
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a bioactive hyaluronic acid hydrogel functionalised with antimicrobial peptides for the treatment of chronic wounds†
Noémie
Petit
ab,
Ana
Gomes
c,
Yu-yin Joanne
Chang
ab,
Jessica
Da Silva
 def,
Ermelindo C.
Leal
efg,
Eugénia
Carvalho
efg,
Paula
Gomes
c and
Shane
Browne
*abh
def,
Ermelindo C.
Leal
efg,
Eugénia
Carvalho
efg,
Paula
Gomes
c and
Shane
Browne
*abh
aTissue Engineering Research Group, Department of Anatomy and Regenerative Medicine, Royal College of Surgeons in Ireland, 123, St Stephen's Green, Dublin 2, Ireland. E-mail: shanebrowne@rcsi.ie
bCÚRAM, Centre for Research in Medical Devices, University of Galway, Galway, H91 W2TY, Ireland
cLAQV-REQUIMTE, Department of Chemistry and Biochemistry, Faculty of Sciences of the University of Porto, Portugal
dUniversity of Coimbra, Institute of Interdisciplinary Research, Doctoral Program in Experimental Biology and Biomedicine (PDBEB), 3004-504 Coimbra, Portugal
eCNC-UC – Center for Neuroscience and Cell Biology, University of Coimbra, 3004-504 Coimbra, Portugal
fCIBB – Centre for Innovative Biomedicine and Biotechnology, University of Coimbra, 3004-504 Coimbra, Portugal
gInstitute of Interdisciplinary Research, University of Coimbra, 3004-504 Coimbra, Portugal
hTrinity Centre for Biomedical Engineering, Trinity College Dublin, Dublin 2, Ireland
First published on 7th May 2025
Abstract
Chronic wounds present significant clinical challenges due to delayed healing and high infection risk. This study presents the development and characterisation of acrylated hyaluronic acid (AcHyA) hydrogels functionalised with gelatin (G) and the antimicrobial peptide (AMP) PP4-3.1 to enhance cellular responses while providing antimicrobial activity. AcHyA-G and AcHyA-AMP hydrogels were formed via thiol–acrylate crosslinking, enabling in situ AcHyA hydrogel formation with stable mechanical properties across varying gelatin concentrations. Biophysical characterisation of AcHyA-G hydrogels showed rapid gelation, elastic behaviour, uniform mesh size, and consistent molecular diffusion across all formulations. Moreover, the presence of gelatin enhanced stability without affecting the hydrogel's degradation kinetics. AcHyA-G hydrogels supported the adhesion and spreading of key cell types involved in wound repair (dermal fibroblasts and endothelial cells), with 0.5% gelatin identified as the optimal effective concentration. Furthermore, the conjugation of the AMP conferred bactericidal activity against Staphylococcus aureus and Escherichia coli, two of the most prevalent bacterial species found in chronically infected wounds. These results highlight the dual function of AcHyA-AMP hydrogels in promoting cellular responses and antimicrobial activity, offering a promising strategy for chronic wound treatment. Further in vivo studies are needed to evaluate their efficacy, including in diabetic foot ulcers.
Introduction
Chronic wounds pose a significant healthcare challenge, affecting millions worldwide due to impaired healing and a heightened risk of infection.1–3 Among these, diabetic foot ulcers (DFUs) are a prevalent example, where long-term hyperglycaemia leads to vascular and immune dysfunction, further exacerbating wound chronicity.4 Other chronic wounds, including venous and pressure ulcers, share similar pathophysiological characteristics, necessitating advanced therapeutic strategies beyond conventional dressings, debridement, and infection management.4,5 Despite the emergence of novel treatments, their clinical adoption remains limited due to cost constraints and variable efficacy, placing a substantial burden on healthcare systems, caregivers, and patients.6,7Acute wound healing typically follows four sequential and interconnected processes: (1) haemostasis, (2) inflammation, (3) cell proliferation and migration, and (4) matrix deposition and remodelling.8 Chronic wounds, however, exhibit dysregulation across these phases, particularly impaired angiogenesis, failing to restore tissue vascularity and inhibiting regeneration.9–11 Biomaterials, and in particular those derived from natural biopolymers, have gained significant attention as potential therapies for chronic wound treatment due to their ability to enhance healing by providing a template that supports cell infiltration and tissue regeneration.12
Hydrogels, three-dimensional, crosslinked hydrophilic polymer networks, that can absorb and retain large amounts of water while maintaining their structure have attracted great interest.13,14 Hydrogels can maintain a moist wound environment and fill irregularly shaped defects, while also supporting tissue regeneration and providing a protective barrier against microbial infections with the appropriate functionalisation.15,16 While a vast array of biopolymers have been assessed as hydrogels, hyaluronic acid (HyA) has been identified as particularly suitable due to its biocompatibility, biodegradability, and ease of chemical modification, key criteria in the development of hydrogel-based strategies for tissue repair.15,17–19 Unlike many native components of the extracellular matrix (ECM), HyA can be chemically modified while preserving its native structure. HyA also promotes fibroblast migration and proliferation, increasing collagen secretion, and contributes to angiogenesis, particularly important given the high amounts of HyA associated with foetal, non-scarring wounds.15,20–22
Chemical modification of HyA with acrylate groups (AcHyA) allows for in situ hydrogel formation via dithiol crosslinkers, as well as the covalent conjugation of thiol-terminated moieties, such as peptides and ECM ligands, to enhance or add further functionality.23–25 AcHyA hydrogels are highly modular and provide an opportunity to independently modify a range of properties including mechanical properties, degradation, and the availability of cell adhesion ligands.26,27 With this in mind, we incorporated thiolated gelatin (G) within AcHyA to support cell adhesion and vascularisation. Gelatin is a natural protein derived from collagen and is also biocompatible and biodegradable.28,29 One main advantage of incorporating gelatin into an AcHyA hydrogel system is the presence of peptide sequences such as RGD and GFOGER in the gelatin which promote cell attachment, fibroblast and endothelial cell growth, aiding in tissue regeneration and vascularisation, all of which are crucial for chronic wound healing.28–32
Beyond tissue regeneration, infection remains a major obstacle in chronic wound treatment, where bacterial colonisation sustains inflammation and delays healing.33,34 While antibiotics remain the standard of care, the rise of antibiotic resistance necessitates alternative approaches. Strategies to combat bacterial infections include (i) addressing antimicrobial resistance mechanisms (e.g. RNA silencing, enzyme inhibitors), (ii) enhancing antimicrobial drug delivery, (iii) applying physico-chemical inactivation (e.g. photoinactivation), and (iv) directly combating antimicrobial resistant bacteria through the use of antimicrobial peptides (AMPs).35,36 Amongst these alternative strategies, AMPs show great promise as they effectively target both Gram-positive and Gram-negative bacteria and are less prone to induce the development of resistance.37,38 Furthermore, AMPs can have indirect effects by modulating host immune defences in response to pathogen infection and by enhancing migration and proliferation of cells.39,40
However, the clinical application of AMPs is limited due to their poor stability and rapid degradation in protease-rich wound environments, leading to short half-lives and burst release when physically entrapped in biomaterials.41 To mitigate these limitations, AMPs have been covalently conjugated to biomaterials. While covalent tethering can protect AMPs from enzymatic degradation and limit cytotoxicity, this method can reduce the antimicrobial efficacy of the AMPs due to steric hindrance or restricted mobility.41 Another challenge in AMP-functionalised biomaterials is the potential loss of antimicrobial efficacy due to structural changes upon conjugation. To overcome this, we engineered a versatile hydrogel based on acrylated hyaluronic acid (AcHyA). This hydrogel is functionalised with thiolated gelatin (AcHyA-G) to enhance cell adhesion and covalently conjugated with a cysteine-terminated AMP (Cys-PP4-3.1) via thiol–acrylate Michael addition (AcHyA-AMP). Moreover, we incorporate an Ahx spacer to preserve the AMP's amphipathic nature and optimise its interaction with bacterial membranes. This strategic design maintains antimicrobial potency while minimising steric hindrance. This dual-functional hydrogel provides sustained antimicrobial activity, preventing burst release and improving protease resistance while promoting cell adhesion, all while maintaining the biophysical properties of the hydrogel.42–44 In this study, we selected PP4-3.1, a hybrid AMP combining the sequences of cosmeceutical pentapeptide-4 (PP4) and a cationic, amphipathic α-helical AMP (3.1), as it effectively disrupts bacterial membranes and inhibits both Gram-positive and Gram-negative bacteria, making it ideal for chronic wound healing applications.45
Our approach integrates several key elements: (i) dual functionality through the combination of gelatin for improved cell adhesion and AMPs for sustained antimicrobial effects, (ii) covalent AMP conjugation via thiol–acrylate chemistry to ensure controlled release and prolonged antimicrobial efficacy, and (iii) the incorporation of gelatin at minimal concentrations to enhance cell adhesion while preserving the hydrogel's biophysical properties. For this, HyA was modified with acrylate groups to facilitate the conjugation of thiolated gelatin and the cysteine-terminated AMP, i.e. Cys-PP4-3.1, while also enabling chemical crosslinking with PEG-dithiol. The effects of gelatin concentration on the biophysical and mechanical properties of AcHyA and AcHyA-G hydrogels were assessed in a concentration-dependent manner, focusing on the optimal formulation to support fibroblast and endothelial cell adhesion and spreading without altering key hydrogel properties. Finally, the antimicrobial activity of the AcHyA-AMP conjugate was evaluated by testing its ability to inhibit the growth of Staphylococcus aureus and Escherichia coli, bacterial pathogens widely found in non-healing wounds like DFU.46–49 Ultimately, we aim to develop an AcHyA hydrogel decorated with gelatin and AMPs to enhance the cellular response and reduce the risk of bacterial infection, offering a promising solution for chronic wound treatment.
