
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTunable hydrogel networks by varying secondary structures of hydrophilic peptoids provide viable 3D cell culture platforms for hMSCs†
Aldaly
Pineda-Hernandez‡
 ,
David A.
Castilla-Casadiego‡§
,
Logan D.
Morton‡¶
,
Sebastian A.
Giordano-Nguyen
,
Kathleen N.
Halwachs
and
Adrianne M.
Rosales
*
,
David A.
Castilla-Casadiego‡§
,
Logan D.
Morton‡¶
,
Sebastian A.
Giordano-Nguyen
,
Kathleen N.
Halwachs
and
Adrianne M.
Rosales
*
Mcketta Department of Chemical Engineering, University of Texas at Austin, Austin, TX 78712, USA. E-mail: arosales@che.utexas.edu
First published on 7th May 2025
Abstract
Hydrogels have excellent ability to mimic the extracellular matrix (ECM) during 3D cell culture, yet it remains difficult to tune their mechanical properties without also changing network connectivity. Previously, we developed 2D culture platforms based on tunable hydrogels crosslinked by peptoids with various secondary structures: helical, non-helical, and unstructured, which allowed control over hydrogel mechanics independent of network connectivity. Here, we extend our strategy to 3D matrices by modifying the peptoids with piperazine and homopiperazine residues to enhance water solubility without altering their secondary structure. Hydrogels crosslinked with helical peptoids exhibited significantly higher stiffness compared to hydrogels crosslinked with non-helical or unstructured peptoids. Human mesenchymal stem cells (hMSCs) encapsulated within these hydrogels were assessed for viability, proliferation, and immunomodulatory potential. The stiffest hydrogels promoted the highest rates of proliferation and increased yes-associated protein (YAP) nuclear localization. Softer hydrogels, however, showed enhanced production of indoleamine 2,3-dioxygenase (IDO), both with and without interferon gamma (IFN-γ) stimulation, highlighting their potential in immunomodulatory applications. The biomimetic platform developed here enables the study of how matrix mechanics influence stem cell behavior without confounding factors from network connectivity, leading to insights for hMSC-mediated immunomodulation.
1. Introduction
Hydrogels, polymeric networks capable of retaining large amounts of water, have emerged as essential platforms for biomedical applications, particularly in the fields of tissue engineering and regenerative medicine.1,2 Their biocompatibility, high water content, and tunable mechanical properties make them highly suitable for mimicking the extracellular matrix (ECM) in 3D cell culture environments.2,3 3D cell culture offers several advantages over traditional 2D systems, particularly in replicating the complex in vivo environment of tissues.4 In 2D cultures, cells are grown on flat surfaces, which limits their interactions to a single plane, resulting in unnatural morphology and behavior.5,6 In contrast, 3D cell culture allows cells to grow in all directions, closely mimicking the native ECM, which promotes more accurate cell-to-cell and cell-to-ECM interactions.7 This more realistic environment enhances the expression of genes, proteins, and cellular behaviors relevant to tissue development, differentiation, and disease progression.8,9 Furthermore, the ability to modulate the mechanical properties of 3D hydrogels is critical, as mechanics is closely linked to cell adhesion, proliferation, differentiation, and secretion of soluble factors.10–14Traditional methods for tuning the mechanical properties of hydrogels rely on altering the polymer concentration or the number of crosslinks within the network, which directly impacts the hydrogel stiffness and viscoelastic properties.15 While effective, these approaches couple hydrogel stiffness to other factors that also depend on polymer concentration and network connectivity, such as porosity and permeability.16–19 This strategy also leads to biophysical properties that are different from the native ECM, which is composed of structured biopolymers that exhibit high mechanical stiffness even at low polymer content, offering a delicate balance between structural integrity and transport of soluble factors.20–23
In recent years, synthetic hydrogel systems have been developed to mimic the hierarchical organization of natural ECM, introducing new strategies to control the mechanical properties of the hydrogel while preserving the network's structural features,15,24–29 but there remains a lack of synthetic polymers with defined secondary structures. Sequence-defined synthetic polymers, such as peptoids (N-substituted glycines), offer a promising approach to address this challenge. Peptoids are typically synthesized using a submonomer method that uses primary amines to incorporate side chains, enabling a large chemical diversity and precise control over secondary structure.30–33 Unlike most traditional synthetic polymers, peptoids can be designed to adopt specific conformations—such as helices,32,34,35 fibers,36 sheets,37 or random coils—that influence their molecular rigidity.30
This work employs a synthetic hydrogel system utilizing peptoid-based crosslinkers with varying secondary structures to systematically tune bulk hydrogel mechanics independent of network connectivity. Previous data indicated that peptoid helices have longer persistence lengths (a proxy for molecular rigidity) than non-helical peptoids.30 In our previous investigations, we developed a system for 2D cell culture that leveraged the rigidity of helical peptoid crosslinkers to increase bulk hydrogel stiffness compared to hydrogels with non-helical crosslinkers.24,25,38 However, because peptoid helicity arises from bulky chiral side chains (often aromatic),34,35 these peptoid crosslinkers were not inherently water soluble, and therefore are not appropriate for 3D cell culture applications. Toward this end, we incorporate piperazine and homopiperazine residues into the peptoid sequence, thereby enhancing their hydrophilicity and water solubility, and facilitating hydrogel formation in the presence of human mesenchymal stem cells (hMSCs) for 3D encapsulation.
Herein, we explore the impact of three distinct peptoid crosslinkers—helical, non-helical, and unstructured—on the mechanical properties of the resulting hydrogels, which are then evaluated for their capacity to support hMSC immunomodulatory activity. Secretion of immunomodulatory factors by hMSCs has been shown to depend on ECM properties, especially 3D geometries and matrix mechanics.39–43 We hypothesized that the mechanical properties of our hydrogels, determined by the relative rigidity of the peptoid crosslinkers, would regulate encapsulated hMSC viability, proliferation, and secretion. Specifically, we expected mechanics to influence the production of indoleamine 2,3-dioxygenase (IDO), a key mediator of immune regulation,40,44 potentially offering new insights into the design of 3D immunomodulatory biomaterials. Overall, this work seeks to establish a biomimetic platform for creating tunable hydrogel systems with decoupled mechanical and structural properties, enabling the systematic study of how matrix mechanics influence hMSC behavior in 3D culture.
2. Materials and methods
2.1 Peptoid synthesis
A series of peptoids, helical, non-helical, and unstructured (Table 1), were synthesized using Rink Amide polystyrene resin (0.3–0.6 meq g−1, 100–200 mesh from Chem-Impex International, Inc.) on a Prelude X automated peptide synthesizer (Gyros Protein Technologies) at a scale of 250 μM according to previously published submonomer methods.45,46 The syntheses were conducted as follows: fresh bromoacetylation reagent was prepared by dissolving bromoacetic acid (98+%, Thermo Scientific) in dimethylformamide (DMF) (99.8%, Fisher Chemical) at 1.2 M. Bromoacetic acid and N,N′-diisopropylcarbodiimide (DIC) (99.5%, Chem-Impex International, Inc.) were mixed with the resin and heated for 20 minutes before draining and washing with DMF. Next, the bromine was displaced with the primary amine or secondary amine in the case of boc-piperazine and boc-homopiperazine, mixed and heated for 1 h, except the thiol containing amine that was mixed and heated for 2 h. All displacement steps utilized 2 M primary or secondary amines (Trt-Cysteamine*HCl (>99%, Iris Biotech), L-alanine-tert butyl ester*HCl (>97.5%, Chem-Impex International, Inc.), (S)-(−)-alpha-methylbenzylamine (99.52%, Chem-Impex International, Inc.), DL-alpha-methylbenzylamine (99%, Acros Organics Chemicals), methylamine, 40% w/w aq. soln. (37–43% w/w, Thermo Scientific), homopiperazine (98%, Sigma Aldrich), 1-Boc-piperazine (99.9%, Chem-Impex International, Inc.), and 1-Boc-hexahydro-1,4-diazepine (98%, Sigma Aldrich)) in N-methyl-2-pyrrolidone (NMP) (>99%, Fisher Scientific), after freebasing the HCl-containing amines. To maximize yield, the cysteamine displacement step was repeated an additional time, as it was determined that this reaction had a lower reaction efficiency.|
|
|||||
|---|---|---|---|---|---|
| Entry | Name | Total monomers | Sequence |
M
obs/Mtheor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
Purityb (%) |
| a M obs were verified via LC/MS and Mtheor were calculated using ChemDraw. b Purity (%) was determined by comparing the area of our desired product to the total chromatogram area using analytical HPLC. | |||||
| 1 | H14 | 14 | Nthe(NspeNsceNsce)4Nthe | 1930.6/1929.1 | 91 |
| 2 | H14-pip | 15 | Pip-Nthe(NspeNsceNsce)4Nthe | 2057.4/2055.3 | 95 |
| 3 | H14-hp | 15 | Homopip-Nthe(NspeNsceNsce)4Nthe | 2069.6/2069.3 | 98 |
| 4 | H14-DHP | 16 | (Homopip)2-Nthe(NspeNsceNsce)4Nthe | 2210.8/2209.5 | 93 |
| 5 | N14-DHP | 16 | (Homopip)2-Nthe(NmbNsceNsce)4Nthe | 2211.5/2209.5 | 100 |
| 6 | U14-DHP | 16 | (Homopip)2-Nthe(SarSarSar)4Nthe | 1384.8/1384.7 | 95 |

Once complete, all syntheses were cleaved from resin with 5 mL of either 90:4:1:5 trifluoroacetic acid (>95%, Fisher Scientific):DI water:triisopropylsilane (98%, Fisher Scientific):1,2-ethanedithiol (>98%, Sigma Aldrich) for the helical and non-helical peptoid or 94:1:2.5:2.5 of the same species for the unstructured peptoid for 5 hours and then the peptoid was separated from the resin by vacuum filtration. The ratios of the cocktail cleavage were optimized by doing a series of small-scale test cleavages at different cocktail ratios, and the abundance of the cleaved peptoid was determined from Mass Spectroscopy (MS) data collected from a Liquid Chromatography(LC)/MS instrument (Agilent Technologies 6125B Single Quadrupole LC/MS). Prior to purification, the peptoids were dried and re-dissolved at 10 mg mL−1 in 50:50 acetonitrile (>99.9%, Fisher Scientific):DI water with 0.1% trifluoroacetic acid. The crude peptoids were purified with a C18 column on a Dionex UltiMate 3000 Ultra-High-Performance Liquid Chromatography (UHPLC) using a 50 min gradient of acetonitrile in water (50%–100%, 10 mL min−1). After lyophilization, masses were verified via LC/MS and purity was confirmed via analytical High Performance Liquid Chromatography (HPLC) (Fig. S1 and S2†).
