
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProximity-induced SuFEx increases the potency of cytosolic nucleotidase inhibitors and reveals a rare example of covalently targeted histidine†
Mikolaj Chrominski *a,
Marcin Warminskib,
Mateusz Kozarskiab,
Dorota Kubackab,
Joanna Panecka-Hofmanb,
Tomasz Spiewlaab,
Mikolaj Zmudzinskia,
Jacek Jemeilitya and
Joanna Kowalska*b
*a,
Marcin Warminskib,
Mateusz Kozarskiab,
Dorota Kubackab,
Joanna Panecka-Hofmanb,
Tomasz Spiewlaab,
Mikolaj Zmudzinskia,
Jacek Jemeilitya and
Joanna Kowalska*b
aCentre of New Technologies, University of Warsaw, Banacha 2c, 02-097 Warsaw, Poland. E-mail: m.chrominski@cent.uw.edu.pl
bDivision of Biophysics, Faculty of Physics, University of Warsaw, Pasteura 5, 02-093 Warsaw, Poland. E-mail: jkowalska@fuw.edu.pl
First published on 23rd April 2025
Abstract
Structure-guided design is one of the most validated solutions for targeting proteins with specific ligands for therapeutic purposes. Nevertheless, it remains challenging to target enzymes with low affinity for their natural ligands and specificities that overlap with those of other proteins. Cytosolic 5′-nucleotidases – involved in the metabolism of nucleic acid derivatives – are an example of such a family. Here we illustrate how precisely designed covalent inhibitors represent a potential solution for selective nucleotidase targeting. We employed the sulfur–fluoride exchange (SuFEx) to develop a covalent inhibitor of cytosolic nucleotidase IIIB (cNIIIB). Using the known inhibitor (7-benzylguanosine monophosphate, Bn7GMP) and computational methods, we designed and synthesized a series of SuFExable inhibitors. One compound indeed covalently bound cNIIIB, which increased the inhibition potency by over 100-fold. The formation of a covalent S–N bond with a non-catalytic His110 residue was confirmed through MS and 15N NMR. The selectivity of the compound in the context of other protein that recognises similar ligands was also confirmed. The study expands the principle of covalent inhibition of nucleotide processing enzymes. It also represents a rare example of histidine tagging by SuFEx. This may facilitate the broader application of SuFEx chemistry in biochemistry and medicinal chemistry.
Introduction
Sulfur–fluorine exchange (SuFEx) is a collective name introduced by Sharpless in 2014 to describe selective transformations of functional groups containing the S(VI)–F bond.1 The strong covalent S(VI)–F bond, despite being generally unreactive, under certain conditions or upon the proper trigger/catalyst, can be activated, transforming fluorine into a good leaving group and enabling reactions with nucleophiles. Due to its versatility, SuFEx has been recognized as a novel “click reaction” in general organic chemistry, medicinal chemistry, and polymer science.2 The latent electrophilicity of sulfonyl fluorides and fluorosulfates can be triggered by an appropriate local environment, which has been rapidly integrated into medicinal and biochemical research.3 Particularly, the forced orientation and interactions in the confined space of protein binding pockets can dramatically enhance the ability of fluorine to act as a leaving group and unlock its reactivity towards neighboring nucleophilic residues.4 This has ignited the development of SuFEx-based covalent binders reacting with nucleophilic aminoacids such as serine/threonine,5–7 tyrosine,8–12 and lysine.13,14 By leveraging this phenomenon, a number of covalent inhibitors, activity-based probes, and other molecular tools for studying protein behaviour were developed.15 A rational strategy based on SuFEx chemistry is typically employed to design efficient and selective covalent probes/inhibitors. This approach usually involves the modification of a known non-covalent ligand that is recognized by the target protein. Here, we present such an approach towards discovering a covalent inhibitor of cytosolic nucleotidase IIIB (cNIIIB).Cytosolic nucleotidases catalyze the dephosphorylation of nucleoside 5′-monophosphates and thereby regulate nucleotide levels in cells.16,17 Targeting cytosolic nucleosidases such as cNII18,19 or cNI20,21 with rationally designed small molecule inhibitors has been widely explored in the literature, e.g., as an anticancer strategy.22,23 However, the structure-guided development of inhibitors is challenging because natural nucleotidase substrates have low affinity for the binding sites (with submillimolar KM values). Consequently, most developed inhibitors show inhibitory activity at micro to millimolar concentrations. Another challenge is that cytosolic nucleotidases have flexible binding sites, broad specificity, and overlapping substrate scope, which may cause potent inhibitors to suffer from poor selectivity.17 We envisaged that these challenges could be surmounted by implementing covalent inhibition, and as a model protein to verify this hypothesis we selected cNIIIB. cNIIIB is one of the most recently discovered 5′-nucleotidases with several unique structural and functional features.24,25 It catalyzes the dephosphorylation of multiple purine and pyrimidine nucleosides and has also pyrophosphatase activity towards some nucleoside diphosphates. One of the most preferred cNIIIB substrates is 7-methylguanosine monophosphate (m7GMP), which is the secondary metabolite arising from mRNA decapping, suggesting a possible role in the degradation of mRNA metabolites.