Materials and methods
Materials
Hyaluronic acid (HyA, sodium salt from Streptococcus equi, ∼1.5–1.8 MDa), adipic dihydrazide (ADH), 1-hydroxybenzotriazole (HOBt), 1-ethyl-3-[3-(dimethylamino)propyl] carbodiimide (EDC), dimethyl sulfoxide (DMSO), ethanol, hydrochloric acid (HCl), N-acryloxysuccinimide (NAS), thiol-functionalised gelatin, dithiothreitol (DTT), poly(ethylene glycol) dithiol (PEG-dithiol), Coomassie® Brilliant Blue G 250 (Coomassie), hyaluronidase from bovine testes, high glucose Dulbecco's Modified Eagle Medium (DMEM), 1× Dulbecco's phosphate-buffered saline (PBS), Fluorescein isothiocyanate–dextran (70 kDa), Mueller–Hinton Agar, and Mueller–Hinton broth were purchased from Sigma-Aldrich.Triethanolamine-buffer (TEOA; 0.2 M, pH 8), SnakeSkin™ dialysis membrane (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MWCO), sodium hydroxide (NaOH), 4% paraformaldehyde were purchased from Thermo Fisher Scientific. Hoechst, Alexa Fluor™ 568 Phalloidin, alamarBlue™ were purchased from Invitrogen. Endothelial Cell Growth Medium 2 (EGM-2) was purchased from PromoCell. SB431542 molecule was purchased from TOCRIS.
000 MWCO), sodium hydroxide (NaOH), 4% paraformaldehyde were purchased from Thermo Fisher Scientific. Hoechst, Alexa Fluor™ 568 Phalloidin, alamarBlue™ were purchased from Invitrogen. Endothelial Cell Growth Medium 2 (EGM-2) was purchased from PromoCell. SB431542 molecule was purchased from TOCRIS.
Unless otherwise specified, concentrations are expressed as % w/v.
Methods
Separately, AcHyA-AMP conjugates were synthesised by reacting AcHyA-G (11.8 mg) conjugates with Cys-Ahx-PP4-3.1 (1 mg) overnight at room temperature. The conjugates were then extensively dialysed and the purified product was lyophilized and stored at −20 °C until use. The conjugation of both gelatin and the AMP to AcHyA were confirmed by 1H NMR.
| Conjugate | AcHyA | Gelatin | Cys-Ahx-PP4-3.1 |
|---|---|---|---|
| AcHyA | 1.5 | 0 | 0 |
| AcHyA-G | 1.5 | 0 | 0 |
| 0.1 | |||
| 0.25 | |||
| 0.5 | |||
| AcHyA-AMP | 1.5 | 0.5 | 0.01 |
Hydrogel formation was assessed using the vial inversion method, where no flow (no fluidity) indicates successful gelation.51,52
The release of gelatin from the hydrogels was assessed under hydrolytic (PBS) and enzymatic conditions, namely hyaluronidase (HyAse, 10 U mL−1) at 37 °C. At regular intervals, supernatant was collected, and the absorbance was measured at 595 nm. After 48 h of incubation, 1000 U mL−1 of HyAse was added to the samples in PBS to ensure complete degradation and gelatin release.
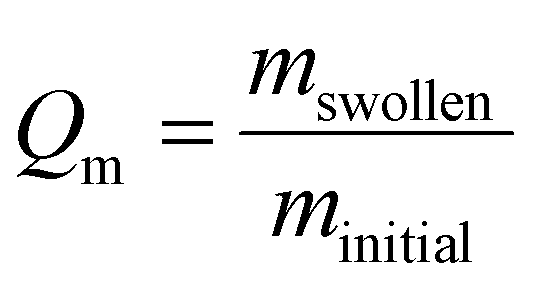 | (1) |
![[M with combining overline]](https://www.rsc.org/images/entities/i_char_004d_0305.gif) c), to then calculate the mesh size (ξ) in eqn (5).56,57
c), to then calculate the mesh size (ξ) in eqn (5).56,57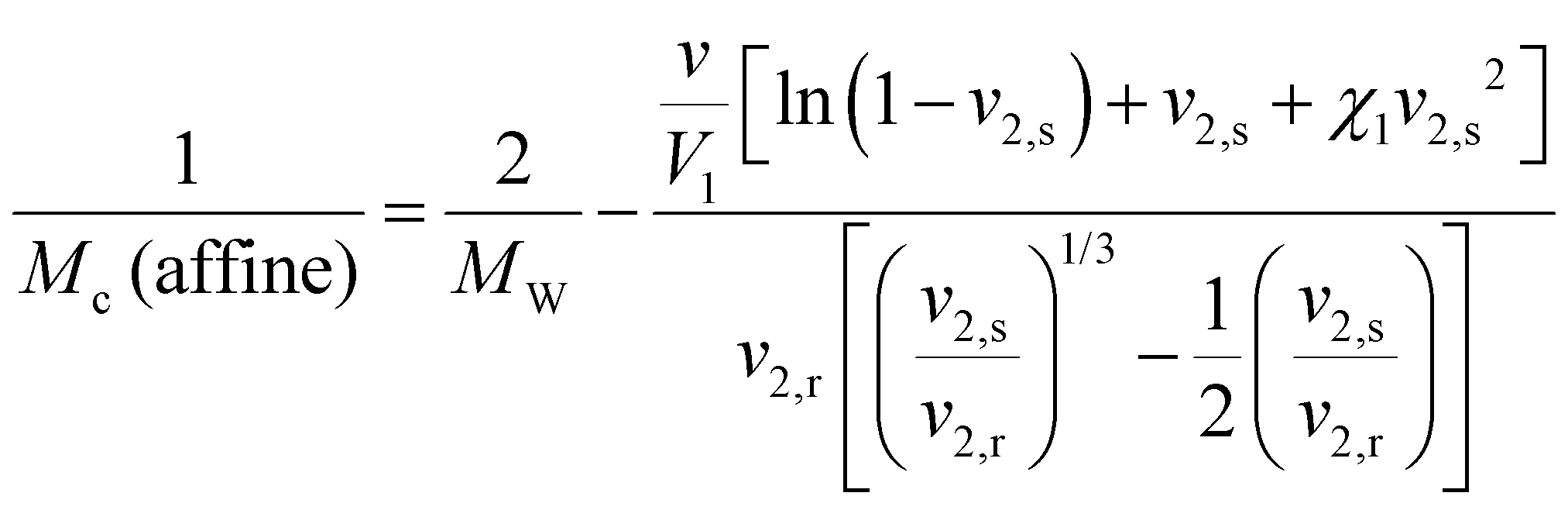 | (2) |
 | (3) |
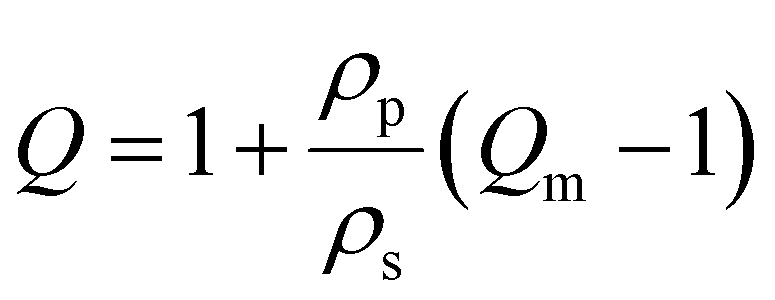 | (4) |
| ξ = 0.1748Mc1/2(v2,s)−1/3 | (5) |
The diffusion coefficient was calculated according to the diffusion model for circular spot using FrapAnalyser Software and is described in eqn (6) and (7).
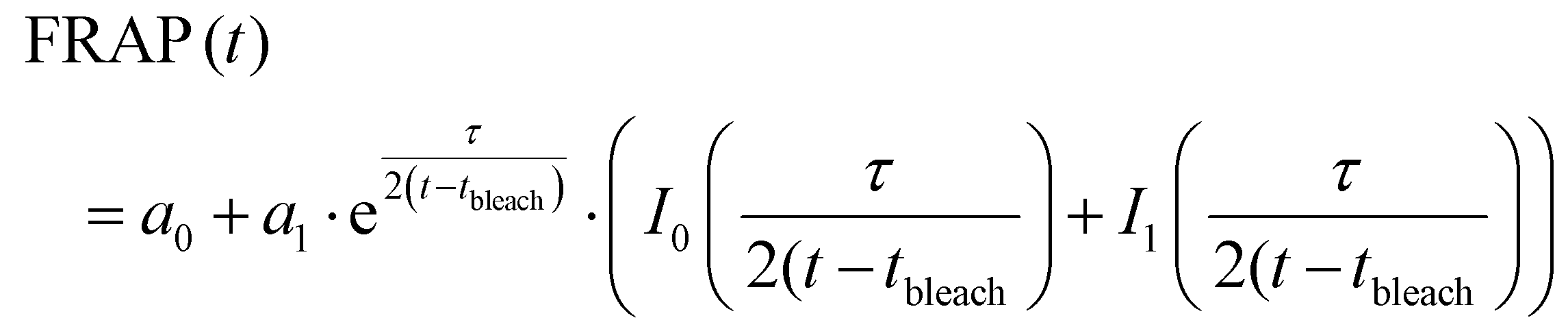 | (6) |
 | (7) |
000 cells per hydrogel in 300 μL of medium. An additional 300 μL of medium was added outside the cell culture inserts. The cell-seeded hydrogels were incubated for 7 days at 37 °C and 5% CO2.