2.2 Peptide synthesis
Two peptide sequences were synthesized: KCGGIQQWGPCK (which was used as a peptide crosslinker control) and GCGYGRGDSPG (which was incorporated in all in vitro studies for cell adhesion). The peptide sequences KCGGIQQWGPCK and GCGYGRGDSPG were selected based on their demonstrated functional roles in hydrogel design and cell–matrix interaction.47–49 The sequence GCGYGRGDSPG incorporates the canonical RGD motif, a well-characterized integrin-binding site that promotes cell adhesion across a wide range of cell types and is widely used in hydrogels to recapitulate bioactivity in otherwise inert synthetic matrices. The flanking sequences in GCGYGRGDSPG additionally confer flexibility and enable site-specific thiol–ene conjugation via the terminal cysteine, facilitating uniform presentation within the hydrogel network. The sequence KCGGIQQWGPCK is a well-studied scramble of the enzymatically degradable KCGPQGIWGQCK derived from collagenase-degradable domains. The flanking cysteine residues once again allow for efficient crosslinking into thiol-reactive networks, ensuring integration into the hydrogel structure.Peptides were synthesized using the same resin and equipment as the peptoids. Standard Fmoc-assisted coupling methods were used at five-fold molar excess of amino acids and O-(1H-6-chlorobenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HCTU) (Chem-Impex International, Inc.) coupling reagent, and a ten-fold excess of N-methylmorpholine (NMM) (99%, Sigma Aldrich). All coupling steps were performed twice.
Once complete, syntheses were cleaved from resin with 5 mL of 92.5:2.5:2.5:2.5 trifluoroacetic acid (>95%, Fisher Scientific):DI water:triisopropylsilane (98%, Fisher Scientific):1,2-ethanedithiol (>98%, Sigma Aldrich) for 4 hours and then the peptides were separated from the resin via vacuum filtration. Prior to purification, the peptides were precipitated into diethyl ether (≥99%, Fisher Scientific), dried, and re-dissolved at 10 mg mL−1 in 20:80 acetonitrile (>99.9%, Fisher Scientific):DI water with 0.1% trifluoroacetic acid. The peptides were purified in a similar manner to the peptoids but used a 40 min gradient of acetonitrile in water (20%–100%, 10 mL min−1). After lyophilization, masses were verified via LC/MS, and purity was checked using an analytical HPLC (Fig. S3†).
2.3 Synthesis of norbornene-functionalized hyaluronic acid (NorHA)
NorHA was synthesized following a modified version of the procedure outlined by Wade et al.50 Initially, hyaluronic acid sodium salt (Na-HA, 72 kDa, Lifecore) was converted to its tetrabutylammonium salt form (HA-TBA). The HA was dissolved in DI water at 2 wt% and stirred with Dowex 50 W ion exchange resin (Sigma Aldrich) at a 5:1 resin-to-HA weight ratio for at least 5 hours. The resin was then filtered out, and the solution was adjusted to a pH of 7–7.1 using tetrabutylammonium hydroxide (Possible Missions), followed by freezing and lyophilization.
Subsequently, the HA-TBA (2 wt%) was dissolved in anhydrous dimethyl sulfoxide (DMSO) (>99%, Fisher Scientific) along with 5-norbornene-2-carboxylic acid (99%, Fisher Scientific) at a 13:1 molar ratio to the calculated HA-TBA repeat units and 4-(dimethylamino)pyridine (Fisher Scientific) at a 3.2:1 ratio to the calculated HA-TBA repeat units. The reaction occurred under argon at 45 °C. Di-tert-butyl dicarbonate (Boc2O, 99%, Fisher Scientific) was then added via syringe in a 1.7:1 ratio to the HA-TBA repeat units, and the reaction proceeded overnight. The reaction mixture was quenched with a five-fold excess of DI water and dialyzed for three days. On the third day, 1 g of NaCl per 100 mL of solution was added, and the solution was precipitated into cold (4 °C) acetone (>99%, Fisher Scientific). The precipitate was collected by centrifugation, re-dissolved in DI water, dialyzed for an additional week, then frozen and lyophilized. Proton nuclear magnetic resonance spectroscopy (1H NMR) confirmed that approximately 28–38% of HA repeat units were functionalized with norbornene (Fig. S4†).
2.4 Solubility in water
Peptoids with and without piperazine, homopiperazine, and two homopiperazine modifications were weighed out to equivalent masses of 1 mg in a microcentrifuge tube. As described by Darapaneni et al.,51 water solubility tests were carried out by the gradual addition of 5 μL distilled water repeatedly. The tubes were regularly vortexed and centrifuged until no solid was visible. This point was taken to be the solubility limit for that peptoid in water. Moreover, plate reader absorbance measurements were conducted at 254 nm, and the absorbance measurements were converted to transmittance using the traditional Beer–Lambert law for Transmittance equation: Absorbance = 2-log(%T). We chose 254 nm because this wavelength is used to detect the presence of organic matter in water with a scattering-independent absorption coefficient below 0.1 m−1 in the ultraviolet spectrum.522.5 Circular dichroism (CD)
CD spectra were acquired from 185 nm to 280 nm using a Jasco J-815 CD Spectropolarimeter at 25 °C. A total of 3 measurements were taken for each sample and averaged to reduce noise. A spectrum of pure water was subtracted from each sample spectrum to remove any baseline. Samples (200 μL) were prepared at 0.05 mM in water for each of the peptoids. A quartz cuvette with a 1 mm path length was used for all samples. Mean residue ellipticity was calculated using the following equation:| [θ] = m° × M/(10 × L × C × n) | (1) |
2.6 Hydrogel preparation
All pre-gel solutions were dissolved in 100% phosphate buffered saline (PBS) with 0.05 wt% lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP). All hydrogels were 3 wt% NorHA crosslinked at an Ellman's corrected 1:1 thiol:ene ratio with either the peptide or peptoid crosslinker. First, the Ellman's correction was determined by quantifying the free thiol content in the peptide or peptoid using an Ellman's test. Serial dilutions of the peptide or peptoid sample between 0 mM and 2 mM were exposed to a 10-fold excess of Ellman's reagent in triplicate. Absorbance was read at 412 nm, and the average absorbance per concentration was compared to a control sample, dithiothreitol (DTT). This calculation was used to determine the amount of free thiols in the synthesized peptides and peptoids (Fig. S6†) and correct the gel formulation stoichiometry to obtain a 1:1 thiol:ene ratio (Table S1†).53–55 The NorHA and peptoid precursors were mixed together after being dissolved separately. Gelation was induced via exposure to 365 nm light (10 mW cm−2, 120 s), as this time ensured that each hydrogel reached its plateau modulus measured by rheological time sweeps. For experiments with live cells, the hydrogels were functionalized with the cell adhesive peptide at 2 mM to allow for attachment, and the pre-gel solution was mixed with the appropriate concentration of cells (∼1 million cells per mL) and mixed well to ensure uniform suspension in the resulting hydrogel.
2.7 Rheometry
A Discovery HR-2 rheometer (TA Instruments) with an 8 mm flat stainless-steel geometry and a UV-transparent quartz plate were used to perform all in situ gelation mechanics measurements. The process included pipetting the precursor solution (15 μL) onto the quartz plate, which was connected via liquid filled light guide to a mercury lamp (Omnicure Series 1500) with a 365 nm filter. The solution was exposed in situ to 365 nm light (10 mW cm−2, 5 min) to induce gelation. Five minutes was chosen as it was long enough to ensure each hydrogel reached its plateau modulus. For time sweeps, 1 rad s−1 and 1% strain were used ensuring that all measurements were conducted in the linear viscoelastic region.2.8 Gel fraction
To determine the hydrogel gel fractions, a procedure was adopted from Meng et al.56 Briefly, each hydrogel (35 μL) was made on a glass bottom dish (Cellvis). The hydrogels were frozen overnight and lyophilized. For each condition, three replicates were weighed to obtain the initial dry weight (mi). The dried samples were subsequently immersed in water to leach out any remaining unreacted precursors. Water was changed every 2.5 h, and a total of three leaching cycles were performed. Finally, the hydrogels were once again frozen and lyophilized. The washed and dried samples were weighed to determine the mass of the cross-linked fraction of the network (mg). From this, the gel fraction was calculated as: | (2) |
2.9 Swelling ratio and calculated swollen modulus
To measure the swelling properties of each condition, hydrogels (35 μL) were formed on a 12 well, flat bottom tissue culture plate (FisherBrand) in triplicate. Each hydrogel was placed in its own well and submerged in 3 mL of PBS to induce swelling. Each hydrogel was then incubated for 3 days before being removed and weighed in a microcentrifuge tube. This was taken to be the mass in the swollen state (ms). Subsequently, the hydrogels were frozen overnight, lyophilized, and weighed again in the dry state (md). Volumetric swelling ratio was calculated using the following formula: | (3) |
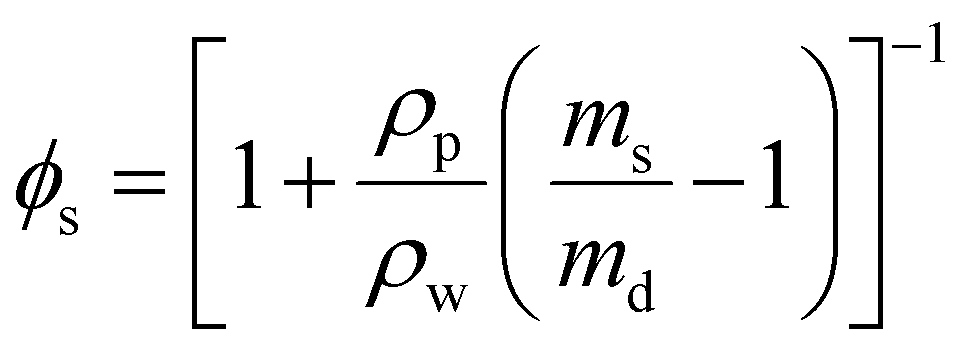 | (4) |
A hypothetical swollen modulus was calculated using:
 | (5) |
 | (6) |
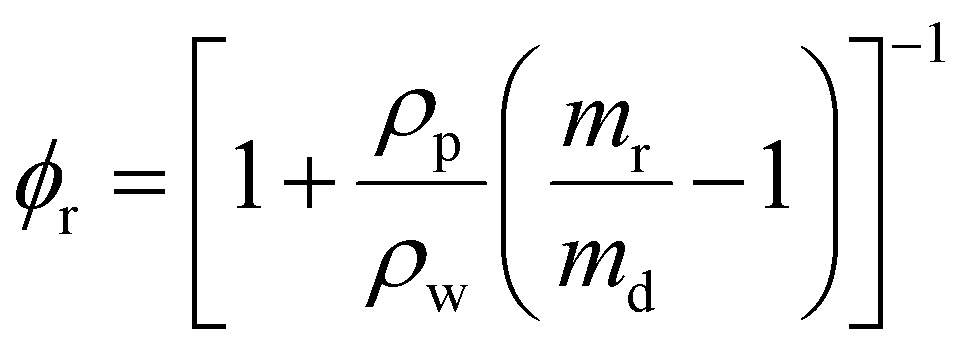 | (7) |
2.10 Cell culture
hMSCs between passages 3–6 from a healthy 29-year-old male (RoosterBio, Cat. #MSC-031, Tissue origin: Human Bone Marrow; Lot. 310277) were used for all experiments. The product information provided by the supplier confirms that these cells preserved differentiation capacity to adipogenic and osteogenic phenotypes and were positive for CD90 and CD166 surface markers, and negative for CD34 and CD45, indicating their stemness. hMSCs were grown in alpha-minimum essential media (1×) (supplemented with L-glutamine, ribonucleosides, and deoxyribonucleosides) (Gibco, Cat. #12561-056) containing 20% fetal bovine serum (Gibco, Cat. #12662029), 1.2% penicillin–streptomycin (Corning, Cat. #30002CI), 1.2% L-glutamine (Corning, Cat. #25005CI) and cultured at 37 °C in a humidified atmosphere of 5% CO2.2.11 Cell viability
hMSCs (cell density: 1000000 cells per mL) were encapsulated within each hydrogel formulation (hydrogel volume: 35 μL) and cultured for 3 days. The pre-gel solution (containing NorHA, peptoid/peptide crosslinker, cell adhesive peptide, and LAP) was used to resuspend the cell pellet. Then, after ensuring the pellet was entirely resuspended, the pre-gel solution was exposed to UV light at 10 mW cm−2 for 2 minutes on glass bottom dishes (Cellvis, Catalog #: NC0794151). Each hydrogel was then suspended in 3 mL of media and allowed to culture for 3 days. Once the culture was complete, the cell viability was assessed via LIVE/DEAD™ assay (Invitrogen, Cat. #L3224). Briefly, a solution containing 4 μM ethidium homodimer-1 and 1 μM calcein-AM in sterile DPBS (without Ca2+ and Mg2+) was added to each dish, followed by incubation for 30 min to allow for the staining process. Next, a Nikon Eclipse Ti2 microscope at a 10× magnification and in the Z-stack mode was used to image the encapsulated cells in each hydrogel. Using ImageJ software, Z-stacks were compiled into a maximum-intensity projection to quantify live and dead cells. Over 5000 cells were counted for each condition across 4 hydrogel replicates.
