimate strength and toughness ranged from 0.89 to 63.76 MPa, 1.99 to 16.09 MPa, and 1.85 to 15.60 MJ m−3, respectively.51 These values are significantly higher than those observed for our HTCC/ALG PEC hydrogels. We hypothesise that this difference arises from the inclusion of a synthetic polymer (PAA), as synthetic PEC hydrogels are highly viscoelastic materials over a wide range of strain rates. On the other hand, fully natural PEC hydrogels are elastic-like materials under small strains, but become plastic-like at large strains, displaying yield, flow, and fracture that are delayed at high strain rates. The transformation of natural PEC hydrogels from elastic- to plastic-like, as well as their differences from synthetic PEC hydrogels is related to the difference in charge density and chain flexibility between natural and synthetic polymers.79 Thus, the mechanical values imparted by the HTCC/ALG hydrogels highlight their suitability for applications where injectability and adaptability are prioritized over mechanical rigidity. This behaviour suggests that the HTCC/ALG hydrogels could represent suitable injectable platforms for soft tissue engineering strategies and drug delivery, as the soft polymeric network would tend to create a more favourable environment for encapsulating and protecting sensitive bioactive molecules, thereby possibly maintaining their bioactivity until release takes place.3.5 In vitro release profile
The structural properties of non-covalent hydrogels, including their mechanical strength, self-healing ability and injectability enable the encapsulation of (bio)active molecules and their localized release, enhancing the efficacy of therapeutic agents following injection. This turns them into an attractive, versatile, and functional platform for minimally invasive therapies.80,81 Thus, based on the microstructure of the HTCC/ALG PEC supramolecular hydrogels, we studied their potential to act as a drug delivery platform using FITC-BSA as a model drug. The in vitro release profile of FITC-BSA from HTCC/ALG PEC hydrogels (Fig. 4) was evaluated using the dialysis method, with the amount of FITC-BSA loaded and released quantified based on a standard calibration curve. The loading capacity of HTCC/ALG PEC hydrogels for FITC-BSA was estimated to be 56.2 ± 2.6%. Notably, the hydrogels effectively mitigated the initial burst release (Fig. 4b) and promoted a more sustained and prolonged release of FITC-BSA over time until a plateau was reached (Fig. 4a). This behaviour enabled a quasi-zero-order release rate during the first 7 days, with a total of 90.4 ± 7.3% of the loaded FITC-BSA being released over a period of 14 days. Fluorescence microscopy images corroborate the observed release trend, revealing that the FITC-BSA release starts from the outer part and progressively moves towards the inner regions of the PEC hydrogel. These results suggest that HTCC/ALG PEC hydrogels can serve as effective vehicles for sustained drug delivery in multiple biomedical applications, enabling drug release at a constant rate over time. Other studies explored the combination of HTCC with polymers of natural or synthetic origin to create hydrogels for controlled drug release. For instance, dopamine and metronidazole were encapsulated in hydrogels made of HTCC and gelatin, and no initial burst release was observed for either drug within the first hours. Both drugs were released in a sustained manner over more than 12 days, demonstrating the hydrogel's effectiveness as a long-term release system,82 and corroborating the release profile of our HTCC/ALG PEC hydrogels. In another approach, the incorporation of magnesium–aluminium layered double hydroxide (LDH)-celecoxib (CLX) complexes into hydrogels encompassing HTCC, gelatin and LAPONITE® resulted in an initial burst release within the first day due to the presence of CLX on the LDH nanoparticles surface. This was followed by a sustained linear release, as CLX intercalated within the LDH layers was gradually released into the hydrogel matrix and then into the release medium over a period of 14 days,83 similar to the release kinetics observed for our PEC hydrogels. | ||