After 7 days in culture, cell-seeded AcHyA hydrogels were fixed with 4% paraformaldehyde at room temperature for 1 hour and subsequently stained with Phalloidin-Atto 488 (cytoskeleton) and DAPI (nuclei). Imaging was performed using a Carl Zeiss LSM 710 confocal microscope. Images were processed using Fiji software.53
Results and discussion
Chronic wounds, including DFUs, pose a significant clinical challenge due to their prolonged healing times and the high risk of infection, representing a significant burden to patients and healthcare systems worldwide. These wounds are often characterised by excessive inflammation, imbalanced protease activity, and bacterial colonisation, all of which contribute to delayed tissue repair.5 This study introduces a dual-functional HyA-based hydrogel, combining the bioactivity-enhancing properties of gelatin (AcHyA-G) with the antimicrobial efficacy of the AMP PP4-3.1 (AcHyA-AMP) for chronic wound treatment (Fig. 1). The conjugation of gelatin and PP4-3.1 to AcHyA via a thiol–acrylate reaction ensures stable incorporation, enabling controlled release and sustained therapeutic effects at the wound site. These conjugation reactions are achieved using an acrylate-modified HyA which supports hydrogel formation through PEG-dithiol crosslinking, resulting in stable, tuneable mechanical properties.25 This approach maintains AcHyA's inherent biocompatibility and provides a versatile platform for functionalisation with thiol-containing bioactive molecules, including ECM moieties and peptides, to promote cellular adhesion, activity, and tissue regeneration.23,24,50 The incorporation of gelatin into the AcHyA matrix enhances cell adhesion, while the incorporation of AMPs offers an effective strategy for infection prevention, ultimately supporting tissue regeneration and improving wound healing.61–66 | ||
| Fig. 1 Schematic representation of the functionalised HyA hydrogels. HyA hydrogels are composed of acrylated HyA chains (AcHyA) along with AcHyA functionalised with thiolated gelatin (AcHyA-G) and PP4-3.1 antimicrobial peptide (AcHyA-AMP), and crosslinked with PEG-dithiol. | ||
The degree of modification of HyA with acrylate groups (AcHyA) was assessed by proton NMR. The presence of two peaks in the 5.5–6.5 ppm region confirmed the successful introduction of acrylate groups onto HyA, with a modification degree of approximately 25%, consistent with previous reports (Fig. 2A).25 The acrylate-thiol reaction was then leveraged to conjugate thiol-functionalised gelatin and the Cys-terminated AMP, PP4-3.1, to AcHyA. A subsequent decrease in the degree of modification to 12% (gelatin, Fig. 2A) and 9.5% (PP4-3.1, Fig. S1†) suggested that fewer acrylate groups were detectable due to their consumption by the conjugation of both gelatin and PP4-3.1 to AcHyA.
 | ||
| Fig. 2 Development and characterisation of AcHyA hydrogels functionalised with gelatin. (A) 1H NMR was used to confirm the modification of HyA with acrylate groups and the conjugation of thiolated gelatin (AcHyA-G). A representative image of an AcHyA hydrogel functionalised with gelatin is shown. (B) The release of gelatin from HyA hydrogels was assessed in hydrolytic conditions (PBS) with the addition of 1000 U mL−1 HyAse at 48 h, and (C) in enzymatic conditions (10 U mL−1 HyAse). Data represents mean ± SD (n = 3). Analysis performed using two-way ANOVA; p < 0.05. | ||
PEG-dithiol was used to crosslink the AcHyA hydrogels via a thiol–acrylate reaction between the acrylate groups of AcHyA and thiol groups in PEG-dithiol.67 PEG-dithiol has demonstrated its biocompatibility and non-immunogenic nature, with minimal risk of an inflammatory response.68–70 Having established the role of PEG-dithiol in crosslinking, we used the protein-specific dye Coomassie to further confirm the conjugation of gelatin to AcHyA to produce AcHyA-G (Fig. 2B and C). We tested gelatin concentrations up to 0.5%, focusing on identifying the optimal concentration required to enhance cellular adhesion while maintaining biophysical properties. Minimal to no gelatin release occurred from AcHyA hydrogels in PBS over the 48 h study period, indicating that gelatin was covalently conjugated to AcHyA via acrylate–thiol bonds. The addition of HyAse at the 48 h timepoint cleaved the β-1,4-glycosidic bonds between the HyA subunits, facilitating AcHyA hydrogel degradation and resulted in rapid gelatin release (Fig. 2B). In the presence of a lower concentration of HyAse, 10 U mL−1, gelatin release was more gradual and complete within 48 h, demonstrating steady release of gelatin as the AcHyA hydrogel degraded (Fig. 2C). This emphasises that gelatin is stably conjugated to the AcHyA hydrogel, with release facilitated only by degradation of the AcHyA hydrogel, in this case mediated by the addition of HyAse.
The impact of gelatin incorporation on the mechanical properties of AcHyA hydrogels was evaluated using rheology. First, the gelation time was observed with the vial inversion method, where no flow was observed after 5 min of incubation for AcHyA hydrogels with and without gelatin, implying rapid crosslinking and hydrogel formation (Fig. 3A). The storage moduli obtained were 928 ± 137, 995 ± 221, 1234 ± 78, and 1210 ± 136 Pa for 0%, 0.1%, 0.25%, and 0.5% gelatin samples, respectively (Fig. 3B). However, no statistically significant differences were observed between samples, demonstrating that the mechanical properties of AcHyA-G hydrogels were independent of gelatin concentration. Next, we investigated the influence of gelatin concentration on the swelling behaviour of the hydrogels to evaluate their moisture retention ability. After 24 h in PBS, AcHyA hydrogels of varying gelatin concentrations swelled to approximately 150%, with no statistically significant differences observed between groups; this indicates that the swelling properties of AcHyA were not affected by gelatin incorporation at concentrations up to 0.5% (Fig. 3C). These findings suggest that gelatin incorporation into AcHyA hydrogels does not significantly alter their mechanical or swelling properties.
 | ||
| Fig. 3 Gelatin incorporation does not significantly affect the mechanical properties or swelling behaviour of AcHyA hydrogels. Characterisation of HyA-derived hydrogels (A) gelation time was assessed with the inversion method for the 0% (top lane) and 0.5% (bottom lane) gelatin formulations, (B) storage modulus, and (C) swelling ratio. Data represents mean ± SD (n = 3). Analysis performed using one-way ANOVA; p < 0.05. | ||
We assessed the stability of AcHyA-G hydrogels at various pH values with and without the addition of HyAse enzyme to assess suitability in the wound environment. The following pH values were chosen to simulate various wound conditions: epithelialized wounds (pH 6), neutral environments (pH 7), and alkaline chronic wounds (pH 8).71 Under hydrolytic conditions, minimal degradation was detected across all conditions. In fact, gelatin-containing hydrogels swelled to a maximum of 130% at pH 6 (Fig. 4A), 125% at pH 7 (Fig. 4B), and 133% at pH 8 (Fig. 4C). In contrast, the 0% gelatin sample only swelled under neutral conditions, losing approximately 20% of its original mass at both pH 6 and pH 8. The differences between the gelatin-containing and gelatin-free samples were pronounced, especially in simulated epithelialized and alkaline wound conditions after 5 days, indicating the enhanced suitability of AcHyA-G hydrogels in the compromised wound environment (Fig. 4A and C). Throughout the study, the 0% and 0.5% samples remained statistically different (Fig. 4A and C). At pH 7, the presence of gelatin had a negligible effect at early time points (1 and 2 h), and all hydrogels remained stable over 5 days (Fig. 4B). This data demonstrates the stability of AcHyA-G hydrogels, and their suitability for exudate absorption, with gelatin enhancing their capacity to maintain a moist environment.
 | ||
| Fig. 4 Gelatin incorporation improves stability of AcHyA hydrogels under varying physiological conditions. Characterisation of HyA-derived hydrogels (A–C) hydrolytic degradation (PBS), and (D–F) enzymatic degradation (10 U mL−1 HyAse). Data represents mean ± SD (n = 3). Analysis performed using two-way ANOVA; p < 0.05. | ||
In addition to hydrolytic degradation, understanding enzymatically-mediated degradation kinetics of wound dressings is crucial as it significantly influences cell and tissue responses, essential for effective wound healing applications.16,72 The controlled degradation of hydrogels is essential for coordinating cell-mediated remodelling with tissue regeneration, supporting effective cell infiltration and vascularisation within the wound site.24 Optimally tuned degradation rates can enhance wound healing by promoting timely cell migration, angiogenesis, and ECM deposition.73–75 During healing, cells secrete a range of enzymes, including HyAse and matrix metalloproteinases (MMPs), that contribute to these processes. Therefore, hydrogels should exhibit a degradation rate that supports cell infiltration and matches the rate of neotissue formation. AcHyA-G hydrogels are susceptible to enzymatic degradation by HyAse, present in the wound environment, making it essential to assess their degradation rates to ensure they maintain integrity while promoting healing.76 Initially, all hydrogels exhibited swelling within the first few hours, likely due to a reduced overall cross-linking density in the hydrogel network as a result of initial degradation.25,77 However, both AcHyA and AcHyA-G hydrogels gradually degraded over 5 days across all pH levels. After 24 h at pH 6 (Fig. 4D) and pH 7 (Fig. 4E), all hydrogels underwent significant degradation, with the mass dropping to approximately one-fifth of the original mass, likely due to increased HyAse activity in these conditions. At pH 8, all hydrogels maintained about 50% of their mass for approximately 24 hours (Fig. 4F). However, across all pH levels, the degradation profiles were not statistically different. These findings indicate that AcHyA-G hydrogels remain susceptible to HyAse-mediated degradation, essential for supporting wound healing, and that the incorporation of gelatin does not significantly impact the overall degradation kinetics. Instead, it enhances hydrogel swelling and moisture retention capabilities without compromising structural integrity. This suggests that AcHyA-G hydrogels are effective in maintaining their properties and remaining biodegradable under the simulated wound conditions assessed.
