
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFabrication of uniform biodegradable microcages with predesigned shape printed from microarrays for sustained release of small hydrophilic molecules†
Jiaxin
Zhang
 *ac,
Rui
Sun
b,
Valeriya
Kudryavtseva
a,
David J.
Gould
c and
Gleb B.
Sukhorukov
ad
*ac,
Rui
Sun
b,
Valeriya
Kudryavtseva
a,
David J.
Gould
c and
Gleb B.
Sukhorukov
ad
aSchool of Engineering and Materials Science, Queen Mary University of London, London, E1 4NS, UK
bCentre for Oral Bioengineering, Bart's and the London, School of Medicine and Dentistry, Queen Mary University of London, Turner Street, London E1 2AD, UK
cBiochemical Pharmacology, William Harvey Research Institute, Queen Mary University of London, London EC1M 6BQ, UK
dLife Improvement by Future Technologies (LIFT) Center, Moscow, 143025, Russia
First published on 27th May 2025
Abstract
Drug delivery vehicles have aroused increasing attention over the years due to their ability to protect and control the release of encapsulated cargo. However, several challenges significantly limit their wide applications including poor size distribution, uncontrollable size and shape, and leakage of loaded small hydrophilic cargos. This work introduces a novel and scalable microarray-based printing technique for preparing uniform biodegradable “microcages” with predesigned shapes for encapsulating and controlling the release of small hydrophilic molecules. The drugs encapsulated in the microcage are centrally located within solid microparticles without being exposed to the surface or dispersed throughout the polymer matrix. Here, 5(6)-carboxyfluorescein (CF) as a small and hydrophilic model drug are successfully loaded into polylactide acid (PLA) microcages with the dry loading method. Additionally, blending polycaprolactone (PCL) with PLA increases the permeability of the microcage polymer shells for controlled release. A higher PCL content results in a faster release rate of the encapsulated drug. Approximately 28 pg of CF particles can be encapsulated within individual microcages. This microcage printing technique provides a novel, scalable method for producing uniform biodegradable microcages, extending microprinting beyond microfilms and microparticles. A unique dry loading approach, independent of drug solubility, further broadens its utility for diverse biomedical applications.
1. Introduction
Microcapsules, with their unique microscale inner hollow structures, have sparked significant interest in various fields such as drug delivery,1 sensing,2 bioimaging,3 microreactors,4 cosmetics.5 Their utility as drug delivery vehicles is paramount, offering protection for the encapsulated cargo, controlled release, enhanced stability and bioavailability, and reduced side effects.6,7 Importantly, the therapeutic efficacy of these vehicles depends on both the drug function and the delivery methods and materials.8 Therefore, compared with non-biodegradable polymers, biopolymers have emerged as ideal candidates for drug delivery due to their excellent biocompatibility and biodegradability, minimizing side effects and unwanted toxicity.9To create biodegradable drug delivery vehicles for biomedical applications, numerous microencapsulation techniques have been explored including the layer-by-layer (LbL) self-assembly approach.10 This method forms microcapsule shell on micro/nanoparticles through the alternate deposition of oppositely charged materials, driven by electrostatic interactions, followed by the removal of a sacrificial core template. While this technique allows for the design of biodegradable microcapsules, it can only manage to encapsulate cargos of high molecular weight or small but hydrophobic molecules, due to the semi-permeability of multilayer shell.11 Since most biologically active molecules have a molecular weight under 500 Da,12 it is of great importance to overcome the challenge of small and hydrophilic cargo encapsulation and release.
Another common technique for preparing microcapsules is bulk emulsification, where droplets of one liquid are dispersed in another immiscible liquid, stabilized by surfactants, and can form double or multiple emulsions with solvent evaporation.13,14 By selecting the appropriate polymer in the external phase, biodegradable microcapsules can be created.15 However, the size of emulsion droplets and resulting microcapsules depends on the emulsification device, surfactants, and applied energy, leading to poorly uniform size distributions with standard batch high-shear mixing.16 This variation in size distribution affects particle surface area and drug release behaviours,17 emphasizing the need for uniform size distribution.
Microfluidic techniques manipulate small fluid volumes (10−9 to 10−18 litres) using lithographed channels, enabling the synthesis of monodispersed microcapsules,18,19 including biodegradable ones.20 Basically, immiscible liquids are driven through microchannels and mixed at junctions, forming droplets due to high shear forces.21 The droplet size is influenced by parameters such as flow rates, fluid viscosities, interfacial tension, and channel dimensions, which can be adjusted in microfluidic devices.22 Although this method offers a versatile route for highly monodispersed microcapsules, it requires the drug-loaded core to be immiscible with the shell material, thus limiting active compound selection. Additionally, microfluidic techniques typically produce spherical or simple non-spherical shapes,23 making it challenging to engineer more complex microcapsule configurations.
Current challenges in fabricating biodegradable drug delivery vehicles highlight the need for an improved approach. Such an approach should non-liquidly encapsulate small, hydrophilic molecules, ensure uniform size distribution, and allow for predetermined shapes. Using hydrophobic and biodegradable polymers for the microcapsule shell leverages their low water permeability.24 Simultaneously, template-based methods can control size distribution by lithographing homogeneous microstructures,25,26 in which the polymers are filled and confined, resulting in uniform particles that mirror the size and shape of the engraved microstructure.27,28 While uniformed biodegradable microparticles have been prepared using similar templates,29 these are merely polymer–drug blends, not microcapsules, leading to potential waste and insufficient protection for drugs near interfaces.
Microprinting is a versatile and efficient method for creating uniform drug delivery systems with submicron resolution. It can be applied to precisely replicate microstructures and fabricate surface patterns on various substrates, making it a convenient technique in a wide range of applications.30 Polydimethylsiloxane (PDMS) is the most widely adopted material for microprinting. As a soft polymer, PDMS can be easily molded and conformed to make full contact with the substrate surface.31 The versatility of microprinting technique extends to printing multiple materials, including polymers, biomolecules, and even cells, thereby enabling diverse applications such as electronic sensors, microfluidics, and cell capture devices.32 Notably, microprinting has been extensively used to prepare microchamber array films for drug delivery.33 However, it has primarily produced drug-loaded microfilms, thus extending this technique for microcapsule printing holds significant potential for broader applications.
In this paper, we propose a method using a combination of hydrophobic and biodegradable polymers as the microcapsule shell, complemented with a template-based microprinting method for size distribution control. Generally, a novel, facile, scalable microprinting technique for the preparation of uniformed biodegradable microcages was first-time proposed. And the biodegradable polymer here is selected as polylactic acid (PLA) and polycaprolactone (PCL), both of which are aliphatic and hydrophobic polymers with numerous advantages including excellent biocompatibility,34,35 exceptional miscibility36,37 and great mechanical properties.38,39 The US Food and Drug Administration (FDA) has approved both polymers for preparing commercial products,40 enabling their wide widespread use in drug deliver41,42 and tissue engineering.43,44 Moreover, an original dry state drug loading method was introduced,45 enabling simple and rapid loading of solid compounds, independent of solubility, molecular weight, or solvent interactions.