In a previous study, we prepared multiple potential substrate-inspired cNIIIB inhibitors to identify the lead structure – 7-benzyl guanosine monophosphate (Bn7GMP).26 The crystal structure for the complex of cNIIIB with one of the strongest inhibitors, N7-(3,4-difluorobenzyl)guanosine 5′-monophosphate (3,4-diFBn7GMP),26 revealed the role of benzyl ring in stabilizing the interaction. The study also identified several nucleophilic sidechains near the benzyl residue, potentially available for covalent targeting. Hence, in this work, we explored the possibility of designing covalent SuFEx-based inhibitors targeting the binding site of cNIIIB.
Results and discussion
Structure design and molecular docking
We anticipated that functionalizing the benzyl ring in Bn7GMP with an S(VI)–F-based electrophilic warhead would create candidates for covalent inhibitors of cNIIIB. In the dense network of direct interactions of 3,4-diFBn7GMP with cNIIIB (Fig. 1), the pivotal role is played by Tyr60 and Trp105 that intercalate the guanine motif and the neighbouring hydrophobic cavity that accommodates difluorophenyl ring of the N7-substituent (Fig. 1).26 The phosphate and ribose moieties are positioned by a net of hydrogen bonds that pulls the phosphate away from the catalytic site preventing dephosphorylation. Although the set of interactions that stabilize this complex is well-defined and rather tight, some residues (for example, Tyr60) leave space for ligand adjustment and search for new interactions and structure optimization. This fact and the presence of many nucleophilic sidechains in the binding pocket of cNIIIB and the closest surrounding (Tyr60, Tyr85, Tyr203, Ser58, Ser231, Lys205, His110) encouraged us to take advantage of recent applications of SuFEx chemistry and create SuFEx based covalent inhibitors.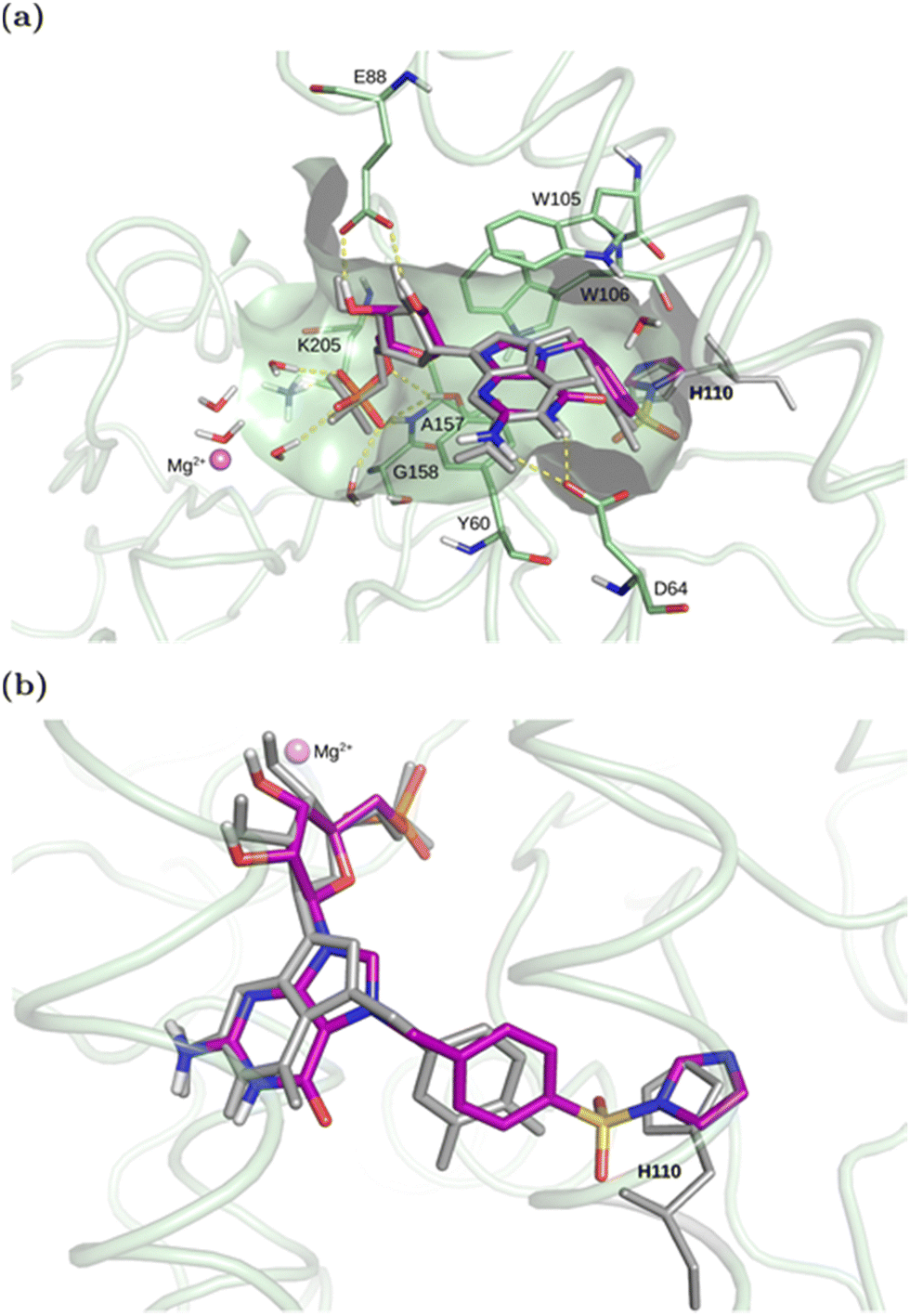 | ||