To further explore the structural characteristics of AcHyA hydrogels and the impact of gelatin incorporation, we calculated the mesh size, which represents the distance between cross-links or physical entanglements within the polymer network. The mesh size is a critical parameter as it directly affects molecular diffusion, material stiffness, degradation kinetics, and cell–material interactions.25,78 Furthermore, in therapeutically loaded hydrogels, the mesh size plays a role in the retention or release rate of therapeutic molecules, including AMPs, as they may be cleaved by native enzymes. Thus, mesh size is a critical parameter in assessing therapeutic release kinetics as well as ensuring sufficient nutrient transport to support the viability of invading cells.79,80 Mesh size may be altered by adjusting crosslinking density and HyA molecular weight (MW) which modifies the number of chemical and physical crosslinks, including chain entanglements.25,81,82 The mesh size of AcHyA (0% gelatin) and AcHyA-G (0.5% gelatin) hydrogels were estimated using both the Phantom network and Affine network models based on the storage modulus (G′) (Fig. 5A and B). The Phantom model yielded mesh sizes on the order of 80–90 nm, while the Affine model estimated mesh sizes of approximately 110–130 nm, with no statistical difference detected between groups regardless of gelatin concentration (Fig. 5A and B). These values are consistent with previously reported measurements, and confirm the formation of a nanoporous hydrogel network.83,84
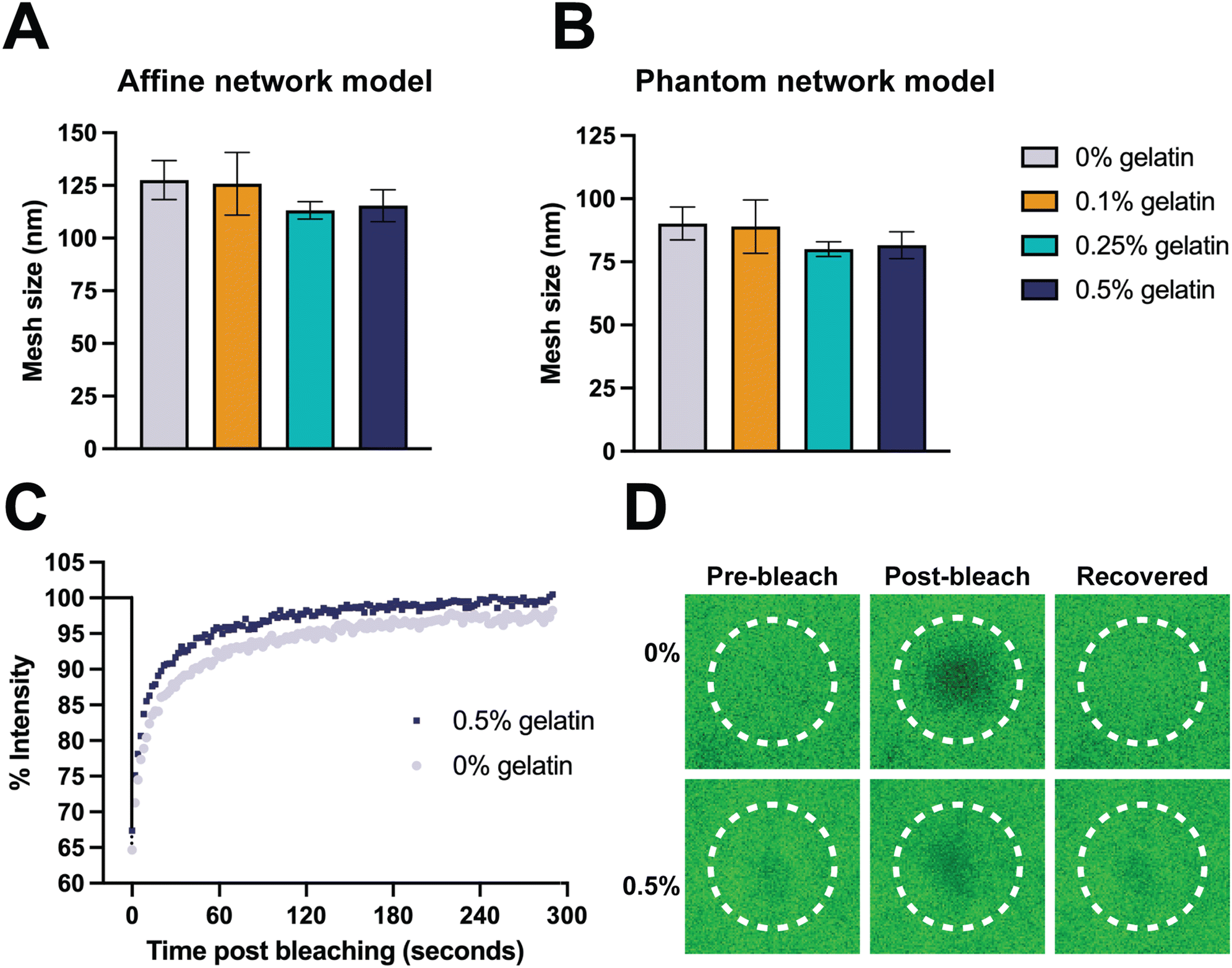 | ||
| Fig. 5 Gelatin concentration has minimal impact on mesh size and molecular diffusion in AcHyA hydrogels. Mesh size was calculated using (A) affine and (B) phantom network models. (C) Recovery of fluorescence after photobleaching with 0% and 0.5% gelatin hydrogels incubated with 70 kDa FITC-Dextran and (D) confocal images of the bleached spot during treatment. Data represents mean ± SD (n = 3). Analysis performed using one-way ANOVA; p < 0.05. | ||
FRAP was employed as a technique to assess molecular mobility and diffusion within the 0% and 0.5% AcHyA-G hydrogels, (Fig. 5C and D). FITC-dextran (70 kDa) was used as a representative biomolecule to assess the molecular diffusion of nutrients and other bioactive molecules through AcHyA hydrogels. FITC-Dextran loaded hydrogels were bleached to approximately 65–70% of their initial fluorescence, and the fluorescence recovery was monitored for 300 s post-bleaching as unbleached molecules diffused back into the region of interest (Fig. 5C and D).85,86 Both AcHyA and AcHyA-G hydrogel formulations, regardless of the presence of gelatin, demonstrated a rapid recovery of fluorescence, reaching 90% of initial intensity within the first 60 seconds and nearly complete recovery by the end of the 300 seconds, indicative of a mobile fraction φmob of 1.87 This rapid recovery of fluorescence indicates a dominant mobile phase favourable for molecular transport and release from the AcHyA and AcHyA-G hydrogels.26 Diffusion coefficients were calculated to be around 0.5 μm2 s−1 for both samples, with no significant difference detected between them. This aligns with data from HyA-based hydrogels, which report diffusion coefficients for smaller FITC-dextran (4 kDa) and mesh sizes ranging from 0.66 to 1.06 μm2 s−1.87 The similar recovery dynamics and molecular release profiles demonstrated between groups revealed that there is no impact of the incorporation of gelatin in AcHyA hydrogels on molecular diffusion. Additionally, the complete fluorescence recovery indicates a mobile fraction allowing molecules with MW up to 70 kDa to readily diffuse within the AcHyA-G hydrogel network. In contrast, hydrogels with incomplete fluorescence recovery indicate immobile fractions (φmob < 1), where molecules become trapped within the network, as demonstrated in HyA-based hydrogels loaded with varying dye charges.87 With a MW of 1940.5 Da, PP4-3.1 peptide would be expected to readily diffuse through and indeed out of the AcHyA-G hydrogels. However, since it is directly conjugated to AcHyA-G by thiol–acrylate reaction, it is therefore not free to diffuse through the hydrogel network. Instead, its release is anticipated to occur only upon degradation of the hydrogel, similar to the behaviour observed with conjugated gelatin (Fig. 2B and C). Furthermore, our results align with the calculated mesh sizes, as expected since the molecular diffusivity is inherently correlated with the hydrogel mesh size, which was similar for both 0% and 0.5% gelatin AcHyA hydrogels (Fig. 5A and B). These results can also be correlated with the degradation observed in Fig. 4D–F, since the mesh size and molecular diffusion were found to be similar across the groups, this means similar diffusion of degrading enzymes (i.e. HyAse) and access to target cleavage sites throughout the hydrogel network.88,89 Overall, molecular mobility and diffusion appeared independent of the gelatin concentrations used in this study, with both AcHyA and AcHyA-G hydrogel formulations demonstrating comparable molecular dynamics. This characteristic is of important consideration since it allows for independent tuning and optimising of the AcHyA-G hydrogel's biophysical and biological properties. Gelatin incorporation did not significantly alter the biophysical properties of AcHyA hydrogels in this study, for several possible reasons. The MW of gelatin used in this study is relatively small (50–100 kDa) compared to the MW of HyA used to form the hydrogel system (1.5 MDa). In addition, the relatively low concentrations of gelatin incorporated (≤0.5%) may not have been sufficient to create significant changes in the hydrogel's mechanical properties. The impact of gelatin on the hydrogel structure is likely dependent on both the concentration and MW of the gelatin, with higher MW or greater gelatin concentrations potentially leading to stronger molecular interactions and greater alterations in stiffness or elasticity.90 In our hydrogel system, the selected formulations allowed for the introduction of biochemical cues without affecting the overall physical properties of the AcHyA hydrogel.
To enhance bioactivity and support cellular adhesion, thiolated gelatin was incorporated into the AcHyA hydrogel. Gelatin, derived from the hydrolysis of collagen, is biocompatible, biodegradable, and contains key peptide sequences known to promote cell adhesion. These sequences, particularly the RGD motif, interact with integrins such as α5β1, α4β1, and αvβ3, which are involved in regulating cell activity, including adhesion, proliferation, and migration.91 By varying the concentration of gelatin, we aimed to tune the cellular response and optimise AcHyA hydrogel composition to support adhesion and spreading of dermal fibroblasts (HDFs) and endothelial cells (iECs) – two critical cell types involved in wound repair and vascularisation.92 As shown in Fig. 6A, gelatin concentration significantly influenced the morphology and behaviour of the HDFs. Lower gelatin concentrations led to fewer, rounded cells, suggesting poor adhesion and limited cell spreading. In contrast, higher gelatin concentrations resulted in enhanced cell spreading and elongation, demonstrating improved adhesion of HDFs. The average cumulative cell area also varied across all formulations: 159.2 ± 869.6, 9901.7 ± 5341.5, 7476.6 ± 1415.3, and 11240.4 ± 3140.2 μm2 for 0%, 0.1%, 0.25%, and 0.5% gelatin formulations, respectively (Fig. 6B). Moreover, the presence of stress fibres in the 0.1%, 0.25%, and 0.5% gelatin formulations indicated that fibroblasts were able to establish cell–matrix interactions (Fig. 6A).93 A similar pattern was observed in iECs (Fig. 6C), albeit with a greater level of cell coverage, where higher gelatin concentrations facilitated the adhesion of a greater number of iECs and the formation of dense cellular networks with greater cell spreading, while lower concentrations of gelatin showed fewer iECs, and minimal network formation. This was evidenced by the average cumulative cell area which ranged from 428.8 ± 294.1, 31813.6 ± 26166.6, 31149.5 ± 15072.8, 27280.2 ± 4778.2 μm2 for 0%, 0.1%, 0.25%, and 0.5% gelatin formulations, respectively (Fig. 6D).