| Fig. 4 (a) Cumulative mass release profile (%) of FITC-BSA in PBS at pH 7.4 from HTCC/ALG hydrogels over 14 days. The inset fluorescence microscopy micrographs show the fluorescence of FITC-BSA within the HTCC/ALG hydrogels over 14 days. Scale bars represent 500 μm. (b) Zoom-in on the cumulative release of FITC-BSA for the first 8 h (represented with a red dotted line in a). Data are presented as mean ± SD of three independent experiments (n = 3) performed in triplicates. | ||
3.6 Extrudability and injectability of HTCC/ALG PEC hydrogels
The development of dynamic and injectable hydrogels that can be introduced into the human body via minimally invasive procedures and delivered on-demand at target-specific sites is highly desirable for biomedical applications. In this regard, we assessed the extrusion properties of the HTCC/ALG PEC supramolecular hydrogels using a microfluidic pump and three different needle gauges (27, 25 and 22 G). Fig. 5a and Movies S5–S7† demonstrate that the HTCC/ALG hydrogels were successfully extruded through all tested needle gauges, exhibiting good injectability and volume stability, and a well-defined needle-shaped structure. In addition, Fig. 5b shows a red-stained filament produced using a 25 G needle, which was the one that yielded the longest filament, and, therefore, it was selected for subsequent assays. Based on these findings, we hypothesised that the HTCC/ALG hydrogels could be injected into a hollow microchannel embedded within larger bulk hydrogels (Fig. 5c), enabling the simulation of a defect and facilitating the study of cell–material interactions in a controlled 3D microenvironment aiming for soft tissue engineering and regenerative medicine strategies. This study would allow us to understand how materials and biochemical factors influence cell behaviour, as well as analyse the biomaterial response and the biocompatibility in more realistic 3D environments. To test this hypothesis, a 10% (w/v) GelMA aqueous solution was placed in a PDMS mould with a needle positioned in the centre. After photopolymerization of the GelMA pre-hydrogel aqueous solution, and subsequent removal of the needle, a well-defined hollow microchannel was created within the GelMA hydrogel (Fig. 5d). Then, the HTCC/ALG-RGD PEC hydrogels were injected into the microchannel, effectively filling the hollow environment inside the channel (Fig. 5e), which demonstrated its suitability for injection. | ||
| Fig. 5 (a) Maximum filament length achieved during the extrudability assay of HTCC/ALG hydrogels using needles with gauges of 27, 25 and 22. Scale bars represent 1 cm. (b) Optical image of a red-stained filament obtained using a 25 G needle. (c) Design of (1) biocompatible and biodegradable GelMA bulk hydrogel constructs featuring a (2) hollow microchannel for the (3) injection of cell-laden HTCC/ALG-RGD PEC hydrogel. Representative optical and confocal bright-field microscopy images of the microchannel within the GelMA hydrogels (d) before and (e) after the injection of the HTCC/ALG hydrogels. Scale bars represent 100 μm. | ||
3.7 In vitro biological performance
Natural polymers have been employed to encapsulate cells in 3D microenvironments.84 HTCC/ALG-RGD PEC hydrogels encapsulating hASCs were injected into the hollow microchannel encapsulated within the GelMA supporting hydrogel to assess the cytocompatibility of the PEC hydrogels, as well as their ability to support cell growth over 7 days of culture. Fig. 6a shows representative fluorescence microscopy images of live/dead assays performed after 1, 3 and 7 days of culture, revealing that the hASCs remained viable up to 7 days of culture with noticeable cell spreading starting at day 3. The morphological features of the encapsulated cells were further assessed by staining the F-actin filaments (phalloidin, stained in red) and nuclei (DAPI, stained in blue) (Fig. 6b), revealing that by day 3 hASCs began elongating, and by day 7 the cells were completely stretched and spread across the entire channel. In addition, the cellular metabolic activity was investigated via an MTS assay after 1, 3 and 7 days of culture (Fig. 6c). The metabolic activity significantly increased from day 1 to day 3 and remained constant by day 7, revealing no significant differences between the culture on days 3 and 7. We hypothesise that the latter might be due to the cells fully occupying the available space within the channel. Nevertheless, it was possible to confirm that the hASCs remained viable for up to 7 days, thus corroborating the cell viability assays. These findings demonstrate the cytocompatibility of the PEC hydrogels and their ability to support stem cell culture and direct stem cell fate, highlighting their potential for soft tissue engineering and regenerative medicine strategies. | ||