Given the unique structure and properties of the biodegradable microstructures we have developed, a new terminology: “microcage” is proposed here. Unlike traditional core–shell microcapsules, these microcages are filled structures with drug crystals located centrally but not exposed to the surface, offering unique advantages. Specifically, uniform PLA microcages are fabricated via a microprinting technique using a microwell-patterned template for uniform size and shape. We investigate the controlled release behaviours of the microcages by adjusting the blending ratios of PCL with PLA. To observe and characterize encapsulation and release behaviours, we selected small, hydrophilic molecules 5(6)-carboxyfluorescein (CF, 376 g mol−1) as model drug to be encapsulated using the dry loading method. In general, this work presents a novel microprinting technique for preparing uniform biodegradable microcages, thereby extending microprinting beyond microfilms and microparticles, and further enhancing drug encapsulation possibilities and advancing biodegradable drug delivery vehicles for biomedical applications.
2. Materials and methods
2.1. Materials
Polycaprolactone (PCL, Sigma 704105, molecular weight ∼45 kDa), Polylactic acid biopolymer (PLA, GoodFellow 346310), 5(6)-carboxyfluorescein (CF, Sigma 21877), Nile Red (Sigma 72485), Gelatin, from bovine skin (Sigma G9382). All the chemicals above were used as received without further purification.Poly (dimethylsiloxane) (PDMS) kit (Sylgard 184) was purchased from Dow-Corning, Midland, USA. Moreover, the temperature of the release incubation experiment was maintained at 37 °C in climate chamber (Memmert, HPP 110). Chloroform was purchased from VWR, Darmstadt, Germany. In addition, the ionic strength and pH of incubation solutions of capsules were maintained by phosphate buffered saline (PBS tablets, Sigma P4417). Deionised (DI) water from a Milli-Q (Millipore) water purification system, with an 18.2 MΩ cm resistance, was used to prepare all the solutions. Polymethyl methacrylate (PMMA) template for preparing patterned PDMS stamp is provided by Dr Maxim Kiryukhin (A*STAR, Singapore). The period of microwell arrays of the PMMA template is 16.35 μm. The microwell is in the shape of rounded quadrangular frustum pyramid, with the long side, short side, depth as 14 μm, 10 μm, 8 μm respectively. Moreover, the thickness of PMMA template films is about 54 μm including the 8 μm depth of microwells. The SEM images of the PMMA template can be found in Fig. S1.†
2.2. Preparation of microwell-patterned PDMS stamp
As schematically illustrated in Fig. S2,† a PDMS mixture was produced by combining PDMS prepolymer and a curing agent in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, stirred to ensure homogeneity, and then centrifuged to eliminate air bubbles. The PDMS mixture was subsequently cast onto a microwell-patterned PMMA template to fabricate PDMS stamps with micropillars. After casting, the PDMS mixture and the PMMA template were cured at 70 °C for 3 hours, following a 30-minute vacuuming process to further remove trapped air bubbles. The final PDMS stamps, with structures inverse to the PMMA template, were then separated from the template and cut to the desired dimensions.
:1 ratio, stirred to ensure homogeneity, and then centrifuged to eliminate air bubbles. The PDMS mixture was subsequently cast onto a microwell-patterned PMMA template to fabricate PDMS stamps with micropillars. After casting, the PDMS mixture and the PMMA template were cured at 70 °C for 3 hours, following a 30-minute vacuuming process to further remove trapped air bubbles. The final PDMS stamps, with structures inverse to the PMMA template, were then separated from the template and cut to the desired dimensions.
To generate PDMS stamps with a microwell array structure, an additional molding and transferring process was needed. In this secondary casting, the previously prepared PDMS stamp with micropillar arrays served as a PDMS template. The preparation procedure mirrored the initial casting process, with the addition of a gold coating on the PDMS template surface to prevent adhesion of the new PDMS mixture. Following a vacuuming and incubation period, a microwell-patterned PDMS stamp was obtained by removing the micropillar-patterned PDMS template.
2.3. Printing of dye-loaded biodegradable microcages
The biodegradable polymers used in this experiment were PLA and PCL, both of which can be easily dissolved in the organic solvent such as chloroform. Firstly, the prepared microwell-patterned PDMS stamp, as well as a flat PDMS, were dipped into the polymer solution (2% w/w dissolved in chloroform for both PLA and PCL) for 5 seconds with a dip coater (Ossila L2006A1) at the speed of 3 mm s−1. Both were gently lifted to create an evenly coated layer using a dip-coating method. It is noteworthy to mention that the thickness of polymer coating on the PDMS stamp can be precisely controlled by the dip-coating speed as well as the polymer solution concentration.46 The dip-coated film on the PDMS stamp was allowed to dry for 5 minutes in ambient conditions before drug loading process.CF crystals were first processed into submicron-sized particles using Precellys 24 Tissue Homogenizer prior to loading at 5500 rpm for 10 minutes, with a 2-minute break after every 30 seconds of homogenization (Fig. S3†). Then, the milled dye particles were placed onto the pre-coated PDMS stamp and loaded into microwells with dry loading methods. Excess dye crystals were carefully wiped away using with fuzz-free lab wipes. The pre-coated flat PDMS was then combined with the drug-loaded patterned PDMS stamp and heated at 140 °C while pressed at a pressure of 0.25–1 MPa for 1 minute by a homemade fixture (Fig. S4†). After cooling to room temperature, the flat PDMS was removed, leaving microcages embedded in the microwells of the patterned PDMS stamp.
To acquire a microcage suspension, the PDMS stamp was as pressed against a glass slide coated with a 10% gelatin solution and subsequently frozen at −20 °C for 10 minutes. After the freezing step, the PDMS stamp was carefully removed, transferring the individual microcages onto the solidified gelatin. To achieve a suspension, the gelatin-microcage slide was incubated in warm water at 37 °C. The resulting suspension of microcages was then transferred into a 2 ml centrifuge tube containing warm water at 37 °C. The tube was centrifuged at 5000 rpm for 1 minute, which was repeated three times to wash away the gelatin, replacing the supernatant each time. The harvested microcages were stored for subsequent experiments. This method was consistently applicable to both PLA and PLA–polycaprolactone (PLA–PCL) blended microcages.
For the fabrication of PLA–PCL blended microcages, the process remained consistent with previous methods, with the only difference being the polymer solution used for dip-coating. Specifically, PLA solutions were substituted with PLA–PCL blended solutions. Both PLA and PCL were prepared as 2% (w/w) chloroform solutions. The blends were created by mixing 90 g of 2% PLA with 10 g of 2% PCL to achieve a PLA:PCL ratio of 9:1. Similarly, blends with PLA:PCL ratios of 8:2 and 7:3 were prepared by mixing 80 g and 70 g of 2% PLA with 20 g and 30 g of 2% PCL, respectively. The procedures and conditions for printing the microcages were otherwise identical to those described above.