 | ||
| Fig. 6 Increased gelatin concentration in AcHyA hydrogels enhances HDF and iEC adhesion, spreading, and network formation, with 0.5% gelatin showing optimal cell behaviour. Hydrogels with 0%, 0.1%, 0.25%, and 0.5% gelatin were cultured with HDFs and iECs. (A) Images of HDFs and (C) images of iECs, after 7 days in culture, (B) cumulative cell area of HDFs, and (D) cumulative cell area of iECs. Scale bar is 100 μm. Data represents mean ± SD (n = 3). Analysis performed using one-way ANOVA; *p < 0.05, **p < 0.01. | ||
To assess fibroblast bioactivity, we quantified the secretion of bFGF, an essential growth factor involved in tissue repair and angiogenesis.94 bFGF plays a critical role in stimulating fibroblast proliferation, ECM deposition, and tissue regeneration, while also promoting angiogenesis indirectly by activating endothelial cells.95,96 By day 3, bFGF levels varied across all conditions, ranging from 2.32 ± 2.06 pg mL−1 (0.1% gelatin) to 4.17 ± 3.88 pg mL−1 (0.5% gelatin). This decline persisted through day 5, with bFGF levels remaining low in all groups. However, by day 7, bFGF secretion increased again in the 0.5% gelatin formulation (8.54 ± 1.51 pg mL−1), suggesting that higher gelatin content sustains fibroblast activation over time. This increase in bFGF secretion aligns with the improved fibroblast adhesion and spreading observed in the imaging analysis (Fig. 7), reinforcing the role of gelatin in supporting cellular engagement with the hydrogel matrix.
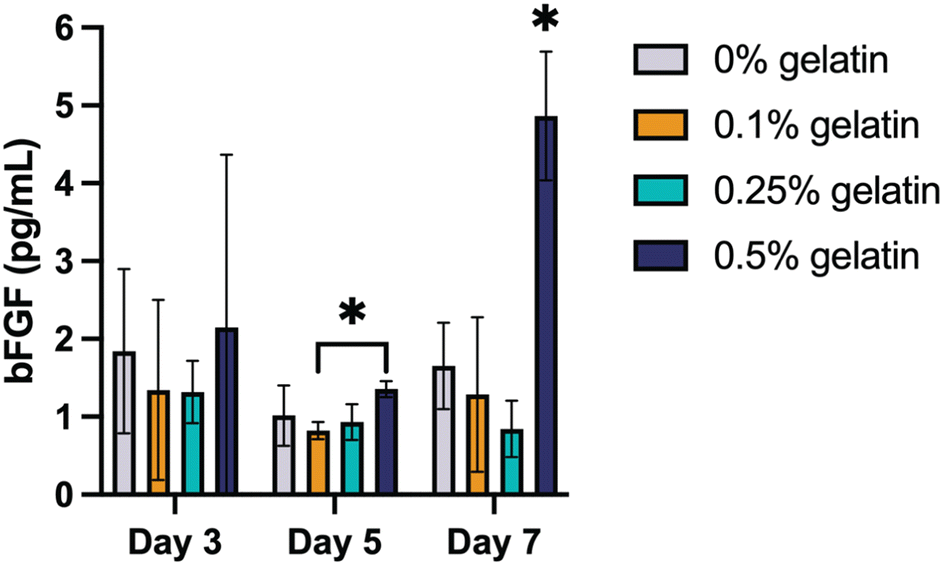 | ||
| Fig. 7 Increased gelatin concentration in AcHyA hydrogels enhances bFGF expression over time in HDFs. bFGF expression was quantified in the supernatant collected at regular intervals from the HDF-seeded AcHyA hydrogels. Data represents mean ± SD (n = 3). Analysis performed using two-way ANOVA; *p < 0.05. | ||
These results suggest that gelatin concentration influences HDFs and iECs behaviour in a concentration-dependent manner. HyA, though known to support cell proliferation, lacks integrin-binding sites, which are essential for cell adhesion.16,97 This absence, combined with the fact that cells were seeded on top of the AcHyA hydrogels rather than encapsulated within them, likely contributed to the poor attachment observed in gelatin-free AcHyA hydrogels. In contrast, the RGD motifs in gelatin enhanced HDF and iEC adhesion and migration.98,99 Our results are consistent with previous studies demonstrating that the incorporation of bioactive components like gelatin significantly improves cell adhesion to AcHyA hydrogels of similar biophysical properties.25 Specifically, the enhanced bFGF secretion observed in the 0.5% gelatin group underscores the role of gelatin in promoting cellular engagement with the hydrogel matrix. The 0.5% gelatin formulation was identified as optimal for supporting the adhesion and spreading of both HDFs and iECs, with minimal impact on the structural characteristics of the AcHyA hydrogel network. This formulation was chosen for further functionalisation, including the incorporation of the AMP PP4-3.1.
As shown in Fig. S2,† tethering the AMP PP4-3.1 onto the hydrogel did not affect the biocompatibility of the latter and maintained support for the adhesion and spreading of both HDFs (Fig. S2A†) and iECs (Fig. S2B†) cells. In terms of biophysical properties, the incorporation of PP4-3.1 into AcHyA hydrogels did not affect their storage modulus (Fig. S3A†), swelling behaviour (Fig. S3B†) or stability across pH levels 6, 7, and 8 (Fig. S4A–C†). However, the peptide-grafted hydrogel showed slower enzymatic degradation at neutral and alkaline pH, likely due to electrostatic interactions of the peptide with HyA that stabilised the hydrogel network, reducing enzyme access to cleavage sites (Fig. S4D–F†).88,89
After confirming the biocompatibility of the AcHyA-AMP hydrogel, we investigated the antimicrobial activity of AcHyA-AMP conjugate against Gram-positive bacterium Staphylococcus aureus and Gram-negative bacterium Escherichia coli. S. aureus and E. coli are common pathogens associated with chronic wounds such as DFUs, often complicating wound healing and increasing infection risk.46,49,100,101 Both bacteria contribute to persistent inflammation and delayed healing in chronic wounds like DFUs, highlighting the need for effective antimicrobial treatments. The antimicrobial potential of AcHyA-AMP conjugate was evaluated using optical density (OD) measurements at 600 nm and culture on agar plates.59 Bacterial suspensions were incubated with either HyA, AcHyA, or AcHyA-AMP for 12 and 24 hours. In the presence of S. aureus, OD600 measurements at 12 hours revealed significant differences in bacterial growth. The OD600 for unmodified HyA reached 0.106 ± 0.004, while AcHyA showed a similar value of 0.120 ± 0.002, with no statistically significant difference between the two. In contrast, the OD600 for the AcHyA-AMP conjugate was markedly lower at 0.065 ± 0.010. After 24 h, the same trend was observed: OD600 values for HyA and AcHyA increased to 0.172 ± 0.008 and 0.157 ± 0.008, respectively, while AcHyA-AMP remained low at 0.069 ± 0.019 (Fig. 8A). To further validate these findings, the resulting solutions (at 24 h) were cultured on agar plates for an additional 24 h. No bacterial colonies formed on the plates treated with the AcHyA-AMP conjugate, contrasting with those treated with HyA and AcHyA, which were covered with bacterial colonies (Fig. 8B). These results suggest that while HyA and AcHyA exhibit a bacteriostatic effect by inhibiting bacterial growth in suspension, AcHyA-AMP demonstrates a bactericidal effect by killing the bacteria.