2.12 Cell proliferation
Proliferative activity was evaluated using a Click-iT™ Plus EdU (5-ethynyl-2-deoxyuridine) Alexa Fluor 488 Imaging Kit (Invitrogen, Cat. #C10337). Like the cell viability study, hMSCs at a concentration of 1000000 cells per mL were encapsulated in hydrogels with each crosslinker of interest and cultured for 3 days (hydrogels were prepared in a glass bottom dish, Cellvis, Cat. # D35-14-1.5-N). After three days of culture, half of the medium was removed, and replaced by fresh medium containing 20 mM of EdU, followed by incubation for 18 hours. Then the samples were washed with DPBS followed by fixation with 3.7% formaldehyde for 15 min at room temperature. Next, permeabilization using 0.5% Triton X-100 was performed at room temperature for 20 min. After the wash with 3% bovine serum albumin (BSA) twice, the Click-iT reaction cocktail was added and incubated for 30 min at room temperature. Following several washes with 3% BSA, nucleus staining was performed with Hoechst 33342 for an additional 30 min of incubation. Finally, samples were washed three more times with DPBS. A Nikon W1 spinning disk confocal microscope at 20× was used to image the cells. Proliferating cells were visualized using a filter with excitation/emission of 495/519 nm. The percentage of EdU-positive nuclei as a measure of proliferation was analyzed using ImageJ software. Over 200 cells were counted for each condition across 4 hydrogel replicates.
2.13 Immunocytochemistry for YAP nuclear localization
Immunofluorescence staining was performed for the detection of yes-associated protein (YAP) nuclear localization in hMSCs (1000000 cells per mL) encapsulated in peptoid-crosslinked hydrogels after 3 days of culture at 37 °C in a humidified atmosphere of 5% CO2 (hydrogels were prepared in a glass bottom dish, Cellvis, Cat. # D35-14-1.5-N). Cells in hydrogel constructs were fixed with 4% paraformaldehyde for 1 h at room temperature and washed 3 × 10 min with PBS. Next, samples were permeabilized with 1% Triton X-100 in PBS for 1 h and blocked with 3% BSA in PBS (Blocking Buffer) for 1 h at room temperature. Later, the blocking buffer was replaced by the primary antibody (mouse monoclonal YAP Antibody (63.7), Santa Cruz Biotechnology, Cat. # sc-101199, 1:200) in 3% BSA and incubated for 24 h at 4 °C. Then, samples were rinsed 3 × 10 min with PBS and incubated with the secondary antibody (AlexaFluor 488 goat anti-mouse, Invitrogen, Cat. # A32723, 1:200) in 3% BSA for 2 h at room temperature. Subsequently, hydrogels were washed 3 × 10 min with PBS and AlexaFluor555 Phalloidin (Invitrogen, Cat. # A34055, 1:100 dilution) and DAPI (Invitrogen, Cat. # A34055, 1:500 dilution) in PBS were added to each well and incubated for 1 h at room temperature. Finally, the samples were rinsed for 3 × 10 min with PBS before being stored at 4 °C in PBS (protected from light) until imaging. Images were acquired using a Nikon AXR confocal microscope with a 40×, 1.25 NA silicone objective, and 3× zoom was used to acquire z-stacks. Imaging was performed by placing the gels in #1.5 glass bottom mini-Petri dishes (Cellvis). YAP nuclear/cytosolic ratio was assessed by measuring the fluorescent intensity of the nucleus and in the cytoplasm domain. The data was normalized to the volumes of the nuclear and cytosolic domains within the cell following the method published by Caliari et al.62 ImageJ software was used to analyze the maximum intensity projection of the images and a DAPI staining image was used to delineate the nuclear regions. At least 20 cells were counted for each condition across 4 hydrogel replicates.
2.14 Secretion activity
IDO (indoleamine 2,3-dioxygenase) release by hMSCs (1000000 cells per mL) encapsulated in each hydrogel condition was measured and normalized per number of cells in the presence and absence of IFN-γ at a concentration of 50 ng mL−1 after 3 days of culture. The protocol as previously described25,63,64 consisted of collecting 100 μL of media from each condition and mixing with 100 μL standard assay mixture (potassium phosphate buffer (50 mM, pH 6.5), ascorbic acid (40 mM, neutralized with NaOH, Sigma, Cat. #50-81-7, molecular weight: 176.12), catalase (200 μg ml−1, Sigma, Cat. #9001-05-2), methylene blue (20 μM, Sigma, Cat. #122965-43-9, molecular weight: 319.85), and L-tryptophan (400 μM, Sigma, Cat. #73-22-3, molecular weight: 204.23)) to allow IDO to convert L-tryptophan to N-formyl-kynurenine. After incubation at 37 °C and 5% CO2 (protected from light) for 30 min, 100 μL of 30% (wt/vol) trichloroacetic acid (Sigma, Cat. #76-03-9, molecular weight: 163.39) was added to stop the reaction by incubating the mixture at 58 °C for 30 min. Next, the solution was centrifuged at 10000 RPM for 10 min. Finally, 100 μL of the supernatant was mixed with 100 μL of Ehrlich's reagent (2% (w/v) p-dimethylaminobenzaldehyde in acetic acid, (Sigma, Cat. #100-10-7, molecular weight: 149.19)) followed by incubation for 10 min. A Synergy H1 Hybrid Multi-Mode Reader was used to measure the absorbance of each sample at a wavelength of 490 nm, which subsequently was converted to a concentration of N-formyl-kynurenine using a standard curve varying the concentration of kynurenine (Sigma, Cat #2922-83-0). IDO secretion was measured for each condition across 4 hydrogel replicates.
2.15 Statistics
The results are presented as a mean ± standard deviation. Figure legends describe the sample size and control for each experimental group. Shapiro-Wilks tests were conducted to ensure normality and Levene's tests were conducted to ensure equal variance between groups (Section S1†). A one-way analysis of variance (ANOVA) with post-hoc Tukey HSD test was performed to establish comparisons among groups. A p-value p < 0.05 was considered statistically significant, and * denotes p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. If the Shapiro-Wilks test indicated the data violated normality, a nonparametric Kruskal–Wallis test was conducted to compare results (avoiding the assumption of normality) and verify the ANOVA results to be appropriate.3 Results and discussion
3.1 Peptoids with increased water solubility maintain their secondary structure
To decouple hydrogel stiffness and network connectivity, while also allowing for 3D hMSC encapsulation, we first engineered helical and non-helical peptoid crosslinkers that maintain a high degree of water solubility. Darapaneni et al. showed that the addition of piperazine or homopiperazine residues at the N-termini of hydrophobic peptoids increased their water solubility by providing hydrogen bond acceptors.51 Thus, we designed a series of helical peptoids (H14) with piperazine (H14-pip), homopiperazine (H14-hp), or two homopiperazines (H14-DHP) at the N-terminus (Table 1 and Fig. 1A). This strategy enabled us to use our previously investigated helical repeat sequence with thiol functional groups for crosslinking. | ||
| Fig. 1 Hydrophobic peptoid structures modified with two homopiperazine monomers provide adequate solubility for use in 3D cell culture. (A) Sequences of helical (H14-DHP), non-helical (N14-DHP), and unstructured (U14-DHP) peptoids with the addition of two homopiperazine monomers at the N-terminus of the structures. (B) Degree of solubility in water of each version of the helical peptoids, along with the non-helical and unstructured peptoids with two homopiperazine monomers (N14-DHP, U14-DHP). The dashed red line represents the minimum concentration needed to form hydrogels in purely aqueous environments (15 mM) while the shaded red area represents solubility that does not satisfy this requirement. (C) Representative images of all peptoids at a concentration of 15 mM in PBS, showing the degree of solubility at such concentration. (D) Transmittance percentage from each peptoid determined using a plate reader at a wavelength of 254 nm. The error bars are presented as means ± standard deviations of n = 3. | ||
To enable aqueous hydrogel formulation, a water solubility of 15 mM was desired. H14 only showed a water solubility of approximately 2.3 mM (Fig. 1B). Addition of a single piperazine or homopiperazine monomer (H14-pip and H14-hp, respectively) slightly increased the water solubility, but not above 15 mM (Fig. 1B). However, when we added two homopiperazine monomers to the sequence (H14-DHP), we achieved a water solubility of 22.9 mM (Fig. 1B). Two homopiperazines were also added to the non-helical (N14-DHP) and unstructured peptoid sequences (U14-DHP) (Fig. 1A). N14-DHP showed similar solubility to H14-DHP (Fig. 1B), perhaps unsurprisingly, as it has the same sequence as the helical peptoid but in racemic form. Although the two homopiperazine monomers were not needed for the unstructured peptoid to be water soluble at the desired concentration, they were added as a control. Visual inspection showed that all peptoids with two homopiperazine monomers were soluble at and above 15 mM in PBS, whereas H14, H14-pip, and H14-hp precipitated out of solution at this concentration (Fig. 1C and Fig. S5†). To verify our qualitative results, transmittance measurements were conducted (Fig. 1D). All peptoids with two homopiperazine monomers showed transmittance values close to 100% compared to a water control, whereas transmittance steadily decreased for the more hydrophobic peptoids.