2.4. Release experiment of printed biodegradable microcages
In order to maintain optimal pH, temperature, and ionic strength, all CF-loaded microcage release experiments were conducted in 0.01 M PBS solutions at 37 °C with a pH of 7.4. All CF-loaded microcages, including those from PLA–PCL blends at ratios of 9:1, 8:2, and 7:3, as well as the pure PLA microcages, were incubated in 2 ml PBS within 2 ml centrifuge tubes. Each tube contained 1 million CF-loaded microcages. And each release group consisted of three replicates. The amount of CF release can be determined from the cumulated release curve. Release experiments were performed in a humidified incubator (5% CO2 at 37 °C). Samples were periodically collected by centrifugation with supernatant replaced for further analysis.
2.5. Drug loading capacity
Due to the nature of the special dry loading methods, it is less meaningful to calculate the conventional drug loading efficiency as a large excess of drug particles was physically applied to the surface of the microwell arrays to ensure sufficient loading, and only a small fraction became trapped within the wells. Importantly, the excess drug can be collected and reused in subsequent loading processes. The amount of drug loaded was determined from the accumulated release amount of the release curve of the microcages. Therefore, the drug loading capacity (DLC%) were calculated to describe the drug loading performance:
2.6. Characterization
The surface morphology of each sample was analyzed using scanning electron microscopy (SEM, FEI Inspect-F). Prior to analysis, samples were sputter-coated with about 5 nm gold nanofilm (SC 7620, Quorum, Laughton, UK) for 45 seconds to ensure conductivity. SEM settings were an accelerating voltage of 20 kV, a spot size of 3.0, and a working distance of ∼10 mm. The confocal laser scanning microscopy (CLSM) images were obtained with a Leica TS confocal scanning system (Leica, Heidelberg, Germany) equipped with a 20× objectives in order to confirm the encapsulation and release of fluorescent cargos. The CF-loaded PLA microcages were observed with Nikon confocal scanning system (Nikon CSU-W1 SoRa Spinning Disk Confocal). In addition, fluorescent spectrometry (LS 55, PerkinElmer) was utilized to quantitatively detect the amount of fluorescent substance at the excitation wavelength of 494 nm with the calibration curve of CF in PBS (pH 7.4) shown in Fig. S5.† The thermal properties of applied polymers were evaluated by differential scanning calorimetry (DSC, TA instrument DSC 25).3. Results and discussion
3.1. Preparation and characterization of microcages
Microchamber array films, offering the advantages of uniformly designable sizes and shapes and a wide range of candidate polymers, have been extensively studied over the past decade.47 The preparation of microcapsules by separating microchamber array films is still under exploration. Our previous work reported a straightforward method for preparing PLA microcapsules from microchambers films by physically removing the polymer on the ridge of the microarrays. While this method achieved uniform size and shape, the use of solvent to separate polymers on the ridge can potentially damage the drug, limiting the range of applicable drugs.48 To address these issues, a novel and scalable microprinting technique for preparing uniform biodegradable microcages for sustained drug release is introduced here.The method and process of printing biodegradable microcages is schematically illustrated in Fig. 1. To gain a better understanding of the printing procedures and to elucidate the mechanism of microcage formation and drug encapsulation, SEM was used to check and observe the surface morphology variations at key stages during the printing process of both the PDMS stamp as well as the final printed microcages. Images in Fig. 2 show PLA microcage production.
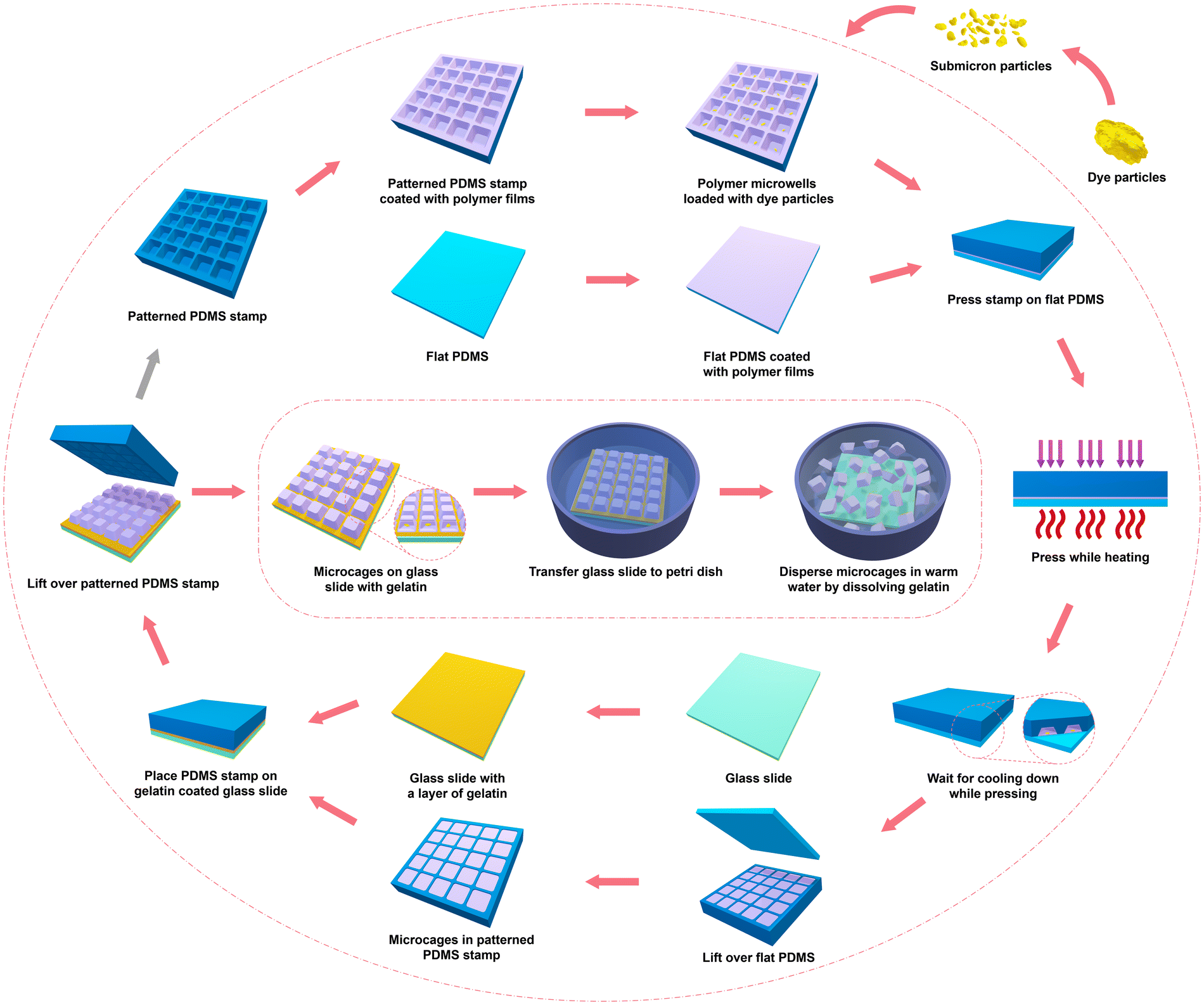 | ||
| Fig. 1 Schematic illustration of printed microcage preparation. | ||
 | ||
| Fig. 2 SEM images of key stages during the preparation of microcages. (a and b) PDMS stamp with microwell array structure. (c and d) A layer of precoated PLA film on the surface of PDMS stamp. (e and f) CF particles loaded into microwell array of PLA films on PDMS stamp. (g and h) Microcages embedded inside microwell array after detaching the flat PDMS. (i and j) Transferred PLA microcages on gelatin. (k and l) Printed PLA microcages after dispersing and washing steps. | ||
Generally, a rubber stamp made of PDMS with microwell array surface structure were necessary for printing biodegradable microcages. The stamp is prepared via double casting and transfer procedures based on a pre-designed PMMA template with microwell array structures on the surface. Fig. 2a and b show the morphology of as-prepared patterned PDMS stamp at different magnifications, highlighting the periodic arrangement of identical microwells on the PDMS surface (specifications can be found in Fig. S1†). The microcage printing process begins by dipping the microwell-patterned PDMS stamp, as well as a flat PDMS, into a PLA polymer solution for 5 seconds. Both the PDMS stamp and flat PDMS are gently and slowly withdrawn and then dried in ambient conditions. This leaves a thin PLA film on the surface after the organic solvent evaporate. Fig. 2c and d shows the surface structure of the precoated PDMS stamp. The PLA thin film uniformly coats the PDMS surface, including the walls and microwells, creating a large number of periodic, shallow microwells where drugs can be loaded. It is worth to note that though PDMS can undergo anisotropic swelling when exposed to chloroform, in the proposed dip-coating process, the contact time between the PDMS stamp and the chloroform solution was less than 10 seconds. Under these conditions, no observable swelling or deformation of the microwell structures occurred.