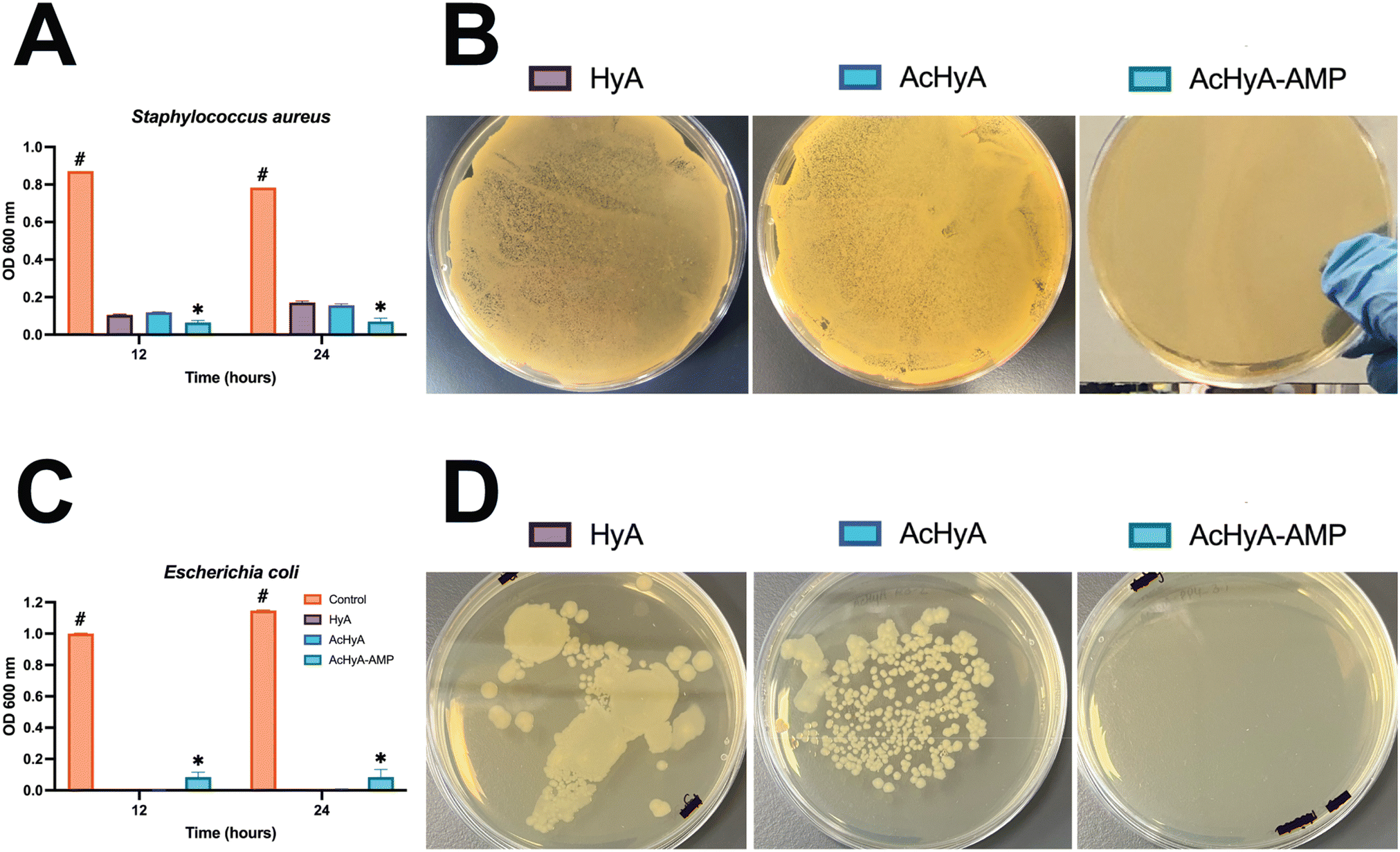 | ||
| Fig. 8 AcHyA-AMP conjugates demonstrate significant bactericidal activity against S. aureus and E. coli, compared to unmodified HyA and AcHyA. The PP4-3.1 AMP was conjugated to AcHyA-G, and the antimicrobial activity of the AcHyA-AMP conjugate was evaluated by (A) OD600 measurements and (B) colony formation after an additional 24 h for S. aureus, and (C) OD600 measurements and (D) colony formation after an additional 24 h for E. coli. Data represent mean ± SD (n = 3). Analysis performed using two-way ANOVA. * indicates statistical differences between groups, and # indicates difference versus the control group. *p < 0.001, #p < 0.001. | ||
This antimicrobial activity of AcHyA-AMP was also assessed against E. coli. At 12 and 24 h, AcHyA-AMP exhibited higher OD600 values (0.084 ± 0.032 and 0.084 ± 0.050) compared to HyA (0.000 ± 0.000 at both time points) and AcHyA (0.001 ± 0.001 and 0.005 ± 0.004) (Fig. 8C). However, despite this apparent increase in OD, no bacterial colonies were recovered from the AcHyA-AMP-treated suspension when plated on agar (Fig. 8D). In contrast, both HyA and AcHyA conditions yielded substantial colony growth (Fig. 8D). Similar to S. aureus, the low OD600 values observed with HyA and AcHyA indicate a bacteriostatic effect, while the absence of viable colonies with AcHyA-AMP suggests a bactericidal action. The discrepancy between OD600 measurements and plating results for AcHyA-AMP likely reflects the presence of cellular debris from membrane disruption and lysis rather than bacterial proliferation.
These findings align with the expected mechanism of action of PP4-3.1 (the AMP used in this study), which exerts antimicrobial effects through direct interactions with bacterial membranes, leading to structural perturbation and cell death. While HyA alone appears to have bacteriostatic effects, as evidenced by the inhibition of bacterial growth in suspension, bacteria were still viable on agar, indicating that HyA alone does not kill bacteria. However, the direct conjugation of the AMP PP4-3.1 to the AcHyA results in the absence of viable bacteria on agar, confirming the bactericidal nature of the AcHyA-AMP conjugate. This study demonstrates a marked increase in the antimicrobial activity of HyA following conjugation of the AMP PP4-3.1, as evidenced by its antimicrobial action against both S. aureus and E. coli. Since the AMP is covalently bound to AcHyA and no hyaluronidase is present in the system, it is unlikely that its antimicrobial activity arises from passive peptide release into the surrounding media. Instead, the observed effect is presumably due to direct contact between the bacteria and the conjugated AMP, where localised interactions with the bacterial membranes lead to disruption and cell death. Indeed, AMPs primarily exert their antimicrobial action through bacterial membrane perturbation or disruption, leading to cell lysis.102,103 In line with this, peptide 3.1 – a cationic, amphipathic, α-helical AMP, has been reported to hypothetically exert its antimicrobial effect by perturbing bacterial membranes, potentially through pore-forming and/or carpet-like mechanisms.104 Previous studies by Gomes et al. demonstrated the effectiveness of the selected AMP, PP4-3.1, against both Gram-positive and Gram-negative bacteria. PP4-3.1 is a hybrid peptide, combining the sequence of the cosmeceutical pentapeptide-4 (PP4) with that of a reported AMP named 3.1.45 Furthermore, PP4-3.1 exhibits MIC values lower than those of most recently reported peptide-based antimicrobials.45
In this study, the AMP was covalently immobilised onto AcHyA via acrylate–thiol Michael addition through the incorporation of a spacer (Ahx) and a cysteine residue. This covalent attachment enhances the AMP's stability and ensures its localisation at the infection site, enabling continuous neutralisation of pathogens and enhanced antimicrobial efficacy over time.42–44 This immobilisation offers several advantages, including reduced peptide aggregation, slower degradation, and enhanced antimicrobial efficacy by maintaining high local concentrations at the hydrogel surface.105 By restricting peptide diffusion, immobilisation optimises its ability to interact with bacterial membranes, which is crucial for sustained antimicrobial action. Unlike free peptides, which can translocate across membranes and target intracellular sites, immobilised peptides primarily exert their antimicrobial effects through surface-driven membrane disruption.106 The presence of the spacer (Ahx) preserves the peptide's amphipathic properties, facilitating its interaction with bacterial membranes, leading to membrane perturbation and subsequent cell lysis.107 This mechanism may also involve changes in bacterial surface electrostatics, where the high concentration of positively charged peptides potentially displaces counterions from the membrane, triggering autolytic enzyme activation and disrupting ionic balance in deeper membrane layers.106
The conjugation of AMP PP4-3.1 into AcHyA hydrogels offers a promising strategy for preventing and treating bacterial infections in chronic wounds. By ensuring the peptide's localisation at infection sites, AcHyA-AMP provides continuous pathogen neutralisation, reducing systemic exposure, thereby reducing any potential side effects. Interestingly, HyA itself has been reported to have antimicrobial properties.108–110 This has been attributed to an ability to disrupt bacterial adhesion and biofilm formation, sequestering essential ions needed for bacterial survival and proliferation, and hindering colonisation through its highly hydrophilic and hydrogel-like properties, which create a physical barrier.108,110,111 Here, the HyA matrix may contribute to the antimicrobial activity by localising bacteria at the AcHyA-AMP conjugate interface, facilitating sustained peptide-bacteria interactions. Our results support the hypothesis that the AMP's antimicrobial activity arises from direct interactions with bacterial membranes, causing membrane destabilisation and cell death. The combined effect of the AMP's electrostatic and hydrophobic interactions, as well as its high local concentration, underscores the potential of AcHyA-AMP conjugates as a potent antimicrobial treatment for chronic wound infections, providing a new approach to improving wound healing and infection management.
Conclusion
We have developed and characterised AcHyA hydrogels functionalised with both gelatin and PP4-3.1 peptide to enhance cell adhesion while providing antimicrobial activity for the treatment of chronic wounds. The addition of gelatin and the AMP was achieved without compromising the biophysical and mechanical properties of the AcHyA hydrogels, which retained their structural integrity and mechanical stability. Gelatin incorporation enhanced swelling behaviour and provided a favourable environment for cellular activity, namely by supporting the adhesion and spreading of HDFs and iECs, with the 0.5% gelatin concentration showing the highest bioactivity. This formulation also promoted fibroblast activation, as evidenced by increased secretion of bFGF, a key growth factor involved in tissue regeneration and angiogenesis. The sustained bFGF production observed in the 0.5% gelatin hydrogel suggests that this formulation supports long-term fibroblast engagement, which may contribute to improved wound healing outcomes. Meanwhile, the conjugation of the AMP PP4-3.1 delivered an AcHyA formulation with bactericidal action on S. aureus and E. coli, two of the most prevalent bacterial pathogens in chronic wounds. Thus, this gelatin and peptide-tethered AcHyA hydrogel demonstrates a local antimicrobial effect, providing a protective barrier against infection while supporting cell adhesion. Given the challenges associated with chronic wound healing, these AcHyA hydrogels, decorated with ECM moieties and AMPs, represent a promising strategy to reduce infection and accelerate recovery in chronic wounds. Future research will involve evaluation in pre-clinical wound models to further assess the efficacy and long-term potential of gelatin and AMP-decorated AcHyA hydrogels in promoting wound healing.Data availability
Data from this study is available from OSF Repository at [https://osf.io/3tyrd/?view_only=950a75755918493395395b93c57fa02e].Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work received support from The European Federation for the Study of Diabetes/Lily European Diabetes Research Programme, Research Ireland – Grant Number 13/RC/2073_P2, and co-funded by the European Union (EU) under MedTrain + Grant Agreement No. 101081457.Thanks are further due to Fundação para a Ciência e a Tecnologia (FCT, Portugal) for funding through project 2023.12331.PEX. The authors would like to thank Dr Brenton Cavanagh for his valuable contributions and technical support for microscopy.
References
- V. Falanga, R. R. Isseroff, A. M. Soulika, M. Romanelli, D. Margolis, S. Kapp, M. Granick and K. Harding, Nat. Rev. Dis. Primers, 2022, 8, 50 CrossRef PubMed.
- K. Raziyeva, Y. Kim, Z. Zharkinbekov, K. Kassymbek, S. Jimi and A. Saparov, Biomolecules, 2021, 11, 700 CrossRef CAS PubMed.
- L. Martinengo, M. Olsson, R. Bajpai, M. Soljak, Z. Upton, A. Schmidtchen, J. Car and K. Järbrink, Ann. Epidemiol., 2019, 29, 8–15 CrossRef PubMed.
- M. Piipponen, D. Li and N. X. Landén, Int. J. Mol. Sci., 2020, 21, 8790 CrossRef CAS PubMed.
- R. Zhao, H. Liang, E. Clarke, C. Jackson and M. Xue, Int. J. Mol. Sci., 2016, 17, 2085 CrossRef PubMed.