To ensure that the incorporation of the homopiperazine residues did not alter helicity, circular dichroism (CD) was used to measure the secondary structure of each peptoid. H14-DHP retained its helicity, as indicated by the negative peak at ∼220 nm corresponding to the n–π* transition of the amide chromophore, another negative peak ∼200 nm, and the positive peak at ∼190 nm, corresponding to the high and low wavelength components of the exciton split π–π* transition (Fig. 2A).35 H14-DHP showed a similar CD profile in comparison to H14; however, H14-DHP showed a slight decrease in the per residue molar ellipticity, which may indicate helices with more conformational flexibility. As expected, neither N14-DHP nor U14-DHP showed absorption profiles indicative of secondary structures (Fig. 2B). It is important to acknowledge, however, that the absence of a CD signal does not conclusively negate structure formation here; instead, it indicates the lack of overall molecular chirality in N14-DHP and U14-DHP. Finally, we examined the peptide control (KCGGIQQWGPCK), which showed a random coil structure with a negative peak at ∼200 nm (Fig. 2B), indicating no defined higher order structure.65
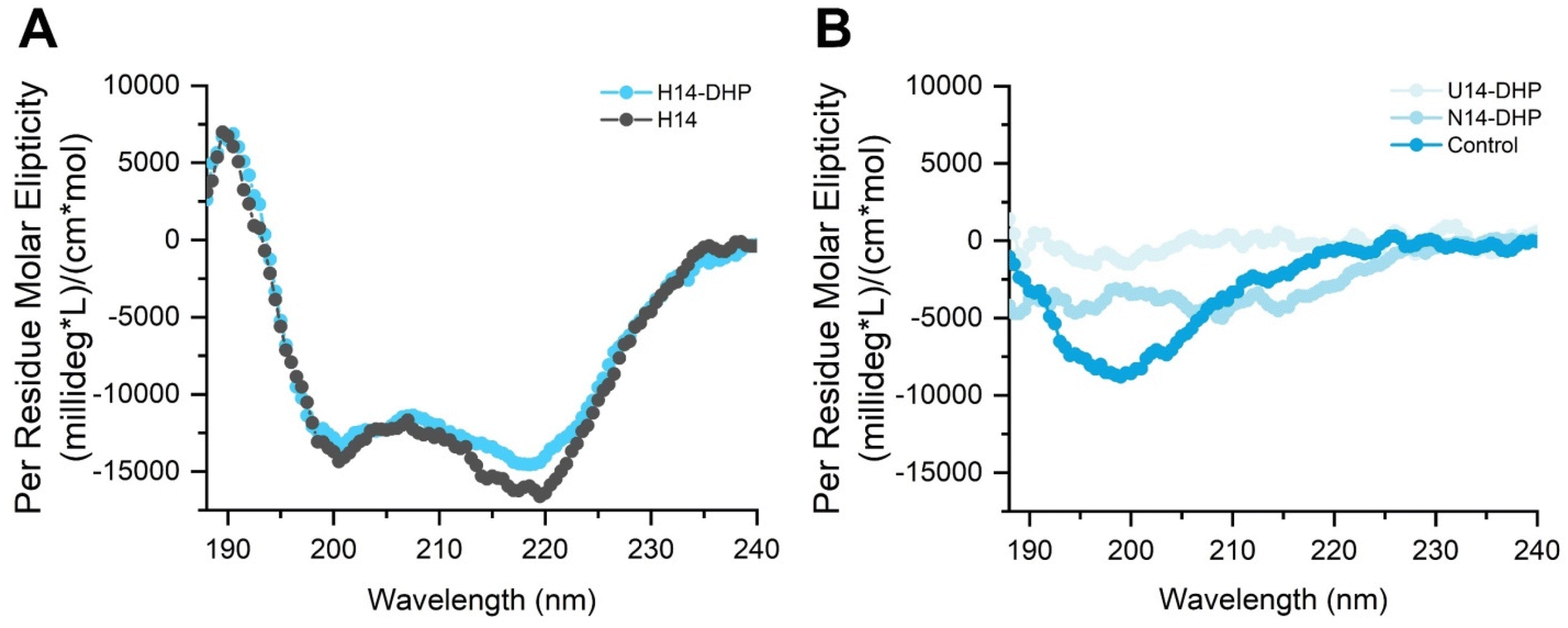 | ||
| Fig. 2 Circular dichroism (CD) spectrum shows secondary structure of helical peptoid is maintained when two homopiperazines are added to the main sequence. (A) Characteristic peaks for helical peptoids are shown with a decrease in signal for the H14-DHP, potentially indicating a decrease in helicity. (B) No secondary structure is seen for N14-DHP and U14-DHP, while the peptide control shows a random coil structure. | ||
3.2 Crosslinker rigidity determines hydrogel bulk stiffness
To determine if hydrogel bulk stiffness can be decoupled from network connectivity in a water soluble hydrogel system, we incorporated our peptoid crosslinkers into norbornene-hyaluronic acid (NorHA) hydrogels via thiol:ene crosslinking.24,66 HA is an attractive starting material for the fabrication of hydrogels because it is inherently biocompatible, biodegradable, and relatively non-immunogenic.67 Before making the hydrogels, we measured the availability of free thiols on the peptoid crosslinkers using Ellman's test (Fig. S6†). All peptoid crosslinkers showed similar thiol availability, where H14-DHP showed 66% thiols available and N14-DHP and U14-DHP showed 63% and 60% availability, respectively. The thiol availability of the peptide control (∼99%) was significantly higher than that of the peptoids, potentially due to residual water in the peptoid mass post-lyophilization. Alternatively, the difference in thiol availability could be attributed to the difference in monomers used between the peptide and peptoid sequences. The peptide control used the cysteine amino acid for the thiol availability (a chiral monomer), whereas the peptoids used cysteamine (an achiral monomer), which may affect thiol presentation. To account for the reduction in thiol availability of the peptoids, the concentration of the crosslinker was increased until 1:1 stoichiometry was obtained. Shear oscillatory rheology tests confirmed that the additional crosslinker increased the storage modulus of the formed hydrogels and added elastically effective linkages (Fig. S7†).