Here, a small and hydrophilic fluorescent dye CF was selected as the model drug to be encapsulated into these microwell areas. CF has been widely accepted as a model drug for delivery systems due to its physicochemical properties including the small molecular weight, the good water solubility as well as the stable fluorescent signal, which offers the possibility to trace the location of labelled sample and analyse the release behaviour of drug delivery vehicles.49 Although the CF model drug could be loaded by the conventional solution evaporation method,50 the loading efficiency was insufficient because of the small microwell space. Additionally, the non-spherical shape of the drug crystals complicates accurate positioning within the microwells.51 Thus, simpler and efficient dry loading method designed for this special microwell array structure was applied, where drug crystals can be physically trapped into microwells without considering the drug solubility. In this approach, CF particles were directly spread over the surface of PLA film precoated PDMS stamp. However, in order to be well accommodated into microwells, the large CF particles were required to be milled and ground into submicron sized crystals. Considering the small molecular weight and hydrophilic properties of CF, the release rate is unlikely to be significantly impacted by the grinding process, as CF particles would dissolve very fast upon contact with water. After the spreading of the crushed CF particles, the surface of the precoated PDMS stamp was gently swiped with fuzz-free tissue to remove excess CF particles. Only those submicron-sized CF particles trapped in the microwells are then left on the surface of precoated PDMS stamp. As demonstrated in Fig. 2e and f, the submicron-sized CF particles were successfully loaded into the periodic microwell array structure. Particles were found exclusively in the microwell regions, with none present on the inter-microwell wall surfaces. Although the number of CF particles loaded into each microwell varied, the cumulative amount of CF loaded across a large population of microwells remained similar.
Following drug loading, the precoated PDMS stamp was covered with a previously dip-coated flat PDMS, forming a sandwich-like structure. The entire assembly was then heated and pressed at 140 °C for 1 minute. The purpose of this step was to stamp and separate the melted polymer films into individual microcages between the PDMS stamp and the flat PDMS. The chosen heating temperature of 140 °C is slightly above the melting temperature of PLA (as shown in the DSC result in Fig. S6†). This choice facilitates the separation of PLA polymer films by the walls of the microwells on the PDMS stamp. Next, when the whole system cooled down to room temperature at ambient conditions, the flat PDMS was lifted off, leaving PLA microcages embedded in microwell array structures on the surface of PDMS stamp, whose surface morphology were displayed in Fig. 2g and h. Apparently, all the identical and uniform drug loaded PLA microcages were perfectly embedded and well-sealed with a smooth surface, occupying the positions that used to be microwells. It is noteworthy that the top of the PLA microcages was lower than the PDMS stamp surface plane owing to the elasticity of PDMS polymer, as the walls of the microwells would bend while pressing and heating the sandwiched structure.
Fig. S7† displayed the morphology of the polymer films of the sandwiched structure after being heated and pressed at various temperatures, and subsequently transferred onto a gelatin-coated glass slide. When the pressing temperature was lower than the polymer melting temperature of 140 °C (Fig. S7a†), the thin films could not be separated into microcages. Instead, PLA microchamber array films would be transferred onto the gelatin-coated glass slide. Conversely, the microcage printing procedures could still succeed with the morphology of the transferred microcages similar to those printed at the melting temperature (Fig. S7b†) if the heated temperature was slightly higher than the melting temperature at 160 °C (Fig. S7c†). However, when the sandwiched structure was heated and pressed at a higher temperature of 180 °C, as shown in Fig. S7d,† some of the melted PLA adhered to the flat PDMS and did not maintain a capsule shape after cooling. Consequently, only some of the microcages were embedded in the PDMS stamp's microwells and transferred onto the glass slide by gelatin.
Once the drug-loaded PLA microcages were prepared, they were stuck inside the microwells and could not be directly dispersed in DI water by regular flushing or rinsing due to the hydrophobic nature of both PLA and PDMS. To detach the microcages from the PDMS stamp, gelatin was used. A 10% gelatin DI water solution was spread over a glass slide, onto which the microcage-embedded PDMS stamp was placed. This combined structure was frozen at −20 °C for 10 minutes to solidify the gelatin. Lifting the PDMS stamp while the gelatin was frozen allowed the PLA microcages to adhere to the surface of the gelatin and thus the cages are transferred onto the glass slide. This technique allows the PDMS stamp to be reused multiple times after simple cleaning steps, showcasing one of the remarkable advantages of this microcage printing method.