- R. G. Frykberg and J. Banks, Adv. Wound Care, 2015, 4, 560–582 CrossRef PubMed.
- E. Darwin and M. Tomic-Canic, Curr. Dermatol. Rep., 2018, 7, 296–302 CrossRef PubMed.
- S. A. Eming, P. Martin and M. Tomic-Canic, Sci. Transl. Med., 2014, 6, 265sr6–265sr6 Search PubMed.
- U. Okonkwo and L. DiPietro, Int. J. Mol. Sci., 2017, 18, 1419 CrossRef PubMed.
- L. Hosty, T. Heatherington, F. Quondamatteo and S. Browne, Mol. Biol. Rep., 2024, 51, 830 CrossRef CAS PubMed.
- M. G. Monaghan, R. Borah, C. Thomsen and S. Browne, Adv. Drug Delivery Rev., 2023, 203, 115120 CrossRef CAS PubMed.
- S. Browne, N. Petit and F. Quondamatteo, Cell Tissue Res., 2024, 395, 133–145 CrossRef CAS PubMed.
- E. Caló and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 65, 252–267 CrossRef.
- S. Correa, A. K. Grosskopf, H. Lopez Hernandez, D. Chan, A. C. Yu, L. M. Stapleton and E. A. Appel, Chem. Rev., 2021, 121, 11385–11457 CrossRef CAS PubMed.
- H. P. Felgueiras, Pharmaceutics, 2023, 15, 258 CrossRef CAS PubMed.
- S. Browne and K. E. Healy, Adv. Drug Delivery Rev., 2019, 146, 155–169 CrossRef CAS PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS PubMed.
- G. D. Prestwich, J. Controlled Release, 2011, 155, 193–199 CrossRef CAS PubMed.
- N. Petit, Y. J. Chang, F. A. Lobianco, T. Hodgkinson and S. Browne, Mater. Today Bio, 2025, 31, 101596 CrossRef CAS PubMed.
- E. L. Pardue, S. Ibrahim and A. Ramamurthi, Organogenesis, 2008, 4, 203–214 CrossRef PubMed.
- D. Park, Y. Kim, H. Kim, K. Kim, Y.-S. Lee, J. Choe, J.-H. Hahn, H. Lee, J. Jeon, C. Choi, Y.-M. Kim and D. Jeoung, Mol. Cells, 2012, 33, 563–574 CrossRef CAS PubMed.
- H. P. Lorenz and N. S. Adzick, West. J. Med., 1993, 159, 350–355 CAS.
- S. Browne, A. K. Jha, K. Ameri, S. G. Marcus, Y. Yeghiazarians and K. E. Healy, PLoS One, 2018, 13, e0194679 CrossRef PubMed.
- A. K. Jha, K. M. Tharp, S. Browne, J. Ye, A. Stahl, Y. Yeghiazarians and K. E. Healy, Biomaterials, 2016, 89, 136–147 CrossRef CAS PubMed.
- S. Browne, S. Hossainy and K. Healy, ACS Biomater. Sci. Eng., 2020, 6, 1135–1143 CrossRef CAS PubMed.
- A. K. Jha, A. Mathur, F. L. Svedlund, J. Ye, Y. Yeghiazarians and K. E. Healy, J. Controlled Release, 2015, 209, 308–316 CrossRef CAS PubMed.
- J. Kim, Y. Park, G. Tae, K. B. Lee, C. M. Hwang, S. J. Hwang, I. S. Kim, I. Noh and K. Sun, J. Biomed. Mater. Res., Part A, 2009, 88A, 967–975 CrossRef CAS PubMed.
- M. N. Collins and C. Birkinshaw, Carbohydr. Polym., 2013, 92, 1262–1279 CrossRef CAS PubMed.
- J. A. Burdick and G. D. Prestwich, Adv. Mater., 2011, 23, H41–H56 CrossRef CAS PubMed.
- J. K. Kim, Y. Xu, X. Xu, D. R. Keene, S. Gurusiddappa, X. Liang, K. K. Wary and M. Höök, J. Biol. Chem., 2005, 280, 32512–32520 CrossRef CAS PubMed.
- H. Wang, O. C. Boerman, K. Sariibrahimoglu, Y. Li, J. A. Jansen and S. C. G. Leeuwenburgh, Biomaterials, 2012, 33, 8695–8703 CrossRef CAS PubMed.
- W.-M. Zhang, J. Käpylä, J. S. Puranen, C. G. Knight, C.-F. Tiger, O. T. Pentikäinen, M. S. Johnson, R. W. Farndale, J. Heino and D. Gullberg, J. Biol. Chem., 2003, 278, 7270–7277 CrossRef CAS PubMed.
- S. Guo and L. A. DiPietro, J. Dent. Res., 2010, 89, 219–229 CrossRef CAS PubMed.
- K. Rahim, S. Saleha, X. Zhu, L. Huo, A. Basit and O. L. Franco, Microb. Ecol., 2017, 73, 710–721 CrossRef PubMed.
- J. Murugaiyan, P. A. Kumar, G. S. Rao, K. Iskandar, S. Hawser, J. P. Hays, Y. Mohsen, S. Adukkadukkam, W. A. Awuah, R. A. M. Jose, N. Sylvia, E. P. Nansubuga, B. Tilocca, P. Roncada, N. Roson-Calero, J. Moreno-Morales, R. Amin, B. K. Kumar, A. Kumar, A.-R. Toufik, T. N. Zaw, O. O. Akinwotu, M. P. Satyaseela and M. B. M. Van Dongen, Antibiotics, 2022, 11, 200 CrossRef CAS PubMed.
- M. McGrath, K. Zimkowska, K. J. Genoud, J. Maughan, J. Gutierrez Gonzalez, S. Browne and F. J. O'Brien, ACS Appl. Mater. Interfaces, 2023, 15, 17444–17458 CrossRef CAS PubMed.
- A. Gomes, L. J. Bessa, I. Fernandes, R. Ferraz, N. Mateus, P. Gameiro, C. Teixeira and P. Gomes, Front. Microbiol., 2019, 10, 1915 CrossRef PubMed.
- C. Ghosh, P. Sarkar, R. Issa and J. Haldar, Trends Microbiol., 2019, 27, 323–338 CrossRef CAS PubMed.
- G. S. Dijksteel, M. M. W. Ulrich, E. Middelkoop and B. K. H. L. Boekema, Front. Microbiol., 2021, 12, 616979 CrossRef PubMed.
- M. L. Mangoni, A. M. McDermott and M. Zasloff, Exp. Dermatol., 2016, 25, 167–173 CrossRef CAS PubMed.
- P. M. Alves, C. C. Barrias, P. Gomes and M. C. L. Martins, Acta Biomater., 2024, 181, 98–116 CrossRef CAS PubMed.
- J. W. Soares, R. Kirby, L. A. Doherty, A. Meehan and S. Arcidiacono, J. Pept. Sci., 2015, 21, 669–679 CrossRef CAS PubMed.
- B. Skerlavaj and G. Boix-Lemonche, Antibiotics, 2023, 12, 211 CrossRef CAS PubMed.
- V. Patrulea, G. Borchard and O. Jordan, Pharmaceutics, 2020, 12, 840 CrossRef CAS PubMed.
- A. Gomes, L. J. Bessa, I. Fernandes, R. Ferraz, C. Monteiro, M. C. L. Martins, N. Mateus, P. Gameiro, C. Teixeira and P. Gomes, Pharmaceutics, 2021, 13, 1962 CrossRef CAS PubMed.
- S. Jain and R. Barman, Indian J. Endocrinol. Metab., 2017, 21, 688 CrossRef CAS PubMed.
- K. E. Macdonald, S. Boeckh, H. J. Stacey and J. D. Jones, BMC Infect. Dis., 2021, 21, 770 CrossRef PubMed.
- R. Edwards and K. G. Harding, Curr. Opin. Infect. Dis., 2004, 91–96 CrossRef PubMed.
- M. Idrees, I. Khan, A. Ullah, S. M. M. Shah, H. Ullah, M. A. Khan, R. Almeer, Z. A. Shah and T. Nadeem, J. King Saud Univ., Sci., 2024, 36, 103320 CrossRef.
- S. L. Natividad-Diaz, S. Browne, A. K. Jha, Z. Ma, S. Hossainy, Y. K. Kurokawa, S. C. George and K. E. Healy, Biomaterials, 2019, 194, 73–83 CrossRef CAS PubMed.
- N. Petit, J. M. Dyer, J. A. Gerrard, L. J. Domigan and S. Clerens, BBA Adv., 2023, 3, 100086 CrossRef CAS PubMed.
- M. Kurisawa, J. E. Chung, Y. Y. Yang, S. J. Gao and H. Uyama, Chem. Commun., 2005, 4312 RSC.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- K. Y. Lee, J. A. Rowley, P. Eiselt, E. M. Moy, K. H. Bouhadir and D. J. Mooney, Macromolecules, 2000, 33, 4291–4294 CrossRef CAS.
- J. E. Mark and B. Erman, Rubberlike elasticity: a molecular primer, Cambridge University Press, 2007 Search PubMed.
- E. I. Altiok, J. L. Santiago-Ortiz, F. L. Svedlund, A. Zbinden, A. K. Jha, D. Bhatnagar, P. Loskill, W. M. Jackson, D. V. Schaffer and K. E. Healy, Biomaterials, 2016, 93, 95–105 CrossRef CAS PubMed.
- S. J. De Jong, B. Van Eerdenbrugh, C. F. Van Nostrum, J. J. Kettenes-van Den Bosch and W. E. Hennink, J. Controlled Release, 2001, 71, 261–275 CrossRef CAS PubMed.
- D. M. Soumpasis, Biophys. J., 1983, 41, 95–97 CrossRef CAS PubMed.
- J. Zhou, H. Zhang, M. S. Fareed, Y. He, Y. Lu, C. Yang, Z. Wang, J. Su, P. Wang, W. Yan and K. Wang, ACS Nano, 2022, 16, 7636–7650 CrossRef CAS PubMed.