Next, the hydrogels were formed in situ on the rheometer to test the gelation kinetics and storage modulus of each formulation with the peptide or peptoid crosslinkers. NorHA and the crosslinker were dissolved in PBS at the desired concentration with LAP photoinitiator. For all rheometric experiments, the precursor solution was exposed to light (365 nm, 10 mW cm−2) at around 20 seconds. All formulations reacted very quickly, with the crossover point of the storage and loss modulus occurring ∼20 seconds after light exposure (Fig. 3A). However, the peptide control condition reached the plateau storage modulus within 60 seconds after light initiation, whereas the peptoid conditions reached the plateau storage modulus within 2 minutes of starting the reaction (Fig. 3A). This difference may be attributable to the presence of the homopiperazine monomers, which may inhibit the thiol reactivity and slow the rate of reaction with the norbornenes on the HA backbone. The peptoid-containing formulations showed no significant difference in reaction kinetics amongst themselves. Altogether, hydrogels were formed within two minutes of light exposure, a time that has previously been shown to facilitate in vitro studies.68,69
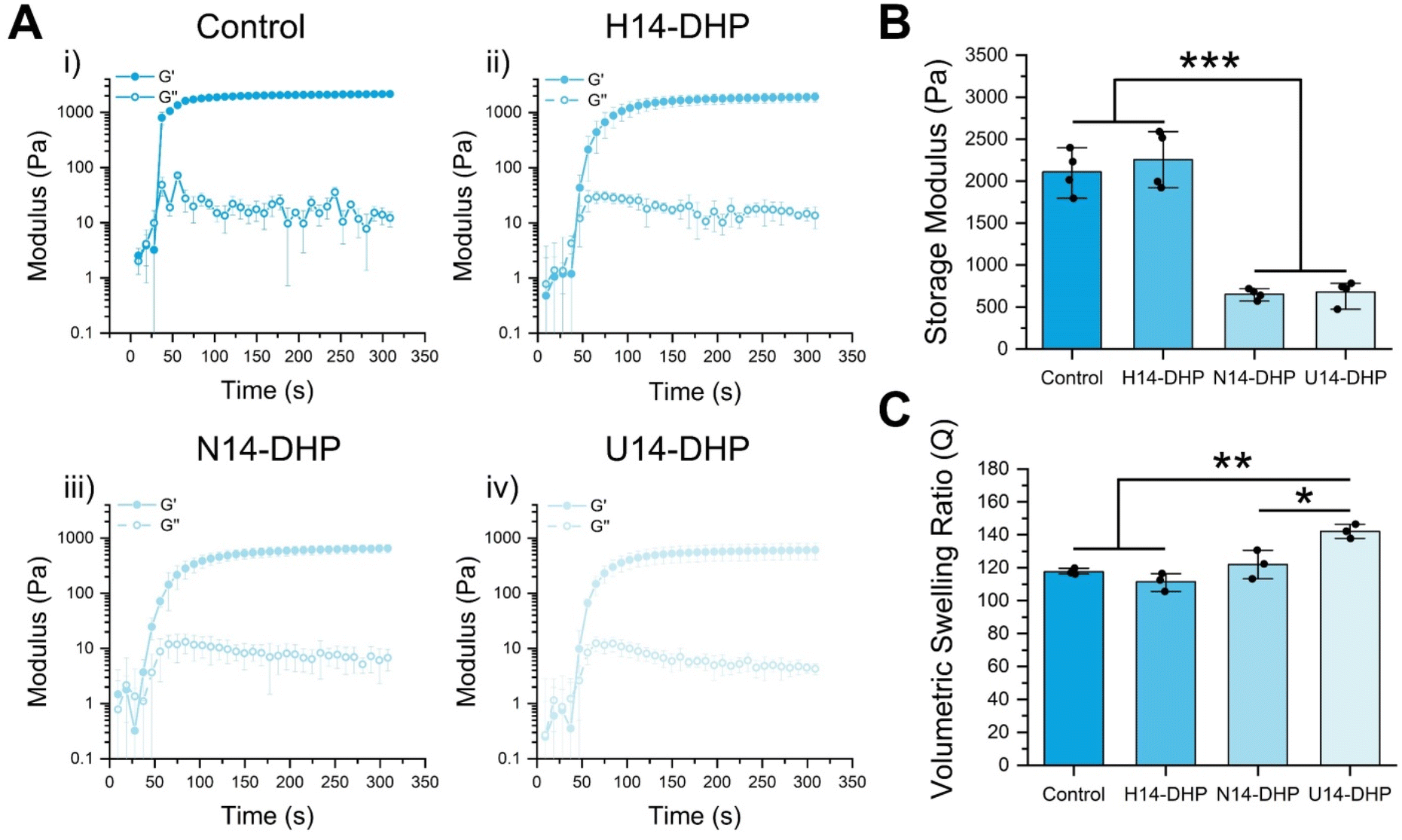 | ||
| Fig. 3 Crosslinker structure determines hydrogel stiffness while keeping similar gelation kinetics and comparable swelling properties. (A) Plateau storage moduli obtained from oscillatory rheology for the three hydrophilic peptoid crosslinked hydrogels and the peptide control. (B) Time sweeps for each condition from subfigure (A) illustrating similar gelation kinetics for the hydrophilic peptoid crosslinked hydrogels and a faster reach to plateau in the peptide control. (C) Volumetric swelling ratios for each hydrogel indicating that the softer hydrogels swell more than the stiffer hydrogels. The storage modulus and time sweeps are presented as means ± standard deviations of n = 5 hydrogel samples from each condition of several independent studies. The volumetric swelling ratio is presented as means ± standard deviations of n = 3 hydrogel samples from each condition of several independent studies.* denotes p < 0.05, ** p < 0.01, and ***p < 0.001. All statistics were calculated by one-way ANOVA with post-hoc Turkey HSD test. | ||
The hydrogel plateau storage moduli (G∞′) were significantly affected by the secondary structure of the peptoid crosslinkers. H14-DHP hydrogels showed a G∞′ of 2500 Pa compared to N14-DHP and U14-DHP hydrogels, which exhibited similar G∞′ of ∼700 Pa (Fig. 3B). To verify this difference was due to secondary structure and not different extents of reaction, gel fraction studies were conducted to determine the amount of sample that forms a system-spanning network. The gel fractions of the peptoid-crosslinked hydrogels were very similar, ∼40% for all three conditions (Fig. S8†). Hydrogels with the peptide crosslinker showed a similar G∞′ (2100 Pa) to the H14-DHP hydrogels (Fig. 3B), but a much higher gel fraction of 70% (Fig. S8†). This result correlates with the higher thiol availability seen via the Ellman's assay compared to that of the H14-DHP crosslinker. Overall, these results suggest that the peptoid-crosslinked hydrogels achieved similar degrees of crosslinking and network connectivity, and thus, hydrogel stiffness was directly controlled by the crosslinker secondary structures.
Due to our interest in exploring these hydrogels for cell encapsulations, we quantified the swelling behavior for each hydrogel condition. Mass swelling ratio was initially obtained by determining the swollen and dry masses before and after freezing and lyophilizing the hydrogels. All hydrogels swelled considerably (Fig. 3C), with the U14-DHP crosslinked hydrogels swelling slightly more than the other conditions. This result aligns with the relatively high water solubility of the U14-DHP crosslinker, which is based on sarcosine repeat units and does not have any chiral side chains. The volumetric swelling ratios were used to calculate the theoretical swollen moduli, and the same trends in G∞′ with secondary structure were observed (Fig. S9†). It was difficult to quantitatively verify the swollen moduli with rheological measurements, due to fact that inertial issues decrease the reliability of the measurements for very soft materials, especially the N14-DHP and U14-DHP hydrogels. However, the estimated stiffness range of the swollen hydrogels (100–500 Pa) has been previously shown to affect hMSC differentiation70,71 and proliferation72,73 in 3D culture; thus, we proceeded with these formulations to in vitro studies.
3.3 hMSC viability and proliferation in peptoid-crosslinked hydrogels
In 2D, peptoid-crosslinked hydrogels were previously demonstrated to be highly compatible substrates for hMSCs, facilitating improved therapeutic efficacy in comparison to tissue culture plastic.24,25,38 To translate our hydrogel design to 3D cell culture applications, we tested the viability and proliferative capacity of encapsulated hMSCs (Fig. 4A). After 3 days of culture, hMSC viability in peptoid-crosslinked hydrogels was assessed using a LIVE/DEAD™ assay (Fig. 4B and C). All peptoid-crosslinked hydrogels indicated high viability (>90%) of encapsulated hMSCs, similar to the peptide-crosslinked hydrogel (positive control), which is known to be a cytocompatible platform.74 At this timepoint, the cells in all conditions remained mostly rounded, indicating that even though hMSCs release hyaluronidase,75,76 the HA degradation was not fast enough to lead to significant cell spreading. The degradability of this platform could be further engineered to support cell spreading by adding proteolytically-degradable sequence motifs to the network,77–79 as the peptoids offer much higher proteolytic stability than peptides.80,81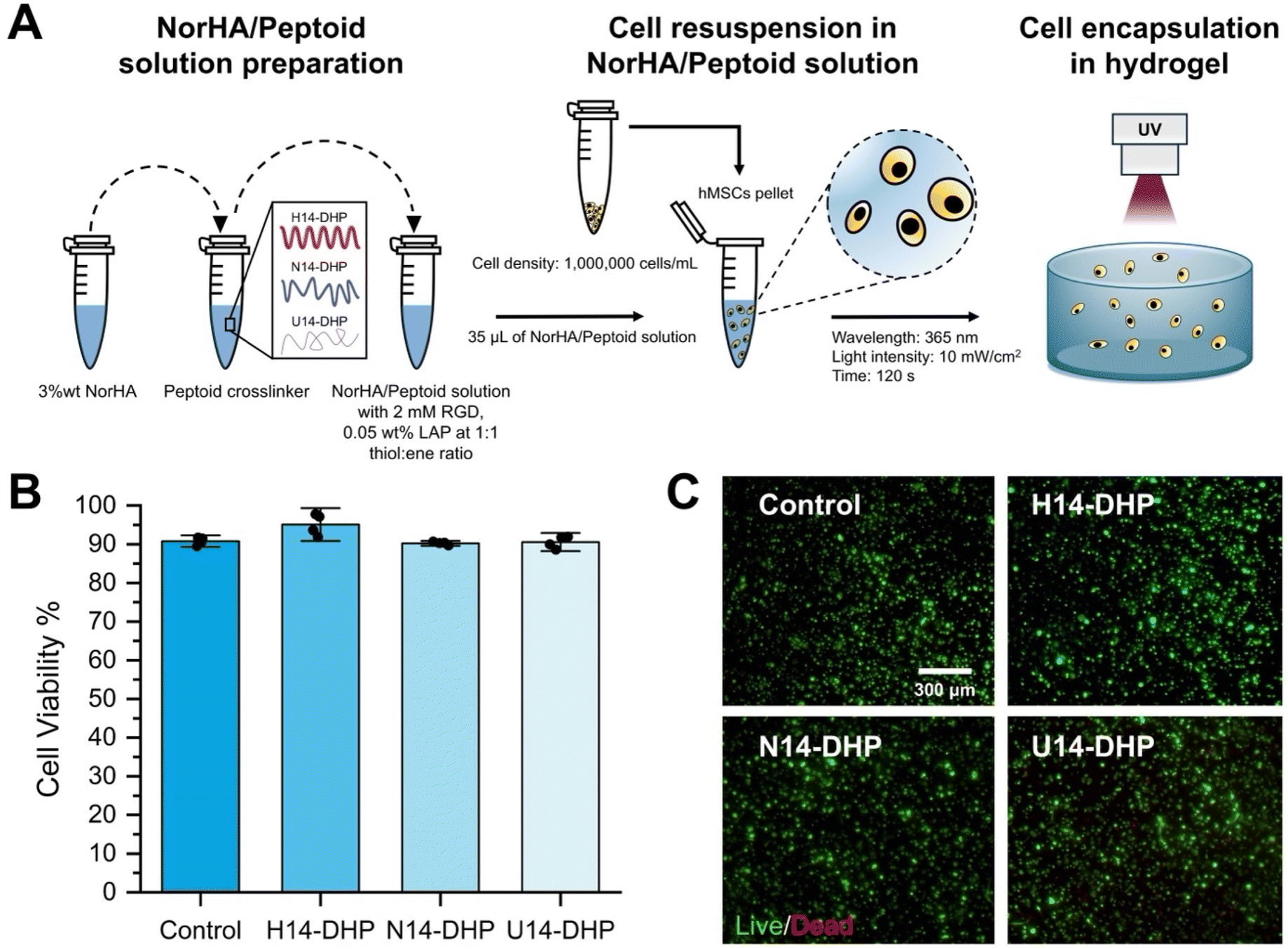 | ||
| Fig. 4 hMSCs in peptoid-crosslinked hydrogels showed high viability. (A) Schematic representation of the hydrogel preparation and hMSC encapsulation in the hydrogel. (B) Viability (assessed by LIVE/DEAD™ assay) of hMSCs encapsulated in peptoid-crosslinked hydrogels after 3 days of culture. The viability data is presented as mean ± standard deviation of n = 4 hydrogel samples from each condition of two independent studies. (C) Representative images of hMSCs in peptoid-crosslinked hydrogels and the peptide control. Green represents live cell staining and red represents dead cells, scale bar = 300 μm. | ||
Next, we measured markers of proliferative ability for the encapsulated hMSCs in our new hydrogel system and evaluated whether the cells showed stiffness-dependent behavior. Multiple studies have highlighted that the relationship between matrix stiffness and hMSC proliferation in 3D is often non-monotonic. Within a moderate range of stiffness values, increasing rigidity can stimulate higher proliferation rates, but when stiffness becomes exceedingly high, stress responses or lineage-commitment pathways may overshadow self-renewal.82,83 In addition, if the matrix is too stiff or too dense, small pore sizes can restrict cell spreading and diffusion of nutrients, ultimately diminishing proliferation rates.84 Here, hMSC proliferation was evaluated by performing an EdU assay after 3 days of culture (Fig. 5). Results showed increased hMSC proliferation with hydrogel stiffness. Specifically, stiffer hydrogels (H14-DHP) significantly increased the percentage of EdU positive cells (∼40%) compared to the softest hydrogels (U14-DHP) (∼26%). Hydrogels crosslinked with N14-DHP showed an intermediate percentage of EdU positive cells. The peptide crosslinked hydrogel (control), which had similar stiffness to the H14-DHP hydrogels, also showed a significant increase (p < 0.05) in the percentage of EdU positive cells compared to the U14-DHP hydrogels. Altogether, these results demonstrate that hMSCs encapsulated in peptoid-crosslinked hydrogels presented significantly different levels of proliferation that depend on the hydrogel's mechanical properties.