| Fig. 1 (a) The top-ranked complex of cNIIIB with compound 1 covalently docked through His110 (Nδ hydrogen from the side of the active site) aligned with the crystallized cNIIIB complex with 3,4-diF-Bn7GMP (the ligand shown in grey, PDB code 7ZEG). The key pocket residues are shown as sticks. Yellow dashed lines mark hydrogen bonds between the docked ligand and the enzyme or water. (b) A close-up view of the covalently-docked compound 1 with His110 exposed to the S–N bond. See ESI† for details. | ||
Based on the preliminary analysis of the crystal structure (PDB: 7ZEG),26 the imidazole ring of histidine 110 was found to be the most optimally oriented nucleophile near the benzyl group of 3,4-diF-Bn7GMP. We found this quite an intriguing opportunity since the selective covalent tagging of histidine residues is generally challenging, regarding both structure design and electrophile selection.27,28 The rarity of this approach can be attributed to the absence of electrophiles that favour histidine over other nucleophilic residues29 and difficulties in characterizing the resulting conjugates. The tameable reactivity of the S(VI)–F bond makes histidine targeting possible30 and is attributed to structural factors of the binding pocket and ligand rather than to the intrinsic reactivity of SuFEx-type electrophilic warheads. The initial reports on the SuFEx-type targeting of histidines were regarded as serendipitous phenomena.31,32 However, more recently, there has been a shift towards the deliberate design of SuFEx and SuFEx-type covalent inhibitors targeting histidines,30,33–35 albeit still rare. We anticipated that the basal structural characteristics (N7 substituted guanosine, benzyl group, 5′ phosphate) would retain a high affinity towards cNIIIB, while a correctly oriented SuFExable group on the phenyl ring would be capable of forming a covalent bond with one of the nucleophilic side chains, preferably His110. For the initial recognition of this concept, we designed a potential covalent inhibitor by introducing a sulfonyl fluoride (–SO2F) to position 4 of the phenyl ring in Bn7GMP (compound 1) and performed covalent docking simulations (Fig. 1, Scheme 1, for details see ESI†).