| Fig. 6 Representative fluorescence microscopy images of hASCs encapsulated in the HTCC/ALG-RGD PEC hydrogels within the GelMA bulk hydrogel, showing (a) live (green) and dead (red) cells, and (b) DAPI (blue) and phalloidin (red) staining at 1, 3, and 7 days of culture. Scale bars represent 200 μm. (c) Metabolic activity (490 nm) of hASCs encapsulated in the hydrogels at 1, 3 and 7 days. Data are presented as mean ± SD of three independent experiments (n = 3) performed in triplicates. | ||
4. Conclusions
In this work, we successfully developed injectable, self-healable, cytocompatible and sustainable PEC hydrogels encompassing water-soluble HTCC and ALG biopolymers under physiological conditions, surpassing the insolubility of native CHT at physiological pH. These supramolecular hydrogels, assembled via polyelectrolyte complexation by resorting to an easy, inexpensive and versatile methodology, revealed a quasi-zero-order release of FITC-BSA for ∼1 week, ensuring a sustained and steady drug release over extended periods. This controlled drug release profile highlights the suitability of the hydrogels for long-term therapeutic strategies which could improve the treatment efficacy while simultaneously reducing the dose frequency and associated side effects. Moreover, the mechanical robustness and self-healing properties, together with the ease of injectability and cytocompatibility towards hASCs, turn the HTCC/ALG PECs into suitable hydrogel biomaterials for biomedical purposes. We envisage that the unique properties imparted by the HTCC, together with its potential to be combined with virtually any type of oppositely charged biopolymer, under mild conditions, to enable the self-assembly of PEC hydrogels, could open new perspectives in the development of size- and shape-tunable complex biomaterials for pursuing advanced therapies in controlled drug delivery and tissue regeneration. For instance, the HTCC/ALG PEC hydrogels hold great promise as platforms for developing 4D hydrogels denoting spatiotemporally controlled regions, assigned by the bonded hydrogel fragments carrying out distinct cargos (e.g., bioactive molecules, cells), owing to the precise control and tunability of the composition and spatial arrangement of each piece over time.Author contributions
J. B. and J. F. M. conceived the original idea and research direction, provided the resources, supervised the project and acquired funding. C. F. V. S., J. B. and J. F. M. designed the experiments and methodology. C. F. V. S performed the experiments and wrote the original draft of the manuscript. All authors analysed the data, discussed the results, and reviewed and edited the manuscript.Data availability
Data for this article are available from DUnAs (institutional research data repository of the University of Aveiro) at https://doi.org/10.48527/YNA9ZG following a 2-year embargo period due to potential IPR issues.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Union's Horizon Europe Research and Innovation Programme under the Grant Agreement No. 101079482 (“SUPRALIFE”). C. F. V. S. and J. B. gratefully acknowledge the Portuguese Foundation for Science and Technology (FCT) for the individual PhD grant (2020.04408.BD, https://doi.org/10.54499/2020.04408.BD), and individual Assistant Researcher contract (2020.00758.CEECIND/CP1589/CT0007, https://doi.org/10.54499/2020.00758.CEECIND/CP1589/CT0007) under the Scientific Employment Stimulus – Individual Call, respectively. This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020 (https://doi.org/10.54499/UIDB/50011/2020), UIDP/50011/2020 (https://doi.org/10.54499/UIDP/50011/2020), and LA/P/0006/2020 (https://doi.org/10.54499/LA/P/0006/2020), financed by national funds through the FCT/MCTES (PIDDAC). The authors gratefully acknowledge Dr Hélène L. Lauzon from Primex ehf (Siglufjordur, Iceland) for kindly providing the chitosan batch used in this work.References
- S. M. Lalwani, C. I. Eneh and J. L. Lutkenhaus, Phys. Chem. Chem. Phys., 2020, 22, 24157–24177 RSC.