 | ||
| Fig. 5 hMSCs in peptoid-crosslinked hydrogels presented significantly different levels of proliferation (EDU-positive cells) that depended on the hydrogel's mechanical properties. (A) Proliferation quantification after 3 days of culture. (B) Representative images from each hydrogel condition with nuclei stained in blue and EDU-positive cells stained in green, scale bar 100 μm. The proliferation data is presented as mean ± standard deviations of n = 4 hydrogel samples from each condition of two independent studies. All conditions were compared to one another and to the peptide crosslinked hydrogel control. * denotes p < 0.05, **p < 0.01, and ***p < 0.001. All statistics were calculated by one-way ANOVA with post-hoc Turkey HSD test. | ||
3.4 YAP nuclear localization is correlated with cell proliferation and hydrogel stiffness
To investigate if the stiffness-dependent behavior of encapsulated hMSCs in peptoid-crosslinked hydrogels is related to known mechanotransduction pathways, we performed immunofluorescence staining for Yes-associated protein (YAP) after 3 days of culture. YAP is known as a mechanosensing molecule in stem cells that plays an important role in cellular proliferation and differentiation.85,86 Previous work has shown that activation of YAP depends on the translocation from the cytoplasm to the nucleus in response to multiple biophysical cues, such as ECM stiffness,62,87 though 2D and 3D culture geometries show conflicting results. In 3D, hydrogel stiffness has been demonstrated to reduce YAP/TAZ nuclear translocation in hMSCs,88,89 whereas hMSCs seeded onto stiff hydrogels in a 2D configuration showed increased nuclear YAP/TAZ.62 In other studies, nuclear localization of YAP directly correlated with cell proliferation.90 For example, a reduced proliferation rate of hMSCs in confined 3D microniches compared to 2D culture was associated with low nuclear localization of YAP or primarily cytoplasmic YAP localization.90Our results demonstrated that stiffer hydrogels (H14-DHP) significantly increased the YAP nuclear localization of encapsulated hMSCs (Fig. 6A and B). The softest hydrogels (U14-DHP) presented the lowest ratio of YAP nuclear-to-cytosolic localization with a ratio of approximately 0.85. These results indicate that the increased levels of proliferative ability observed for hMSCs encapsulated in H14-DHP-crosslinked hydrogels may be due to cells sensing their increased matrix stiffness.
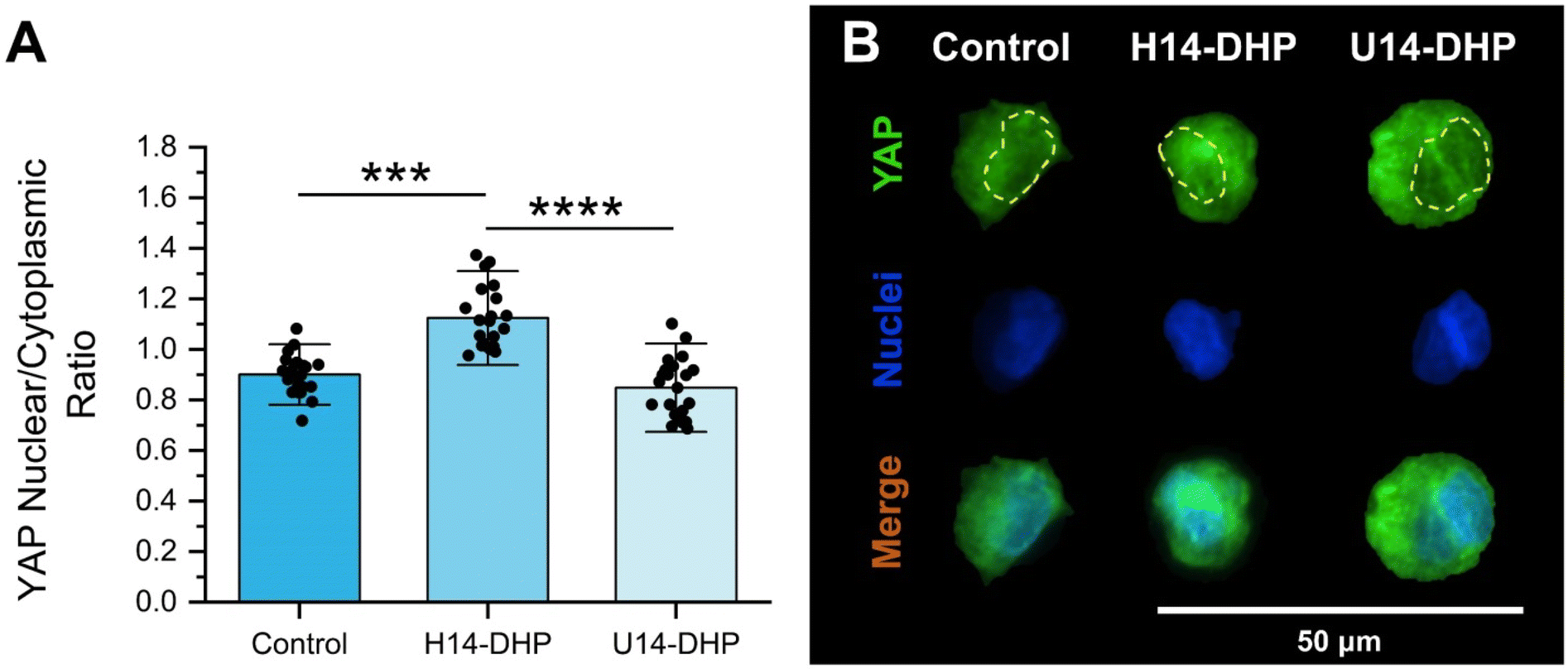 | ||
| Fig. 6 Stiffer hydrogels (H14-DHP) increase the YAP nuclear localization of hMSCs in peptoid-crosslinked hydrogels. (A) Fluorescence intensity of the YAP nuclear localization for hMSCs encapsulated within peptoid-crosslinked hydrogels. Green represents YAP staining and blue represents the nucleus (DAPI), scale bar 50 μm. (B) Representative fluorescent images of YAP nuclear activation in hMSCs after 3 days of culture. All data is presented as mean ± standard deviations of n = 4 hydrogel samples from each condition of two independent studies. H14-DHP hydrogels were compared with U14-DHP hydrogels and both were compared to the peptide crosslinked hydrogel control. ****denotes p < 0.0001 and ***p < 0.001 between conditions. All statistics were calculated by one-way ANOVA with post-hoc Tukey HSD test. | ||
3.5 Immunomodulatory potential of encapsulated hMSCs in peptoid-crosslinked hydrogels
With evidence that encapsulated hMSCs could sense matrix stiffness in our 3D peptoid-crosslinked hydrogels, we studied hMSC secretory activity through IDO production as a measurement of their immunomodulatory potential.44,91 For this study, we focused on the IDO expression of encapsulated hMSCs in the stiffest condition (H14-DHP), the softest condition (U14-DHP), and the peptide control. All conditions were assessed after 3 days of culture in the absence and presence of IFN-γ supplemented in the culture medium. Conditions without IFN-γ supplementation showed that the U14-DHP hydrogels (the softest condition) significantly upregulated IDO production compared to H14-DHP hydrogels and the peptide control hydrogels (Fig. 7A). A similar trend was reported in our previous studies when hMSCs were seeded onto the hydrogel surface.25 However, in the present study, encapsulating the hMSCs in the hydrogel substantially increased the IDO production per cell from approximately 2200 pg per cell (2D)25 to about 9000 pg per cell (3D) for the softest condition. Because IFN-γ is known to promote the immunomodulatory potential of hMSCs,92,93 we looked at the effect of IFN-γ supplementation in the culture media on IDO production (Fig. 7B). As expected, the levels of IDO secretion in the presence of IFN-γ were significantly higher than conditions in the absence of IFN-γ for all conditions (Fig. 7A and B). In addition, these data still showed stiffness-dependent results; the softer U14-DHP-crosslinked hydrogels improved IDO production of encapsulated hMSCs compared to control hydrogels and the H14-DHP-crosslinked hydrogels. | ||
| Fig. 7 Softer peptoid-crosslinked hydrogels improve immunosuppression of encapsulated hMSCs. Quantification of N-formylkynurenine (IDO activity) produced per encapsulated hMSCs in peptoid crosslinked hydrogels and the peptide crosslinked control (A) without IFN-γ supplementation (B) with IFN-γ supplementation. All data is presented as mean ± standard deviations of n = 4 hydrogel samples from each condition of two independent studies. Within a condition, all conditions were compared (H14-DHP was compared with U14-DHP and both were compared to the peptide crosslinked control). Additionally, each condition (including the peptide crosslinked control) were compared with and without IFN-γ supplementation. * denotes p < 0.05 and **** denotes p < 0.0001. All statistics were calculated by one-way ANOVA with post-hoc Turkey HSD test. @ indicates p < 0.01, & indicates p < 0.0001, and $ indicates p < 0.0001 between that condition with and without IFN-γ. | ||
4 Conclusion
In summary, this study illustrates the versatility of peptoid-based hydrogel networks as effective biomimetic platforms for applications in tissue engineering and 3D cell culture. By strategically modulating the secondary structures of peptoid crosslinkers, we achieved control over hydrogel mechanics decoupled from changes in network connectivity or polymer concentration. Our findings highlight the significant impact of hydrogel stiffness on the behavior of hMSCs in 3D cell culture. Stiffer hydrogels crosslinked with helical peptoids promoted higher proliferation rates and increased YAP nuclear localization. In contrast, softer hydrogels crosslinked with non-helical or unstructured peptoids demonstrated slightly enhanced production of IDO, both with and without IFN-γ stimulation. Interestingly, the 3D culture geometry and stimulation with IFN-γ influenced IDO secretion on a per cell basis more strongly than matrix stiffness, which may enable the use of matrix stiffness to increase other cell processes important to stem cell manufacturing, such as proliferative ability. The implications of these findings are far-reaching, as they enable increased understanding of how ECM properties influence the production of highly potent, therapeutically relevant hMSCs. Moreover, the ability to decouple mechanical properties from network connectivity opens new avenues for investigating the complex interplay between mechanical and biochemical cues in the ECM and paves the way for design of advanced hydrogel culture systems. In conclusion, this study not only reinforces the importance of hydrogel mechanics in stem cell biology but also highlights the potential of peptoid-based materials in the design of next-generation biomaterials in the fields of regenerative medicine and immunotherapy.Data availability
The data supporting this article have been included as part of the ESI.† This includes characterization data for peptoids, peptides, as well as mechanical testing results for the hydrogels. Specifically, ESI figures† provide analytical HPLC traces and LC-MS spectra for all peptoids and peptides, 1H NMR of norbornene-functionalized hyaluronic acid (NorHA), Ellman's assay results, gel fraction measurements, calculated swollen moduli of all hydrogels, and statistical analyses to validate the use of ANOVA.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number K99GM151459. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. In addition, this research was supported by the National Institutes of Health (R35GM138193, A.M.R) and the National Science Foundation (NSF MRSEC DMR-2308817, A.M.R.), and L.D.M. was supported through a National Science Foundation Graduate Research Fellowship.References
- J. Patterson, M. M. Martino and J. A. Hubbell, Mater. Today, 2010, 13, 14–22 CrossRef CAS.