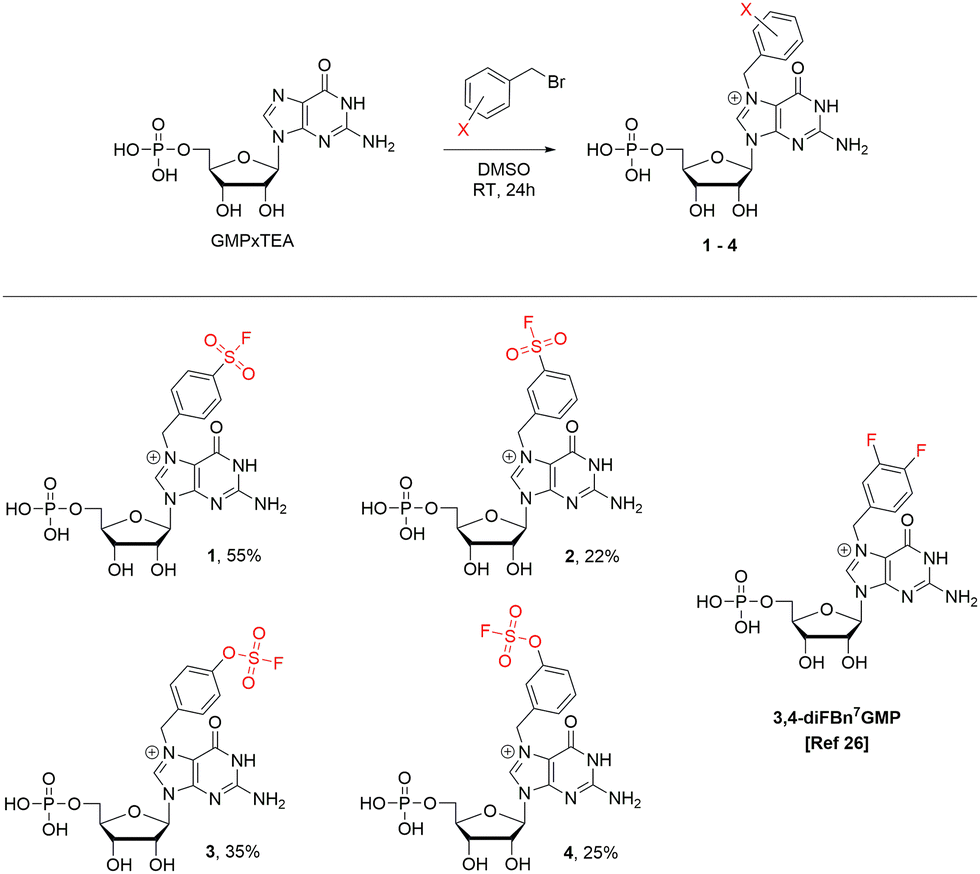 | ||
| Scheme 1 The synthesis of compounds 1–4. | ||
The computations assumed different types of amino acids (see Table S1, ESI†) as attachment points. We found that covalent bonding to Tyr, Ser, Thr, most His (except His110), and Lys residues within the active site is unlikely. In neither of the cases, the docked conformations resembled the binding pose of the crystallized compound 3,4-diFBn7GMP, and the docking scores were very unfavorable when compared to the docked ligand (Fig. S1, ESI†): worse than –6.4 vs. –11.3 kcal mol−1, respectively (Tables S1 and S2, ESI†). The only promising candidate for the covalent binding to ligand 1 was His110. All four possible reaction points on the imidazole ring were considered – Nδ or Nε in the two possible flipped conformations of the imidazole ring (see Fig. S2a, ESI†). The attachment to Nδ from the side of the active site was found to be the most favorable in terms of the docking score (−10.0 kcal mol−1, Table S2, ESI†). In the protein with this His110 state, the compound 1 covalently bonded to His110 adopted a similar conformation as the crystallized compound 3,4-diFBn7GMP, reproducing the key interaction pattern (Fig. 1). The top pose of compound 1 docked to the protein with His110 having Nε hydrogen positioned from the side of the active site (see Fig. S2a, ESI†) also displayed a similar binding mode in the active site (Fig. S2bc, ESI†) and slightly worse docking score (−9.5 kcal mol−1, Table S2, ESI†). For other His 110 states, the docking scores were significantly worse (see Fig. S2a and Table S2 for details, ESI†). Therefore, based on the docking results, His110, with Nδ–H or Nε–H directed towards the active site, was found to be the most likely covalent attachment point of compound 1.