- Y. Ghimire and A. Bhattarai, Int. J. Eng. Res. Tecnhol., 2020, 9, 876–889 Search PubMed.
- E. A. Appel, J. del Barrio, X. J. Loh and O. A. Scherman, Chem. Soc. Rev., 2012, 41, 6195–6214 RSC.
- W. Hu, Z. Wang, Y. Xiao, S. Zhang and J. Wang, Biomater. Sci., 2019, 7, 843–855 RSC.
- H. Senebandith, D. Li and S. Srivastava, Langmuir, 2023, 39, 16965–16974 CrossRef CAS PubMed.
- V. S. Meka, M. K. G. Sing, M. R. Pichika, S. R. Nali, V. R. M. Kolapalli and P. Kesharwani, Drug Discovery Today, 2017, 22, 1697–1706 CrossRef CAS PubMed.
- B. Sim, J. J. Chang, Q. Lin, J. H. M. Wong, V. Ow, Y. Leow, Y. J. Wong, Y. J. Boo, R. Goh and X. J. Loh, Biomacromolecules, 2024, 25, 7563–7580 CrossRef CAS PubMed.
- J. Y. C. Lim, Q. Lin, K. Xue and X. J. Loh, Mater. Today Adv., 2019, 3, 100021 CrossRef.
- M. Neumann, G. di Marco, D. Iudin, M. Viola, C. F. v. Nostrum, B. G. P. v. Ravensteijn and T. Vermonden, Macromolecules, 2023, 56, 8377–8392 Search PubMed.
- J. A. Riback, L. Zhu, M. C. Ferrolino, M. Tolbert, D. M. Mitrea, D. W. Sanders, M. T. Wei, R. W. Kriwacki and C. P. Brangwynne, Nature, 2020, 581, 209–214 CrossRef CAS PubMed.
- H. Shao, K. N. Bachus and R. J. Stewart, Macromol. Biosci., 2009, 9, 464–471 CrossRef CAS PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- J. R. Vieregg, M. Lueckheide, A. B. Marciel, L. Leon, A. J. Bologna, J. R. Rivera and M. V. Tirrell, J. Am. Chem. Soc., 2018, 140, 1632–1638 CrossRef CAS PubMed.
- F. B. Sheinerman, R. Norel and B. Honig, Curr. Opin. Struct. Biol., 2000, 10, 153–159 CrossRef CAS PubMed.
- T. H. Walther and A. S. Ulrich, Curr. Opin. Struct. Biol., 2014, 27, 63–68 CrossRef CAS PubMed.
- R. R. Costa, A. M. S. Costa, S. G. Caridade and J. F. Mano, Chem. Mater., 2015, 27, 7490–7502 CrossRef CAS.
- E. N. Durmaz, M. I. Baig, J. D. Willott and W. M. De Vos, ACS Appl. Polym. Mater., 2020, 2, 2612–2621 CrossRef CAS PubMed.
- E. Jamróz, M. Janik, L. Juszczak, T. Kruk, P. Kulawik, M. Szuwarzyński, A. Kawecka and K. Khachatryan, Carbohydr. Polym., 2021, 274, 118627 CrossRef PubMed.
- M. Balima, I. Morfin, G. Sudre and A. Montembault, Carbohydr. Polym., 2024, 339, 122265 CrossRef CAS PubMed.
- E. S. Dragan, M. V. Dinu and C. A. Ghiorghita, Gels, 2022, 8, 240 CrossRef CAS PubMed.
- J. K. Bediako, C.-R. Lim, E. Repo, S.-H. Choi and Y.-S. Yun, Chem. Eng. Sci., 2023, 274, 118688 CrossRef CAS.