- E. González-Díaz and S. Varghese, Gels, 2016, 2, 20 CrossRef PubMed.
- J. A. Rowley, G. Madlambayan and D. J. Mooney, Biomaterials, 1999, 20, 45–53 CrossRef CAS PubMed.
- M. Kapałczyńska, T. Kolenda, W. Przybyła, M. Zajączkowska, A. Teresiak, V. Filas, M. Ibbs, R. Bliźniak, Ł. Łuczewski and K. Lamperska, Arch. Med. Sci., 2018, 14, 910–919 Search PubMed.
- J. P. Jung, M. K. Bache-Wiig, P. P. Provenzano and B. M. Ogle, BioRes. Open Access, 2016, 5, 37–48 CrossRef CAS PubMed.
- D. Widera, in Handbook of Stem Cell Therapy, Springer, 2022, pp. 565–584 Search PubMed.
- Y. Li and K. A. Kilian, Adv. Healthcare Mater., 2015, 4, 2780–2796 CrossRef CAS PubMed.
- C. Jensen and Y. Teng, Front. Mol. Biosci., 2020, 7 Search PubMed.
- M. Ravi, V. Paramesh, S. R. Kaviya, E. Anuradha and F. D. P. Solomon, J. Cell. Physiol., 2015, 230, 16–26 CrossRef CAS PubMed.
- Z. Zhuang, Y. Zhang, X. Yang, T. Yu, Y. Zhang, K. Sun, Y. Zhang, F. Cheng, L. Zhang and H. Wang, Acta Biomater., 2022, 149, 69–81 CrossRef CAS PubMed.
- H. Yoshii, M. Kajiya, M. Yoshino, S. Morimoto, S. Horikoshi, M. Tari, S. Motoike, T. Iwata, K. Ouhara, T. Ando, T. Yoshimoto, T. Shintani and N. Mizuno, Stem Cell Rev. Rep., 2024, 20, 347–361 CrossRef CAS PubMed.
- F. Liu, D. Lagares, K. M. Choi, L. Stopfer, A. Marinković, V. Vrbanac, C. K. Probst, S. E. Hiemer, T. H. Sisson, J. C. Horowitz, I. O. Rosas, L. E. Fredenburgh, C. Feghali-Bostwick, X. Varelas, A. M. Tager and D. J. Tschumperlin, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2015, 308, L344–L357 CrossRef CAS PubMed.
- D. E. Ingber, FASEB J., 2006, 20, 811–827 CrossRef CAS PubMed.
- D. J. Tschumperlin, Compr. Physiol., 2011, 1, 1057–1073 CrossRef PubMed.
- R. S. Navarro, M. S. Huang, J. G. Roth, K. M. Hubka, C. M. Long, A. Enejder and S. C. Heilshorn, Adv. Healthcare Mater., 2022, 2200011 CrossRef CAS PubMed.
- C. M. Madl, B. L. LeSavage, R. E. Dewi, C. B. Dinh, R. S. Stowers, M. Khariton, K. J. Lampe, D. Nguyen, O. Chaudhuri and A. Enejder, Nat. Mater., 2017, 16, 1233–1242 CrossRef CAS PubMed.
- J. L. Holloway, H. Ma, R. Rai and J. A. Burdick, J. Controlled Release, 2014, 191, 63–70 CrossRef CAS PubMed.
- S. Lee, X. Tong and F. Yang, Acta Biomater., 2014, 10, 4167–4174 CrossRef CAS PubMed.
- H. Omidian, S.-A. Hasherni, F. Askari and S. Nafisi, Iran. J. Polym. Sci. Technol., 1994, 3(2), 115–119 CAS.
- A. D. Theocharis, S. S. Skandalis, C. Gialeli and N. K. Karamanos, Adv. Drug Delivery Rev., 2016, 97, 4–27 CrossRef CAS PubMed.
- M. Aumailley, Composition of the Extracellular Matrix, ed. F. R. A. Maia, J. M. Oliveira and R. L. Reis, Springer International Publishing, Cham, 2023, pp. 1–30 Search PubMed.
- E. Shimshoni, R. Afik, I. Solomonov, I. Adir, A. Shenoy, M. Adler, L. Puricelli, V. Ghini, O. Mouhadeb, N. Gluck, S. Fishman, L. Werner, D. S. Shouval, C. Varol, A. Podestà, P. Turano, T. Geiger, P. Milani, C. Luchinat, U. Alon and I. Sagi, bioRxiv, 2019, 665653 Search PubMed.
- A. Padhi and A. S. Nain, Ann. Biomed. Eng., 2020, 48, 1071–1089 CrossRef PubMed.
- L. D. Morton, A. Hillsley, M. J. Austin and A. M. Rosales, J. Mater. Chem. B, 2020, 8, 6925–6933 RSC.
- L. D. Morton, D. A. Castilla-Casadiego, A. C. Palmer and A. M. Rosales, Acta Biomater., 2023, 155, 258–270 CrossRef CAS PubMed.
- A. P. Dhand, J. H. Galarraga and J. A. Burdick, Trends Biotechnol., 2021, 39, 519–538 CrossRef CAS PubMed.
- K. Liu, S. M. Mihaila, A. Rowan, E. Oosterwijk and P. H. J. Kouwer, Biomacromolecules, 2019, 20, 826–834 CrossRef CAS PubMed.
- P. H. J. Kouwer, M. Koepf, V. A. A. Le Sage, M. Jaspers, A. M. van Buul, Z. H. Eksteen-Akeroyd, T. Woltinge, E. Schwartz, H. J. Kitto, R. Hoogenboom, S. J. Picken, R. J. M. Nolte, E. Mendes and A. E. Rowan, Nature, 2013, 493, 651–655 CrossRef CAS PubMed.
- K. H. Vining, A. Stafford and D. J. Mooney, Biomaterials, 2019, 188, 187–197 CrossRef CAS PubMed.
- A. M. Rosales, H. K. Murnen, S. R. Kline, R. N. Zuckermann and R. A. Segalman, Soft Matter, 2012, 8, 3673–3680 RSC.
- B. C. Lee, R. N. Zuckermann and K. A. Dill, J. Am. Chem. Soc., 2005, 127, 10999–11009 CrossRef CAS PubMed.
- P. Armand, K. Kirshenbaum, A. Falicov, R. L. J. Dunbrack, K. A. Dill, R. N. Zuckermann and F. E. Cohen, Folding Des., 1997, 2, 369–375 CrossRef CAS PubMed.
- T. W. Craven, R. Bonneau and K. Kirshenbaum, ChemBioChem, 2016, 1824–1828 CrossRef CAS PubMed.
- C. W. Wu, T. J. Sanborn, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2001, 123, 2958–2963 CrossRef CAS PubMed.
- T. J. Sanborn, C. W. Wu, R. N. Zuckermann and A. E. Barron, Biopolymers, 2002, 63, 12–20 CrossRef CAS PubMed.
- W. Yang, Y. Zhou, B. Jin, X. Qi, B. Cai, Q. Yin, J. Pfaendtner, J. J. De Yoreo and C.-L. Chen, J. Colloid Interface Sci., 2023, 634, 450–459 CrossRef CAS PubMed.
- M. Zhao, K. J. Lachowski, S. Zhang, S. Alamdari, J. Sampath, P. Mu, C. J. Mundy, J. Pfaendtner, J. J. De Yoreo, C.-L. Chen, L. D. Pozzo and A. L. Ferguson, Biomacromolecules, 2022, 23, 992–1008 CrossRef CAS PubMed.
- D. A. Castilla-Casadiego, L. D. Morton, D. H. Loh, A. Pineda-Hernandez, A. P. Chavda, F. Garcia and A. M. Rosales, Macromol. Biosci., 2024, 24, 2400111 CrossRef CAS PubMed.
- B. Follin, M. Juhl, S. Cohen, A. E. Pedersen, J. Kastrup and A. Ekblond, Tissue Eng., Part B, 2016, 22, 322–329 CrossRef CAS PubMed.
- D. S. Kim, I. K. Jang, M. W. Lee, Y. J. Ko, D.-H. Lee, J. W. Lee, K. W. Sung, H. H. Koo and K. H. Yoo, EBioMedicine, 2018, 28, 261–273 CrossRef PubMed.