- Agilent , E. coli Cell Culture Concentration from OD600 Calculator, https://www.agilent.com/store/biocalculators/calcODBacterial.jsp, (accessed March 3, 2025).
- N. Ahmad, S. N. A. Bukhari, M. A. Hussain, H. Ejaz, M. U. Munir and M. W. Amjad, RSC Adv., 2024, 14, 13535–13564 RSC.
- A. Copling, M. Akantibila, R. Kumaresan, G. Fleischer, D. Cortes, R. S. Tripathi, V. J. Carabetta and S. L. Vega, Int. J. Mol. Sci., 2023, 24, 7563 CrossRef CAS PubMed.
- E. R. Cross, S. M. Coulter, S. Pentlavalli and G. Laverty, Soft Matter, 2021, 17, 8001–8021 RSC.
- Y. Guo, F. Gao, M. Rafiq, B. Yu, H. Cong and Y. Shen, Int. J. Biol. Macromol., 2024, 274, 133494 CrossRef CAS PubMed.
- C. Liang, H. Wang, Z. Lin, C. Zhang, G. Liu and Y. Hu, Front. Bioeng. Biotechnol., 2023, 11, 1310349 CrossRef PubMed.
- E. G. Wiita, Z. Toprakcioglu, A. K. Jayaram and T. P. J. Knowles, ACS Appl. Mater. Interfaces, 2024, 16, 46167–46176 CrossRef CAS PubMed.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- J. Zhu, Biomaterials, 2010, 31, 4639–4656 CrossRef CAS PubMed.
- X. Kong, Q. Tang, X. Chen, Y. Tu, S. Sun and Z. Sun, Neural Regener. Res., 2017, 12, 1003 CrossRef CAS PubMed.
- Z. Xiao, S. Zhao, X. Zhang, G. Wei and Z. Su, Macromol. Mater. Eng., 2022, 307, 2200385 CrossRef CAS.
- S. Tao, S. Zhang, K. Wei, K. Maniura-Weber, Z. Li and Q. Ren, Adv. Healthcare Mater., 2024, 2400921 CrossRef CAS PubMed.
- L. I. F. Moura, A. M. A. Dias, E. Carvalho and H. C. De Sousa, Acta Biomater., 2013, 9, 7093–7114 CrossRef CAS PubMed.
- R. Yang, X. Liu, Y. Ren, W. Xue, S. Liu, P. Wang, M. Zhao, H. Xu and B. Chi, Acta Biomater., 2021, 127, 102–115 CrossRef CAS PubMed.
- D. C. West, I. N. Hampson, F. Arnold and S. Kumar, Science, 1985, 228, 1324–1326 CrossRef CAS PubMed.
- H. Yang, L. Song, Y. Zou, D. Sun, L. Wang, Z. Yu and J. Guo, ACS Appl. Bio Mater., 2021, 4, 311–324 CrossRef CAS PubMed.
- K. L. Aya and R. Stern, Wound Repair Regen., 2014, 22, 579–593 CrossRef PubMed.
- J. L. West and J. A. Hubbell, Macromolecules, 1999, 32, 241–244 CrossRef CAS.
- J. Xia, Z.-Y. Liu, Z.-Y. Han, Y. Yuan, Y. Shao, X.-Q. Feng and D. A. Weitz, Acta Biomater., 2022, 141, 178–189 CrossRef CAS PubMed.
- N. Raina, R. Pahwa, J. Bhattacharya, A. K. Paul, V. Nissapatorn, M. de Lourdes Pereira, S. M. R. Oliveira, K. G. Dolma, M. Rahmatullah, P. Wilairatana and M. Gupta, Pharmaceutics, 2022, 14, 574 CrossRef CAS PubMed.
- B. Liu and K. Chen, Gels, 2024, 10, 262 CrossRef CAS PubMed.
- M. S. Rehmann, K. M. Skeens, P. M. Kharkar, E. M. Ford, E. Maverakis, K. H. Lee and A. M. Kloxin, Biomacromolecules, 2017, 18, 3131–3142 CrossRef CAS PubMed.
- E. Hoch, C. Schuh, T. Hirth, G. E. M. Tovar and K. Borchers, J. Mater. Sci.: Mater. Med., 2012, 23, 2607–2617 CrossRef CAS PubMed.
- B. Ananthanarayanan, Y. Kim and S. Kumar, Biomaterials, 2011, 32, 7913–7923 CrossRef CAS PubMed.
- M. N. Collins and C. Birkinshaw, J. Appl. Polym. Sci., 2008, 109, 923–931 CrossRef CAS.
- K. Braeckmans, L. Peeters, N. N. Sanders, S. C. De Smedt and J. Demeester, Biophys. J., 2003, 85, 2240–2252 CrossRef CAS PubMed.
- J. E. Mealy, C. B. Rodell and J. A. Burdick, J. Mater. Chem. B, 2015, 3, 8010–8019 RSC.
- J. Parlow, A. Rodler, J. Gråsjö, H. Sjögren and P. Hansson, Int. J. Pharm., 2024, 664, 124628 CrossRef CAS PubMed.
- Q. Luo, J. W. Borst, A. H. Westphal, R. M. Boom and A. E. M. Janssen, Food Hydrocolloids, 2017, 66, 318–325 CrossRef CAS.
- N. Petit, J. M. Dyer, M. Richena, L. J. Domigan, J. A. Gerrard and S. Clerens, Int. J. Food Sci. Technol., 2024, 59, 2927–2942 CrossRef CAS.
- S. G. Zambuto, S. S. Kolluru, E. Ferchichi, H. F. Rudewick, D. M. Fodera, K. M. Myers, S. P. Zustiak and M. L. Oyen, J. Mech. Behav. Biomed. Mater., 2024, 154, 106509 CrossRef CAS PubMed.
- P. Dhavalikar, A. Robinson, Z. Lan, D. Jenkins, M. Chwatko, K. Salhadar, A. Jose, R. Kar, E. Shoga, A. Kannapiran and E. Cosgriff-Hernandez, Adv. Healthcare Mater., 2020, 9, 2000795 CrossRef CAS PubMed.
- J. Karam, B. J. Singer, H. Miwa, L. H. Chen, K. Maran, M. Hasani, S. Garza, B. Onyekwere, H.-C. Yeh, S. Li, D. D. Carlo and S. K. Seidlits, Acta Biomater., 2023, 169, 228–242 CrossRef CAS PubMed.
- T. Yeung, P. C. Georges, L. A. Flanagan, B. Marg, M. Ortiz, M. Funaki, N. Zahir, W. Ming, V. Weaver and P. A. Janmey, Cell Motil. Cytoskeleton, 2005, 60, 24–34 CrossRef PubMed.
- J. Li, Y. Zhang and R. S. Kirsner, Microsc. Res. Tech., 2003, 60, 107–114 CrossRef CAS PubMed.
- X. Zhang, X. Kang, L. Ji, J. Bai, W. Liu and Z. Wang, Int. J. Nanomed., 2018, 13, 3897–3906 CrossRef CAS PubMed.
- S. Kanazawa, T. Fujiwara, S. Matsuzaki, K. Shingaki, M. Taniguchi, S. Miyata, M. Tohyama, Y. Sakai, K. Yano, K. Hosokawa and T. Kubo, PLoS One, 2010, 5, e12228 CrossRef PubMed.
- S. Ouasti, P. J. Kingham, G. Terenghi and N. Tirelli, Biomaterials, 2012, 33, 1120–1134 CrossRef CAS PubMed.
- G. Camci-Unal, D. Cuttica, N. Annabi, D. Demarchi and A. Khademhosseini, Biomacromolecules, 2013, 14, 1085–1092 CrossRef CAS PubMed.
- S. Poveda-Reyes, V. Moulisova, E. Sanmartín-Masiá, L. Quintanilla-Sierra, M. Salmerón-Sánchez and G. G. Ferrer, Macromol. Biosci., 2016, 16, 1311–1324 CrossRef CAS PubMed.
- F. L. Sapico, J. L. Witte, H. N. Canawati, J. Z. Montgomerie and A. N. Bessman, Clin. Infect. Dis., 1984, 6, S171–S176 CrossRef PubMed.
- N. Djahmi, N. Messad, S. Nedjai, A. Moussaoui, D. Mazouz, J.-L. Richard, A. Sotto and J.-P. Lavigne, Clin. Microbiol. Infect., 2013, 19, E398–E404 CrossRef CAS PubMed.
- Y. Luo and Y. Song, Int. J. Mol. Sci., 2021, 22, 11401 CrossRef CAS PubMed.
- V. Nizet, Curr. Issues Mol. Biol., 2006, 8, 11–26 CAS.
- S. Kang, H. Won, W. Choi and B. Lee, J. Pept. Sci., 2009, 15, 583–588 CrossRef CAS PubMed.
- F. Costa, I. F. Carvalho, R. C. Montelaro, P. Gomes and M. C. L. Martins, Acta Biomater., 2011, 7, 1431–1440 CrossRef CAS PubMed.
- B. Bechinger and S.-U. Gorr, J. Dent. Res., 2017, 96, 254–260 CrossRef CAS PubMed.
- S. Ghosh, S. Chatterjee and P. Satpati, J. Chem. Inf. Model., 2023, 63, 5823–5833 CrossRef CAS PubMed.
- A. Ardizzoni, R. G. Neglia, M. C. Baschieri, C. Cermelli, M. Caratozzolo, E. Righi, B. Palmieri and E. Blasi, J. Mater. Sci.: Mater. Med., 2011, 22, 2329–2338 CrossRef CAS PubMed.
- F. Zamboni, C. K. Wong and M. N. Collins, Bioact. Mater., 2023, 19, 458–473 CAS.
- G. A. Carlson, J. L. Dragoo, B. Samimi, D. A. Bruckner, G. W. Bernard, M. Hedrick and P. Benhaim, Biochem. Biophys. Res. Commun., 2004, 321, 472–478 CrossRef CAS PubMed.
- P. Pirnazar, L. Wolinsky, S. Nachnani, S. Haake, A. Pilloni and G. W. Bernard, J. Periodontol., 1999, 70, 370–374 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5bm00567a |
| This journal is © The Royal Society of Chemistry 2025 |