- A. A. Altam, L. Zhu, W. Huang, H. Huang and S. Yang, Carbohydr. Polym. Technol. Appl., 2021, 2, 100100 CAS.
- D. N. Carvalho, C. Gonçalves, J. M. Oliveira, D. S. Williams, A. Mearns-Spragg, R. L. Reis and T. H. Silva, Green Chem., 2021, 23, 7016–7029 RSC.
- J. R. A. Romal and S. K. Ong, Coord. Chem. Rev., 2023, 482, 215054 CrossRef.
- X. Lin, J. Wang, X. Wu, Y. Luo, Y. Wang and Y. Zhao, Adv. Funct. Mater., 2023, 33, 2211323 CrossRef CAS.
- L. Spitzer, A. Adrien, U. T. Veettil, S. Olza, A. Alonso-Varona and S. C. M. Fernandes, Carbohydr. Polym. Technol. Appl., 2024, 8, 100541 CAS.
- Y. Sun, Y. Bai, W. Yang, K. Bu, S. K. Tanveer and J. Hai, Front. Chem., 2022, 10, 915648 CrossRef PubMed.
- A. Das, T. Ringu, S. Ghosh and N. Pramanik, Polym. Bull., 2023, 80, 7247–7312 CrossRef CAS PubMed.
- R. K. Manivannan, N. Sharma, V. Kumar, I. Jayaraj, S. Vimal and M. Umesh, Carbohydr. Polym. Technol. Appl., 2024, 8, 100536 Search PubMed.
- G. Satchanska, S. Davidova and P. D. Petrov, Polymers, 2024, 16, 1159 CrossRef CAS PubMed.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106–126 CrossRef CAS PubMed.
- P. Sher, C. A. Custódio and J. F. Mano, Small, 2010, 6, 2644–2648 CrossRef CAS PubMed.
- R. R. Costa, E. Castro, F. J. Arias, J. C. Rodríguez-Cabello and J. F. Mano, Biomacromolecules, 2013, 14, 2403–2410 CrossRef CAS PubMed.
- S. Petroni, I. Tagliaro, C. Antonini, M. D'Arienzo, S. F. Orsini, J. F. Mano, V. Brancato, J. Borges and L. Cipolla, Mar. Drugs, 2023, 21, 147 CrossRef CAS PubMed.
- S. G. Caridade, C. Monge, F. Gilde, T. Boudou, J. F. Mano and C. Picart, Biomacromolecules, 2013, 14, 1653–1660 CrossRef CAS PubMed.
- J. Borges, S. G. Caridade, J. M. Silva and J. F. Mano, Macromol. Rapid Commun., 2015, 36, 405–412 Search PubMed.
- J. M. Silva, C. A. Custódio, R. L. Reis and J. F. Mano, ACS Biomater. Sci. Eng., 2016, 2, 2304–2314 CrossRef CAS PubMed.
- N. I. Martins, M. P. Sousa, C. A. Custódio, V. C. Pinto, P. J. Sousa, G. Minas, F. Cleymand and J. F. Mano, Acta Biomater., 2017, 57, 313–323 CrossRef CAS PubMed.
- C. Ribeiro, J. Borges, A. M. S. Costa, V. M. Gaspar, V. de Zea Bermudez and J. F. Mano, Molecules, 2018, 23, 625 CrossRef PubMed.
- C. F. V. Sousa, C. A. Saraiva, T. R. Correia, T. Pesqueira, M. I. Rial-hermida, J. Borges and J. F. Mano, Biomolecules, 2021, 11, 863 CrossRef CAS PubMed.
- M. M. A. Sacramento, J. Borges, F. J. S. Correia, R. Calado, J. M. M. Rodrigues, S. G. Patrício and J. F. Mano, Front. Bioeng. Biotechnol., 2022, 10, 1041102 Search PubMed.