Fig. 2i and j illustrated the surface morphology of PLA microcages after transfer by gelatin on a glass slide. It is worth to note that all the PLA microcage were periodically arranged on the surface of solidified gelatin right after detaching the PDMS stamp, while the prepared SEM sample were microcages fixed in melted and further dried solidified gelatin. Although Fig. 2i and j may not precisely reflect the exact position and array order of microcages on frozen gelatin, it does indicate that the evenly coated PLA film on the PDMS stamp was separated by the walls of the microwells after the heating and pressing process, leading to the formation of individual drug-loaded PLA microcages as anticipated. Finally, to create a water suspension of printed PLA microcages, the microcage-fixed, gelatin-coated glass slide was placed into a Petri dish. The slide was rinsed with 37 °C DI water to dissolve the gelatin and disperse the PLA microcages, followed by transferring into 2 ml centrifuge tube and DI water three times washing to remove the dissolved gelatin.
The final PLA microcage morphology after washing and drying steps are presented in Fig. 2k and l, with all microcages identical and uniform both in shape and size determined by the applied template without any aggregation. The specification of printed microcages was detailed in Table S1.† The length of long side, short side and height were about 12 μm, 9 μm and 7 μm, respectively. It should be noted that the final dimensions of the microcages are slightly different from the original microwell dimensions because they were pressed and embedded within microwells with smaller size owing to the elasticity of PDMS polymer, as shown in Fig. 2g and h. It is worth to note that the uniform size and shape of the microcages allow for smooth suspension in aqueous media and facilitate delivery through fine-gauge needles without clogging, thus reducing the risk of injection-related discomfort or blockages. Additionally, it is evident that a small “tail” was attached at the edge of the individual PLA microcage at a higher magnification view. The “tail” came from the crossing ridge part of the walls on PDMS stamp after heating and pressing, caused by the distortion of the PDMS while pressing.
CLSM was employed to observe and confirm the encapsulation of CF model drug particles in PLA microcages, with the results shown in Fig. 3. To differentiate the components, the shell polymer PLA was labelled with a red fluorescent dye, Nile red, while the encapsulated CF particles emitted their own green fluorescent signal. Bright field channel CLSM images of each sample were also captured, providing another perspective to confirm the encapsulation of the model drug particles.
 | ||
| Fig. 3 CLSM images of PLA microcages confirm the encapsulation of CF model drug. (a–d) CF loaded PLA microcages printed and embedded inside the microwell array of patterned PDMS stamp. (e–h) CF loaded printed PLA microcages dispersed in DI water. (i–l) Higher magnification of (e–h) focused on a single PLA microcage. PLA is labelled with red fluorescent dye Nile Red and the green color represents the encapsulated CF. | ||
CLSM was first applied at the preparation stage when CF loaded PLA microcages were printed and embedded inside the microwell array of patterned PDMS stamp after detaching the flat PDMS, whose results were demonstrated in Fig. 3a–d. The red channel (Fig. 3a) showed the empty space inside PLA polymers where located the CF particles exhibited in Fig. 3b, which was confirmed in the overlayed channel. Besides, it is clearly displayed in the bright field channel (Fig. 3c) that dye particles were fixed inside the area of microwells without settling on walls in between. In addition, the CF loaded printed PLA microcages dispersion was also characterized via CLSM (Fig. 3e–h), and confirmed the sealing of printed microcages. It is worth mentioning that these CF loaded PLA microcages had been incubated in DI water overnight and washed three times to ensure the non-encapsulated hydrophilic CF particles had been dissolved and removed. All the microcages were imaged while floating and suspending in solution. In general, Fig. 3e showed that every PLA microcage had a lumen occupied by CF particles (Fig. 3f) as confirmed in the red–green overlayed channel of Fig. 3g. Especially, Fig. 3h evidently demonstrated that the CF model drug crystals, displayed as dark dots, were located at the position of each microcage, indicating the robust sealing and absence of leakage in the printed PLA microcages. Moreover, one of the side-position-up microcages in Fig. 3e–h was magnified and focused as Fig. 3i–l presented. The red channel (Fig. 3i) showed that the empty area was located in the middle of the microcage structure where also the green particles (Fig. 3j) stayed. In addition, the overlayed channel (Fig. 3k) proved that the encapsulated CF particles emerged only inside the core of PLA microcages instead of appearing on the surface of the polymer shell or randomly distributed in the bulk polymer. However, the fluorescent channels were only a thin cross-section of the microcage, while the bright field channel (Fig. 3l) further revealed that a significant number of model drug particles can be encapsulated in each individual microcage, as indicated by the dark and dense area.
3.2. Release kinetics of microcages made of PLA and PCL blends
Drawing inspiration from previous work that demonstrated the capability of hydrophobic polymer PLA to assist PCL in better retaining hydrophilic small molecules for enzymatic drug release in the microfilms,45 this study further developed previous finding by the novel hot press printing techniques that separate flat microfilm into microcages to expand its applicability. Thus, this work investigates the passive release kinetics of novel printed microcages made of different blends of PLA and PCL. The biodegradable and biocompatible polymer PCL was chosen to blend with PLA due to its wide adoption and its higher water permeability compared to PLA.52The preparation of PLA–PCL blended polymer solutions was straightforward, given that both PLA and PCL readily dissolve in the organic solvent chloroform. By varying the blend ratios, the chosen polymer solutions for microcage printing were obtained at PLA:PCL ratios of 9:1, 8:2, and 7:3. Although it was technically possible to increase the PCL content beyond these ratios, it was found that achieving smooth and homogeneous dip-coated films on the PDMS stamp became challenging when the PCL component exceeded 40%. Here, CF was encapsulated into printed microcages in order to examine the release kinetics of microcages made with different polymer blends. Fig. 4a–h showed the SEM images of printed microcages made of PLA and PLA–PCL blended polymers at different ratios.
 | ||
| Fig. 4 The SEM images (a–h), DSC curves (i) and release kinetics (n = 3) (j) of printed microcages made of PLA and PLA–PCL blended polymers at different ratios. | ||
The printed microcages made of different polymers were nearly identical both in size and shape. Upon closer examination, the microcages composed of pure PLA displayed a smooth and complete surface as Fig. 4a and b showed, while for the microcages comprising 10% of PCL (Fig. 4c and d), minor bumps emerged due to the phase separation between PLA and PCL, but the surface remained flat and intact. When the PCL content reached 20% (Fig. 4e and f), the surface morphology of microcage resembled the PLA:PCL = 9:1 one, with complete surface and small pores. However, the microcages made of 70% PLA and 30% PCL exhibited a somewhat uneven surface (Fig. 4g and h), though still complete, indicating that the PCL content had melted and flowed on the surface due to phase separation. Generally, the SEM results suggested that identical microcages made of various polymers were successfully prepared, and even though the detailed surface morphology differed, the model drug CF particles were still entirely encapsulated within the microcages without direct exposure to the surface.