Synthesis
To verify if 1 is capable of covalently binding to His110 in cNIIIB we synthesized it by treating GMP trietylammonium salt with 1.2 equiv. of respective benzyl bromide in DMSO (Scheme 1).Additionally, using a similar approach, we have prepared compound 2, which is a regioisomer of 1 with sulfonyl fluoride moved to position 3 of the phenyl ring, along with compounds 3 and 4, which are their respective analogs possessing the fluorosulfate group (Scheme 1).
We obtained the expected compounds 1–4 in reasonable yields, although the conversions were not full. Due to the reactive nature of SO2F units, the compounds required some non-standard but facile workup/purification procedures (see ESI,† for details). Nonetheless, the purified compounds were robust and stable for months when stored as freeze-dried powders. They were also stable in aqueous solutions at pH range of 6.5–7.5 at r.t.
Biochemical characterization
The ability of 1–4 to inhibit cNIIIB-catalyzed dephosphorylation of m7GMP was investigated using colorimetric detection of phosphate ion (Malachite Green Phosphate Assay, MGP). 3,4-diFBn7GMP was employed as a reference non-covalent inhibitor.26 In the initial experiment, the reaction was started by adding the enzyme to the mixture of the substrate (m7GMP) and inhibitor under investigation. Such conditions enable competition between the substrate and the potential inhibitor to occupy the enzyme′s active site. The results were plotted as a function of ligand concentration (Fig. 2 and Fig. S3, ESI†) and IC50 values were calculated (Table 1 column 3, see ESI,† for details). The tested compounds (except for 2) showed very good inhibitory properties with 4 being the strongest in the series (IC50 = 1.6 ± 0.6 μM), much stronger than the reference compound (IC50 = 10.8 ± 2.8 μM).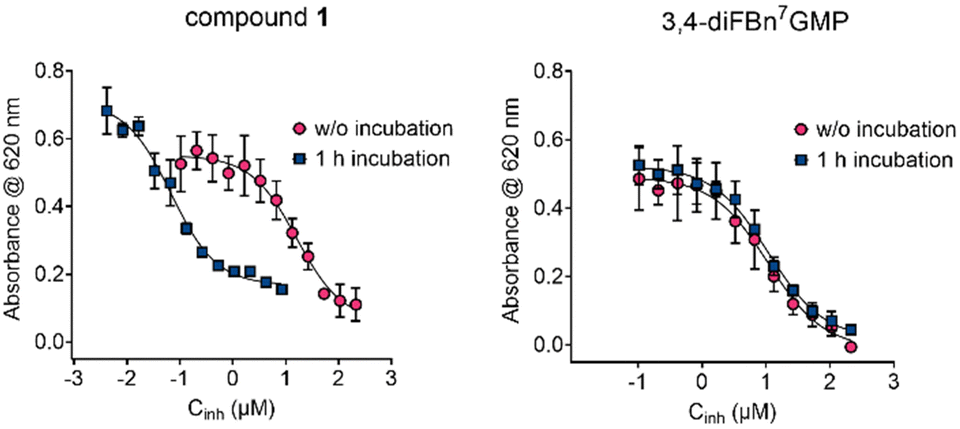 | ||
| Fig. 2 Inhibition curves of cNIIIB for 1 and 3,4-diFBn7GMP recorded with and without preincubation (see ESI,† for details). | ||
| Entry | Compound | IC50 ± SD (μM) w/o incubationb | IC50 ± SD (μM) with 1 h incubationc |
|---|---|---|---|
| a The IC50 experiments were performed in the presence of cNIIIB (80 nM), m7GMP (100 μM), and a 12-point dilution series of the inhibitor. Reactions were run for 45 min at 30 °C in 20 mM HEPES buffer (pH 7.5, 50 mM KCl, 5 mM MgCl2). The phosphate released during incubation was determined using an MGP assay, and IC50 values were determined by fitting a standard dose–response equation to the experimental data. The data present mean values ± SD from triplicate experiments.b m7GMP has been mixed with potential inhibitor followed by cNIIIB addition to start the enzymatic reaction.c cNIIIB enzyme was pre-incubated together with inhibitor for 1 h at 30 °C followed by the addition of m7GMP to start the enzymatic reaction. See text and ESI for details. | |||
| 1 | 1 | 16.2 ± 4.0 | 0.067 ± 0.011 |
| 2 | 2 | >50 | >50 |
| 3 | 3 | 16.9 ± 5.7 | 16.3 ± 2.9 |
| 4 | 4 | 1.6 ± 0.6 | 0.9 ± 0.3 |
| 5 | 3,4-diFBn7GMP (reference) | 10.8 ± 2.8 | 10.9 ± 1.9 |
Since compounds 1–4 were designed to function as covalent competitive inhibitors, we hypothesized that excluding the kinetic factor might alter the results. Hence, in the second set of experiments, the tested compounds were incubated with cNIIIB for 1 h at 30 °C followed by the addition of m7GMP. For compounds 2–4 and the reference, switching the addition order and preincubation with cNIIIB had almost no influence on the IC50 values (Table 1, entries 2–5, Fig. 2 and Fig. S3, ESI†). However, for compound 1, there was a notable shift in the inhibition curve towards lower concentrations (Fig. 2). The calculated IC50 value decreased by over two orders of magnitude, reaching the nanomolar range (Table 1, entry 1, 0.067 ± 0.011 μM).