- C. F. V. Sousa, L. P. G. Monteiro, J. M. M. Rodrigues, J. Borges and J. F. Mano, J. Mater. Chem. B, 2023, 11, 6671–6684 Search PubMed.
- M. A. Pujana, L. Pérez-Álvarez, L. C. C. Iturbe and I. Katime, Polymer, 2012, 53, 3107–3116 Search PubMed.
- B.-I. Andreica, X. Cheng and L. Marin, Eur. Polym. J., 2020, 139, 110016 CrossRef CAS.
- K. Kamiński, K. Szczubiałka, K. Zazakowny, R. Lach and M. Nowakowska, J. Med. Chem., 2010, 53, 4141–4147 CrossRef PubMed.
- L. Wang, C. Qin, W. Wang and W. Li, Carbohydr. Polym., 2011, 84, 1289–1292 CrossRef CAS.
- B. Lorkowska-Zawicka, K. Kamiński, J. Ciejka, K. Szczubiałka, M. Białas, K. Okoń, D. Adamek, M. Nowakowska, J. Jawień, R. Olszanecki and R. Korbut, Mar. Drugs, 2014, 12, 3953–3969 CrossRef PubMed.
- E. D. Freitas, C. F. Moura, J. Kerwald and M. M. Beppu, Polymers, 2020, 12, 2878 CrossRef CAS PubMed.
- V. A. Izumrudov, I. F. Volkova and M. Y. Gorshkova, Eur. Polym. J., 2013, 49, 3302–3308 CrossRef CAS.
- J. You, S. Xie, J. Cao, H. Ge, M. Xu, L. Zhang and J. Zhou, Macromolecules, 2016, 49, 1049–1059 CrossRef CAS.
- S. Vasiliu, S. Racovita, M. Popa, L. Ochiuz and C. A. Peptu, in Cellulose-Based Superabsorbent Hydrogels, 2018, pp. 1–31 Search PubMed.
- M. B. Oliveira, H. X. S. Bastos and J. F. Mano, Biomacromolecules, 2018, 19, 2742–2749 CrossRef CAS PubMed.
- X. Li, L. Shang, D. Li, W. Wang, S. Chen, H. Zhong, Y. Huang and S. Long, Polym. Test., 2022, 109, 107547 CrossRef CAS.
- Y. Luo and Q. Wang, Int. J. Biol. Macromol., 2014, 64, 353–367 CrossRef CAS PubMed.
- G. Conzatti, D. Faucon, M. Castel, F. Ayadi, S. Cavalie and A. Tourrette, Carbohydr. Polym., 2017, 172, 142–151 CrossRef CAS PubMed.
- R. J. Hunter, Zeta potential in colloid science. Principles and applications, Academic Press, London, UK, 1988 Search PubMed.
- X. Zhao, Q. Lang, L. Yildirimer, Z. Y. Lin, W. Cui, N. Annabi, K. W. Ng, M. R. Dokmeci, A. M. Ghaemmaghami and A. Khademhosseini, Adv. Healthcare Mater., 2016, 5, 108–118 CrossRef CAS PubMed.
- D. Nikolova, M. Simeonov, C. Tzachev, A. Apostolov, L. Christov and E. Vassileva, Polym. Int., 2022, 71, 668–678 CrossRef CAS.
- F. Kurayama, S. Suzuki, T. Oyamada, T. Furusawa, M. Sato and N. Suzuki, J. Colloid Interface Sci., 2010, 349, 70–76 CrossRef CAS PubMed.
- B. Sarker, D. G. Papageorgiou, R. Silva, T. Zehnder, F. Gul-E-Noor, M. Bertmer, J. Kaschta, K. Chrissafis, R. Detsch and A. R. Boccaccini, J. Mater. Chem. B, 2014, 2, 1470–1482 RSC.
- M. Tirrell, ACS Cent. Sci., 2018, 4, 532–533 CrossRef CAS PubMed.