This phase separation phenomenon aligns with previous reports on PLA–PCL blends, where such phase separation was shown to influence both microstructure and drug release behaviour. In earlier work investigating a microchamber array film composed of a PLA–PCL blend (2:1 ratio), PCL domains were dispersed throughout the matrix and formed pores following enzymatic degradation, enabling sustained drug release for 26 hours. As the PLA content increased above 2:1 ratio, the release duration was significantly extended, reaching more than 28 days and suggesting that higher PLA composition enhances structural integrity. These findings are consistent with the morphological features observed from SEM, where blend composition influences the surface texture and likely plays a role in controlling drug release kinetics.
One of the vital requirements for successful PLA and PCL blended microcages was controlling the heating and pressing temperature during the printing process. Thus, the thermal behavior of the prepared PLA and PCL blended polymer, as key parameters for microcage printing, were evaluated and compared with pure PLA and PCL via DSC. After the thermal pre-treatment in the first heating cycle, the thermal properties of all these polymers were obtained (Fig. 4i). In general, the PLA–PCL blended polymers had a similar melting temperature as PLA. PCL showed a unique curve that has a melting point at around 55 °C without any heat flow after the melting point. In contrast, pure PLA demonstrated the melting point at around 135 °C while no thermal effect at 55 °C. With higher blending composition of PCL, the PLA–PCL blended polymer tended to have higher melting temperature for PLA content but lower melting point for PCL content. Base on the DSC curves, a heating temperature at around 140 °C with excess pressure would be suitable for all the polymer blends. Thus, it is feasible to apply the same printing procedures and conditions as preparing PLA microcages for synthesizing PLA–PCL microcages after simply replacing the polymer solution from pure PLA to PLA–PCL blended polymer solutions.
After confirming the preparation feasibility of printed biodegradable microcages as well as the good encapsulation of model drug CF particles, the release kinetics of the microcages were further studied. Fig. 4j demonstrated the release behaviours of CF encapsulated microcages made of biodegradable polymers over a period of 28 days, and the corresponding percentage release curves were shown in Fig. S8.† Generally, as expected, the printed microcages made of polymers with a higher PCL component tended to release the encapsulated CF particles quicker. Based on the dye release curve in Fig. 4j, the CF particle encapsulation amount was calculated as approximately 28 μg per million microcages, indicating that, on average, 28 pg of CF particles were loaded into individual microcages. The calculated weight of individual microcage is about 950 pg (Table S1†), thus the drug loading capacity is about 3.0 ± 1.1%. This low standard deviation suggests that, at the macroscale, the total amount of drug loaded into the microcage remained similar, despite variability of drug loading at the individual microwell level.
There are some reasons that result in this low drug loading capacity. First, the size and shape of the template has a huge impact of the loading capacity. During the dip-coating process, the majority of the microwell space was occupied by the polymer (Fig. 2c and d) due to polymer liquid entrapment, leaving little space for drugs. Adjusting the polymer solution concentration could reduce the influence, but only a narrow concentration range (1.75%–2.25%) could be used for dip-coating. Specifically, a minimum concentration is required to maintain complete and smooth film formation for optimal drug sealing while a maximum concentration is required to keep empty space within microwells. Although this is not a high loading capacity, the loading potential of this microcage preparation technique is huge and the novelty of preparing uniform microcages via printing is not compromised. For example, a larger size or a shallower incline of microwell structure template can significantly reduce the polymer volume portion in the microwell. Thus the advantage of the special loading method of this technique can be maximized.
During the first 24 hours of incubation, a quick release of encapsulated CF was observed in all groups, which could be caused by multiple factors. On the one hand, the poorly sealed CF from some flawed microcages may not be fully washed out, even after all the samples of the four groups had been incubated at 37 °C for 1 hour in water and three washing steps. On the other hand, the intensive incubation solution replacement would accelerate the release rate as well. While for the microcage incubation group of PLA–PCL = 7:3, the quick release was, to a large extent, contributed by the permeability of the designed polymer shell itself. Moreover, the release rate of this group was significantly higher than the other three groups with almost 80% encapsulated dye particles diffused out.
Following the initial day's release, the release curves of all four groups gradually increased steadily since the dye release rate tended to be governed by the permeability of the microcage shell constitution. However, the PLA:PCL = 7:3 group was an exception as the decrease in release rate resulted from an inadequate amount of encapsulated CF particles, i.e., the drug diffusion rate was unsaturated. Essentially, from day 1 till day 7, samples from all four groups continued releasing cargos, albeit at different speeds. The PLA:PCL = 8:2 group released around 22% of the overall encapsulated amount, while the PLA:PCL = 7:3, PLA:PCL = 9:1, and pure PLA groups had 11.15%, 9.64%, and 3.55% of the loaded dye released within 6 days, respectively.
From day 7 to day 28, the release curve of all samples became almost linear as the dye release rate became nearly constant, indicating a predominant influence from the diffusion activity and less effect from flawed microcages and manual operation inaccuracies. The release curve of the PLA:PCL = 7:3 group during this period was almost level, with 98% of CF released by day 28, as most of the loaded cargos had been released in the first week due to the highest polymer permeability. Therefore, the PLA:PCL = 8:2 group, with the second-highest PCL content, released CF particles at the highest rate, with 35.8% of encapsulated drugs released within 3 weeks. However, as incubation time increased, the release rate slightly declined as the encapsulated crystal amount became less. Additionally, when the blending ratio of PLA to PCL was 9:1, the CF release rate became moderate, with 17.8% released from day 7 to day 28, and a total of about 43% released after 4 weeks of incubation. The slowest release increment of the pure PLA group denoted the lowest diffusion rate of CF model drug as expected, with only 9% of particles released during the 3 weeks and overall 30.46% following a 4-week period.
Above observations demonstrated the influence of the PCL proportion in the PLA–PCL blend on the release rate of encapsulated hydrophilic molecules, which is a critical factor in the design of controlled drug delivery systems.