- T. J. Bartosh, J. H. Ylöstalo, A. Mohammadipoor, N. Bazhanov, K. Coble, K. Claypool, R. H. Lee, H. Choi and D. J. Prockop, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13724–13729 CrossRef CAS PubMed.
- A. Gonzalez-Pujana, K. H. Vining, D. K. Y. Zhang, E. Santos-Vizcaino, M. Igartua, R. M. Hernandez and D. J. Mooney, Biomaterials, 2020, 257, 120266 CrossRef CAS PubMed.
- D. A. Castilla-Casadiego, D. H. Loh, A. Pineda-Hernandez and A. M. Rosales, Biomacromolecules, 2024, 25, 6319–6337 CrossRef CAS PubMed.
- J. C. Mbongue, D. A. Nicholas, T. W. Torrez, N.-S. Kim, A. F. Firek and W. H. R. Langridge, Vaccines, 2015, 3, 703–729 CrossRef CAS PubMed.
- K. Kirshenbaum, A. E. Barron, R. A. Goldsmith, P. Armand, E. K. Bradley, K. T. V. Truong, K. A. Dill, F. E. Cohen and R. N. Zuckermann, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4303–4308 CrossRef CAS PubMed.
- R. N. Zuckermann, J. M. Kerr, W. H. Moosf and S. B. H. Kent, J. Am. Chem. Soc., 1992, 114, 10646–10647 CrossRef CAS.
- S. P. Singh, M. P. Schwartz, J. Y. Lee, B. D. Fairbanks and K. S. Anseth, Biomater. Sci., 2014, 2, 1024–1034 RSC.
- B. V. Sridhar, J. L. Brock, J. S. Silver, J. L. Leight, M. A. Randolph and K. S. Anseth, Adv. Healthcare Mater., 2015, 4, 702–713 CrossRef CAS PubMed.
- C. O. Crosby, A. Hillsley, S. Kumar, B. Stern, S. H. Parekh, A. Rosales and J. Zoldan, Acta Biomater., 2021, 122, 133–144 CrossRef CAS PubMed.
- R. J. Wade, E. J. Bassin, W. M. Gramlich and J. A. Burdick, Adv. Mater., 2015, 27, 1356–1362 CrossRef CAS PubMed.
- C. M. Darapaneni, P. J. Kaniraj and G. Maayan, Org. Biomol. Chem., 2018, 16, 1480–1488 RSC.
- M. Spangenberg, J. I. Bryant, S. J. Gibson, P. J. Mousley, Y. Ramachers and G. R. Bell, Sci. Rep., 2021, 11, 3682 CrossRef CAS PubMed.
- A. Aitken and M. Learmonth, Estimation of Disulfide Bonds Using Ellman's Reagent, ed. J. M. Walker, Humana Press, Totowa, NJ, 2009, pp. 1053–1055 Search PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- P. W. Riddles, R. L. Blakeley and B. Zerner, in Enzyme Structure Part I, Academic Press, 1983, vol. 91, pp. 49–60 Search PubMed.
- Z. Meng, L. Löser, K. Saalwächter, U. Gasser and H.-A. Klok, Macromolecules, 2024, 57, 3058–3065 CrossRef CAS PubMed.
- P. J. Flory and J. Rehner Jr., J. Chem. Phys., 1943, 11, 512–520 CrossRef CAS.
- P. J. Flory and J. Rehner Jr., J. Chem. Phys., 1943, 11, 521–526 CrossRef CAS.
- N. R. Richbourg and N. A. Peppas, Prog. Polym. Sci., 2020, 105, 101243 CrossRef CAS.
- N. A. Peppas and C. T. Reinhart, J. Membr. Sci., 1983, 15, 275–287 CrossRef CAS.
- C. T. Reinhart and N. A. Peppas, J. Membr. Sci., 1984, 18, 227–239 CrossRef CAS.
- S. R. Caliari, S. L. Vega, M. Kwon, E. M. Soulas and J. A. Burdick, Biomaterials, 2016, 103, 314–323 CrossRef CAS PubMed.
- M. Haseli, D. A. Castilla-Casadiego, L. Pinzon-Herrera, A. Hillsley, K. A. Miranda-Munoz, S. Sivaraman, A. M. Rosales, R. R. Rao and J. Almodovar, Mater. Today Bio, 2022, 13, 100194 CrossRef CAS PubMed.
- D. A. Castilla-Casadiego, H. Timsina, M. Haseli, L. Pinzon-Herrera, Y.-H. Chiao, S. R. Wickramasinghe and J. Almodovar, ACS Biomater. Sci. Eng., 2020, 6, 6626–6651 CrossRef CAS PubMed.
- L. J. Smith, K. M. Fiebig, H. Schwalbe and C. M. Dobson, Folding Des., 1996, 1, R95–R106 CrossRef CAS PubMed.
- P. M. Kharkar, M. S. Rehmann, K. M. Skeens, E. Maverakis and A. M. Kloxin, ACS Biomater. Sci. Eng., 2016, 2, 165–179 CrossRef CAS PubMed.
- X. Xu, A. K. Jha, D. A. Harrington, M. C. Farach-Carson and X. Jia, Soft Matter, 2012, 8, 3280–3294 RSC.
- B. D. Fairbanks, M. P. Schwartz, C. N. Bowman and K. S. Anseth, Biomaterials, 2009, 30, 6702–6707 CrossRef CAS PubMed.
- S. J. Bryant, C. R. Nuttelman and K. S. Anseth, J. Biomater. Sci., Polym. Ed., 2000, 11, 439–457 CrossRef CAS PubMed.
- V. Moulisová, S. Poveda-Reyes, E. Sanmartín-Masiá, L. Quintanilla-Sierra, M. Salmerón-Sánchez and G. Gallego-Ferrer, ACS Omega, 2017, 2, 7609–7620 CrossRef PubMed.
- R. K. Das, V. Gocheva, R. Hammink, O. F. Zouani and A. E. Rowan, Nat. Mater., 2016, 15, 318–325 CrossRef CAS PubMed.
- S. Y. Cheon, J. S. Park, Y. Lee, C. Lee, H. Jeon, D. Lee, S. H. Kim, S. G. Lim and H. Koo, Adv. Sci., 2024, 11, 2304861 CrossRef CAS PubMed.
- A. Pangjantuk, P. Kaokaen, P. Kunhorm, N. Chaicharoenaudomrung and P. Noisa, Sci. Rep., 2024, 14, 4436 CrossRef CAS PubMed.
- J. L. Leight, D. L. Alge, A. J. Maier and K. S. Anseth, Biomaterials, 2013, 34, 7344–7352 CrossRef CAS PubMed.
- E. D. Goldberg, A. M. Dygai, G. N. Zyuz'kov, V. V. Zhdanov, E. V. Simanina and L. A. Gur'yantseva, Bull. Exp. Biol. Med., 2007, 143, 548–551 CrossRef CAS PubMed.
- M. K. Paap and R. Z. Silkiss, Plast. Aesthetic Res., 2020, 7, 36 CrossRef.
- M. V. Turturro, M. C. Christenson, J. C. Larson, D. A. Young, E. M. Brey and G. Papavasiliou, PLoS One, 2013, 8, e58897 CrossRef CAS PubMed.
- L. D. Amer and S. J. Bryant, Ann. Biomed. Eng., 2016, 44, 1959–1969 CrossRef PubMed.
- K. B. Fonseca, D. B. Gomes, K. Lee, S. G. Santos, A. Sousa, E. A. Silva, D. J. Mooney, P. L. Granja and C. C. Barrias, Biomacromolecules, 2014, 15, 380–390 CrossRef CAS PubMed.
- A. M. Clapperton, J. Babi and H. Tran, ACS Polym. Au, 2022, 2, 417–429 CrossRef CAS PubMed.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zuckermann, J. M. Kerr and W. H. Moos, Bioorg. Med. Chem. Lett., 1994, 4, 2657–2662 CrossRef CAS.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- S. R. Peyton and A. J. Putnam, J. Cell. Physiol., 2005, 204, 198–209 CrossRef CAS PubMed.
- N. Huebsch, E. Lippens, K. Lee, M. Mehta, S. T. Koshy, M. C. Darnell, R. M. Desai, C. M. Madl, M. Xu, X. Zhao, O. Chaudhuri, C. Verbeke, W. S. Kim, K. Alim, A. Mammoto, D. E. Ingber, G. N. Duda and D. J. Mooney, Nat. Mater., 2015, 14, 1269–1277 CrossRef CAS PubMed.
- N. Damkham, S. Issaragrisil and C. Lorthongpanich, Int. J. Mol. Sci., 2022, 23, 14634 CrossRef CAS PubMed.
- A. Husari, T. Steinberg, M. P. Dieterle, O. Prucker, J. Rühe, B. Jung and P. Tomakidi, Cell. Signal., 2019, 63, 109382 CrossRef CAS PubMed.
- K. E. Scott, S. I. Fraley and P. Rangamani, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(20), e2021571118 CrossRef CAS PubMed.
- T. Luo, B. Tan, L. Zhu, Y. Wang and J. Liao, Front. Bioeng. Biotechnol., 2022, 10, 817391 CrossRef PubMed.
- Y. Li, Z. Zhong, C. Xu, X. Wu, J. Li, W. Tao, J. Wang, Y. Du and S. Zhang, Natl. Sci. Rev., 2023, 10(8), nwad165 CrossRef CAS PubMed.
- O. Y. Dudaryeva, A. Bucciarelli, G. Bovone, F. Huwyler, S. Jaydev, N. Broguiere, M. Al-Bayati, M. Lütolf and M. W. Tibbitt, Adv. Funct. Mater., 2021, 31, 2104098 CrossRef CAS.
- W. Chen, Nat. Immunol., 2011, 12, 809–811 CrossRef CAS PubMed.
- J. R. García, M. Quirós, W. M. Han, M. N. O'Leary, G. N. Cox, A. Nusrat and A. J. García, Biomaterials, 2019, 220, 119403 CrossRef PubMed.
- S. J. Cifuentes, P. Priyadarshani, D. A. Castilla-Casadiego, L. J. Mortensen, J. Almodóvar and M. Domenech, J. Biomed. Mater. Res., Part A, 2021, 109, 951–965 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5bm00433k |
| ‡ These authors contributed equally to this work. |
| § Present address: Department of Biomedical Engineering, The University of Miami, Coral Gables, Florida 33146, USA. |
| ¶ Present address: Department of Biomedical Engineering, Tufts University, Medford, Massachusetts 02155, USA. |
| This journal is © The Royal Society of Chemistry 2025 |