- J. M. Townsend, E. C. Beck, S. H. Gehrke, C. J. Berkland and M. S. Detamore, Prog. Polym. Sci., 2019, 91, 126–140 Search PubMed.
- L. del-Mazo-Barbara and M. P. Ginebra, J. Eur. Ceram. Soc., 2021, 41, 18–33 CrossRef CAS.
- J. Hao and R. A. Weiss, Macromolecules, 2016, 49, 6687–6693 Search PubMed.
- X. Yan, Y.-R. Chen, Y.-F. Song, J. Ye, M. Yang, B.-B. Xu, J. Zhang, X. Wang and J.-K. Yu, Front. Bioeng. Biotechnol., 2020, 8, 847 CrossRef PubMed.
- V. Sousa, A. J. R. Amaral, E. J. Castanheira, I. Marques, J. M. M. Rodrigues, V. Félix, J. Borges and J. F. Mano, Biomacromolecules, 2023, 24, 3380–3396 Search PubMed.
- J. Zheng, X. Song, Z. Yang, C. Yin, W. Luo, C. Yin, Y. Ni, Y. Wang and Y. Zhang, J. Controlled Release, 2022, 350, 898–921 CrossRef CAS PubMed.
- M. Wu, L. Han, B. Yan and H. Zeng, Supramol. Mater., 2023, 2, 100045 Search PubMed.
- J. Xu and S. Hsu, J. Biomed. Sci., 2023, 30, 43 CrossRef CAS PubMed.
- C. D. Spicer, Polym. Chem., 2020, 11, 184–219 Search PubMed.
- J. Liu, C. Du, W. Huang and Y. Lei, Biomater. Sci., 2024, 12, 8–56 RSC.
- C. Xie, G. Liu, L. Wang, Q. Yang, F. Liao, X. Yang, B. Xiao and L. Duan, Pharmaceutics, 2024, 16, 430 CrossRef CAS PubMed.
- D. Lee, H. Zhang and S. Ryu, in Cellulose-Based Superabsorbent Hydrogels, 2019, pp. 865–884 Search PubMed.
- E. Rybacki, A. Reinicke, T. Meier, M. Makasi and G. Dresen, J. Pet. Sci. Eng., 2015, 135, 702–722 CrossRef CAS.
- T. Su, M. Xu, F. Lu and Q. Chang, RSC Adv., 2022, 12, 24501–24510 Search PubMed.
- K. Princen, N. Marien, W. Guedens, G. J. Graulus and P. Adriaensens, ChemBioChem, 2023, 24, e202300149 CrossRef CAS PubMed.
- M. Balima, I. Morfin, G. Sudre and A. Montembault, Carbohydr. Polym., 2024, 339, 122265 CrossRef CAS PubMed.
- R. Shi, T. L. Sun, F. Luo, T. Nakajima, T. Kurokawa, Y. Z. Bin, M. Rubistein and J. P. Gong, Macromolecules, 2018, 51, 8887–8898 CrossRef CAS PubMed.
- M. J. Cardoso, R. R. Costa and J. F. Mano, Mar. Drugs, 2016, 14, 1–27 CrossRef PubMed.
- M. I. Rial-Hermida, A. Rey-Rico, B. Blanco-Fernandez, N. Carballo-Pedrares, E. M. Byrne and J. F. Mano, ACS Biomater. Sci. Eng., 2021, 7, 4102–4127 CrossRef CAS PubMed.
- Y. Ren, X. Zhao, X. Liang, P. X. Ma and B. Guo, Int. J. Biol. Macromol., 2017, 105, 1079–1087 Search PubMed.
- M. Nezadi, H. Keshvari, F. Shokrolahi and P. Shokrollahi, Int. J. Biol. Macromol., 2024, 266, 131337 Search PubMed.
- L. Gasperini, J. F. Mano and R. L. Reis, J. R. Soc., Interface, 2014, 11, 20140817 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5bm00072f |
| This journal is © The Royal Society of Chemistry 2025 |