4. Conclusions
In this study we reported on a successful fabrication and application of biodegradable polymer-based microcages as competent drug delivery vehicles. The microcages were able to efficiently encapsulate small and hydrophilic cargos and demonstrate a controlled release rate by varying the blend ratios of PLA and PCL. The size and shape of the microcages, governed by lithographed templates and microarray structures, can be pre-determined, allowing for broad and diverse customization. The drug loading capacity has huge potential to be increased by adjusting the template microwell structure. The novelty of this approach arises from two major advancements. First, individual uniform microcages can be reliably fabricated at scale, developing the scope of microprinting technique beyond microfilms and microparticles. Second, a unique drug-loading technique is introduced that remains compatible with this microcage printing method and does not need to consider drug solubility.It is noteworthy to address that the cargo we choose in this work is just an example, the microcages production methods introduced is an versatile technique with a wide range of pharmaceutical compounds or formulations can be encapsulated and sustained released, such as antimicrobial agents53 or antibiotics.54 In addition, the appearance of the microcages prepared in this work is also a demonstration, as the size and the shape of printed microcages are determined not only by various predesigned lithographed template, but also by the casted microarray structure on the surface of PDMS stamp with the resolution limit in the range of 100–200 nm.55 Larger microarray structures are expected to produce microcages with increased drug loading capacity and higher drug-to-polymer ratios. To enable broader therapeutic applications, especially the treatments that requiring higher doses, further optimization of microwell geometry and drug loading strategies will be essential. Besides, high temperature used during the fabrication process limits the applicability of this technology for heat-sensitive drugs. To broaden the applicability of this platform, alternative fabrication strategies that either eliminate or significantly reduce thermal exposure should be further explored, enabling the safe and effective encapsulation of heat-sensitive drugs Moreover, this versatility extends its applicability across diverse fields, including lab-on-chip,56 sensing,57 electronics,58 optics,59 and so on.
It is anticipated that this uniform microcage printing technique will have industrially relevant benefits in reproducibility, scalability, and consistency. When prepared manually in the laboratory, approximately 90% yield of intact microcages across the template area can be achieved. With further process optimization and automation, it is expected that the fabrication could produce nearly 100% yield at scale. The most advantageous route will be injectable suspensions, particularly for localized and targeted drug delivery.60,61 Future research should focus on optimizing the fabrication parameters further and validating this microcage drug delivery system in more complex biological settings. This can pave a novel way for substantial impacts in the field of controlled drug delivery and extend the adaptability of the method to encapsulate a wide range of pharmaceutical compounds.
Author contributions
Jiaxin Zhang: Conceptualization, methodology, investigation, formal analysis, writing – original draft. Rui Sun: Investigation, visualization, methodology, writing – original draft. Valeriya Kudryavtseva: Methodology, investigation. David J. Gould: Methodology, writing – review & editing. Gleb B. Sukhorukov: Supervision, resources, project administration, funding acquisition, writing – review & editing.Data availability
All relevant data are within the paper and the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by China Scholarship Council no. 201706630010 (J. Z.). We would like to express our gratitude to Dr Maxim Kiryukhin for generously providing the master microwell array template.References
- M. Lengyel, N. Kállai-Szabó, V. Antal, A. J. Laki and I. Antal, Sci. Pharm., 2019, 87, 20 CrossRef CAS
.
- X. Xie, W. Zhang, A. Abbaspourrad, J. Ahn, A. Bader, S. Bose, A. Vegas, J. Lin, J. Tao, T. Hang, H. Lee, N. Iverson, G. Bisker, L. Li, M. S. Strano, D. A. Weitz and D. G. Anderson, Nano Lett., 2017, 17, 2015–2020 CrossRef CAS PubMed
- J. T. Hou, W. X. Ren, K. Li, J. Seo, A. Sharma, X. Q. Yu and J. S. Kim, Chem. Soc. Rev., 2017, 46, 2076–2090 RSC
- A. R. Longstreet and D. T. McQuade, Acc. Chem. Res., 2013, 46, 327–338 CrossRef CAS PubMed
- J. Song and H. Chen, Flavour Fragrance J., 2018, 33, 160–165 CrossRef CAS
- X. Yu, Z. Zhao, W. Nie, R. Deng, S. Liu, R. Liang, J. Zhu and X. Ji, Langmuir, 2011, 27, 10265–10273 CrossRef CAS PubMed
- H. Bysell, R. Månsson, P. Hansson and M. Malmsten, Adv. Drug Delivery Rev., 2011, 63, 1172–1185 CrossRef CAS PubMed
- Y. Ding, W. Li, F. Zhang, Z. Liu, N. Zanjanizadeh Ezazi, D. Liu and H. A. Santos, Adv. Funct. Mater., 2019, 29, 1–35 Search PubMed
- S. Bin Park, E. Lih, K. S. Park, Y. K. Joung and D. K. Han, Prog. Polym. Sci., 2017, 68, 77–105 CrossRef
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS
- R. Georgieva, S. Moya, M. Hin, R. Mitlöhner, E. Donath, H. Kiesewetter, H. Möhwald and H. Bäumler, Biomacromolecules, 2002, 3, 517–524 CrossRef CAS PubMed
- D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed
- A. Yaqoob Khan, S. Talegaonkar, Z. Iqbal, F. Jalees Ahmed and R. Krishan Khar, Curr. Drug Delivery, 2006, 3, 429–443 CrossRef PubMed
- B. K. Lee, Y. Yun and K. Park, Adv. Drug Delivery Rev., 2016, 107, 176–191 CrossRef CAS PubMed
- B. B. C. Youan, T. L. Jackson, L. Dickens, C. Hernandez and G. Owusu-Ababio, J. Controlled Release, 2001, 76, 313–326 CrossRef CAS PubMed
- M. F. Thorne, F. Simkovic and A. G. Slater, Sci. Rep., 2019, 9, 1–7 CrossRef CAS PubMed
- A. R. Pohlmann, F. N. Fonseca, K. Paese, C. B. Detoni, K. Coradini, R. C. Beck and S. S. Guterres, Expert Opin. Drug Delivery, 2013, 10, 623–638 CrossRef CAS PubMed
- O. Seok Kwon, J. Jang and J. Bae, Curr. Org. Chem., 2013, 17, 3–13 CrossRef
- G. M. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS PubMed
- T. Y. Lee, M. Ku, B. Kim, S. Lee, J. Yang and S. H. Kim, Small, 2017, 13, 1–11 Search PubMed
- S. Lignel, A. V. Salsac, A. Drelich, E. Leclerc and I. Pezron, Colloids Surf., A, 2017, 531, 164–172 CrossRef CAS
- R. K. Shah, H. C. Shum, A. C. Rowat, D. Lee, J. J. Agresti, A. S. Utada, L. Y. Chu, J. W. Kim, A. Fernandez-Nieves, C. J. Martinez and D. A. Weitz, Mater. Today, 2008, 11, 18–27 CrossRef CAS
- J. T. Wang, J. Wang and J. J. Han, Small, 2011, 7, 1728–1754 CrossRef CAS PubMed
- V. Morillon, F. Debeaufort, M. Capelle, G. Blond and A. Voilley, J. Agric. Food Chem., 2000, 48, 11–16 CrossRef CAS PubMed
- A. Pimpin and W. Srituravanich, Eng. J., 2012, 16, 37–55 CrossRef
- D. A. Canelas, K. P. Herlihy and J. M. DeSimone, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 391–404 CAS
- G. Acharya, C. S. Shin, K. Vedantham, M. McDermott, T. Rish, K. Hansen, Y. Fu and K. Park, J. Controlled Release, 2010, 146, 201–206 CrossRef CAS PubMed
- J. J. Kim, K. W. Bong, E. Reátegui, D. Irimia and P. S. Doyle, Nat. Mater., 2017, 16, 139–146 CrossRef CAS PubMed
- Y. Lu, M. Sturek and K. Park, Int. J. Pharm., 2014, 461, 258–269 CrossRef CAS PubMed
- S. Qiu, J. Ji, W. Sun, J. Pei, J. He, Y. Li, J. J. Li and G. Wang, Smart Mater. Med., 2021, 2, 65–73 CrossRef
- A. Perl, D. N. Reinhoudt and J. Huskens, Adv. Mater., 2009, 21, 2257–2268 CrossRef CAS
- C. Sakib-Uz-Zaman and M. A. H. Khondoker, in 1st International Conference on Industrial, Manufacturing, and Process Engineering (ICIMP-2024), MDPI, Basel Switzerland, 2024, p. 42.
- J. Zhang, J. E. Read, G. Mittal, R. N. Poston, J. Reilly, G. Howling, B. Golland, G. B. Sukhorukov and D. Gould, J. Controlled Release, 2025, 381, 113590 CrossRef CAS PubMed
- H. Y. Kweon, M. K. Yoo, I. K. Park, T. H. Kim, H. C. Lee, H. S. Lee, J. S. Oh, T. Akaike and C. S. Cho, Biomaterials, 2003, 24, 801–808 CrossRef CAS PubMed
- J. M. Anderson and M. S. Shive, Adv. Drug Delivery Rev., 2012, 64, 72–82 CrossRef
- M. J. K. Chee, J. Ismail, H. W. Kammer and C. Kummerloöwe, Polymer, 2001, 43, 1235–1239 CrossRef
- A. M. Torres-Huerta, D. Palma-Ramírez, M. A. Domínguez-Crespo, D. Del Angel-López and D. De La Fuente, Eur. Polym. J., 2014, 61, 285–299 CrossRef CAS
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed
- D. W. Hutmacher, T. Schantz, I. Zein, K. W. Ng, S. H. Teoh and K. C. Tan, J. Biomed. Mater. Res., 2001, 55, 203–216 CrossRef CAS PubMed
- M. Herrero-Herrero, J. A. Gómez-Tejedor and A. Vallés-Lluch, Eur. Polym. J., 2018, 99, 445–455 CrossRef CAS
- S. H. Chang, H. J. Lee, S. Park, Y. Kim and B. Jeong, Biomacromolecules, 2018, 19, 2302–2307 CrossRef CAS PubMed
- B. Tyler, D. Gullotti, A. Mangraviti, T. Utsuki and H. Brem, Adv. Drug Delivery Rev., 2016, 107, 163–175 CrossRef CAS PubMed
- A. Grémare, V. Guduric, R. Bareille, V. Heroguez, S. Latour, N. L'heureux, J. C. Fricain, S. Catros and D. Le Nihouannen, J. Biomed. Mater. Res., Part A, 2018, 106, 887–894 CrossRef PubMed
- M. K. Ahmed, S. F. Mansour, R. Al-Wafi, M. Afifi and V. Uskoković, Int. J. Pharm., 2020, 577, 118950 CrossRef CAS PubMed
- J. Zhang, R. Sun, A. O. Desouza-Edwards, J. Frueh and G. B. Sukhorukov, Soft Matter, 2020, 16, 2266–2275 RSC
- M. Gai, J. Frueh, T. Tao, A. V. Petrov, V. V. Petrov, E. V. Shesterikov, S. I. Tverdokhlebov and G. B. Sukhorukov, Nanoscale, 2017, 9, 7063–7070 RSC
- A. S. Sergeeva, D. A. Gorin and D. V. Volodkin, Bionanoscience, 2014, 4, 1–14 CrossRef
- V. Kudryavtseva, S. Boi, J. Read, R. Guillemet, J. Zhang, A. Udalov, E. Shesterikov, S. Tverdokhlebov, L. Pastorino, D. J. Gould and G. B. Sukhorukov, ACS Appl. Mater. Interfaces, 2021, 13, 2371–2381 CrossRef CAS PubMed
- H. Bi, S. Ma, Q. Li and X. Han, J. Mater. Chem. B, 2016, 4, 3269–3277 RSC
- M. Gai, J. Frueh, V. L. Kudryavtseva, A. M. Yashchenok and G. B. Sukhorukov, ACS Appl. Mater. Interfaces, 2017, 9, 16536–16545 CrossRef CAS PubMed
- K. Koh, H. Hwang, C. Park, J. Y. Lee, T. Y. Jeon, S. H. Kim, J. K. Kim and U. Jeong, ACS Appl. Mater. Interfaces, 2016, 8, 28149–28158 CrossRef CAS PubMed
- R. Shogren, J. Environ. Polym. Degrad., 1997, 5, 91–95 CrossRef CAS
- R. Sun, J. Zhang, R. A. Whiley, G. B. Sukhorukov and M. J. Cattell, Pharmaceutics, 2021, 13, 1–16 CrossRef PubMed
- K. Saha, K. Dutta, A. Basu, A. Adhikari, D. Chattopadhyay and P. Sarkar, Nano-Struct. Nano-Objects, 2020, 21, 100418 CrossRef CAS
- G. Acharya, C. S. Shin, M. McDermott, H. Mishra, H. Park, I. C. Kwon and K. Park, J. Controlled Release, 2010, 141, 314–319 CrossRef CAS PubMed
- X. Chen, D. F. Cui, C. C. Liu and H. Li, Sens. Actuators, B, 2008, 130, 216–221 CrossRef CAS
- J. Zhang, M. Gai, A. V. Ignatov, S. A. Dyakov, J. Wang, N. A. Gippius, J. Frueh and G. B. Sukhorukov, ACS Appl. Mater. Interfaces, 2020, 12, 19080–19091 CrossRef CAS PubMed
- R. Prehn, L. Abad, D. Sánchez-Molas, M. Duch, N. Sabaté, F. J. Del Campo, F. X. Muñoz and R. G. Compton, J. Electroanal. Chem., 2011, 662, 361–370 CrossRef CAS
- S. Reitzenstein and A. Forchel, J. Phys. D: Appl. Phys., 2010, 43, 033001 CrossRef
- R. Xue, H. Wu, S. Li, N. Pu, D. Wei, N. Zhao, Y. Cui, H. Li, Z. Song and Y. Tao, Mater. Today Bio, 2024, 27, 101126 CrossRef CAS PubMed
- P. Yadav, Y. Singh, D. Chauhan, P. K. Yadav, A. S. Kedar, A. K. Tiwari, A. A. Shah, J. R. Gayen and M. K. Chourasia, Expert Opin. Drug Delivery, 2024, 21, 639–662 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5bm00154d |
| This journal is © The Royal Society of Chemistry 2025 |