This dramatic change in the inhibitory activity of 1 under different experimental conditions strongly suggested that it works by a distinct mechanism compared to its analogs 2–4 and the reference. Hence, we used MS experiments to verify if this difference resulted from the SuFEx covalent ligation. cNIIIB was incubated with a 10-fold excess of 1 for 60 minutes at room temperature; the remaining ligand was removed by ultrafiltration (10 kDa cutoff), and the MS spectrum under denaturing conditions of such sample was recorded (Fig. 3, see ESI,† for experimental details).
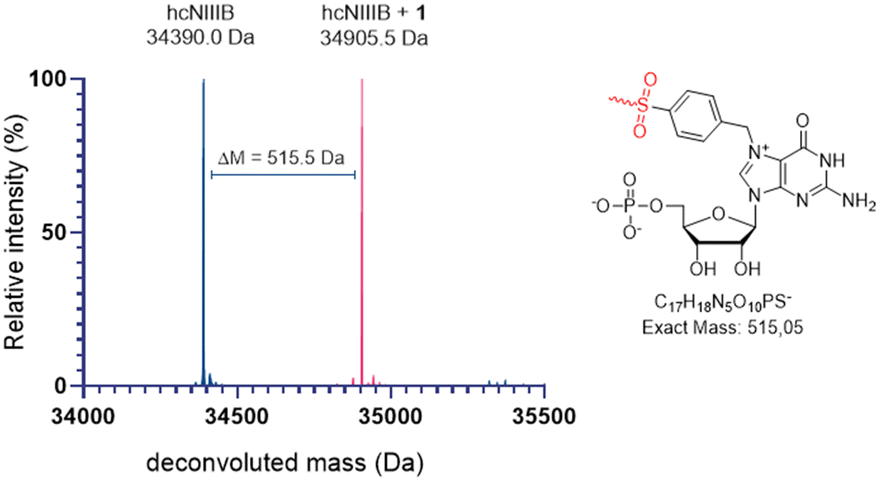 | ||
| Fig. 3 Comparison of deconvoluted MS spectra of apo cNIIIB (blue) and cNIIIB incubated with 1 (pink). | ||
The comparison of MS spectra for apo-cNIIIB and cNIIIB incubated with 1 (34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 390 Da vs. 34905 Da) revealed a clear shift of + 515 Da, which is consistent with covalent attachment of 1 in a SuFEx reaction with the loss of fluoride. An additional experiment in which the MS spectrum of the cNIIIB and 1 mixture was recorded at 10-minute intervals was next conducted (Fig. S4, ESI†). The resulting data indicated that the reaction fits to first-order kinetics, with an estimated rate constant of k ≈ 0.054 min−1 and a half-life of approximately 13 min (Fig. S5, ESI†). In analogous experiments conducted for ligands 2–4, no changes in MS spectra were observed over time. Similar MS experiments were performed to verify the selectivity of 1 towards cNIIIB. N7-Benzyl derivatives of GMP have been previously shown to bind eukaryotic translation initiation factor 4E (eIF4E).36 Thus, we tested the selectivity of SuFEx ligation of 1 in the context of this protein. The MS spectrum recorded for the sample obtained by 30 min incubation of eIF4E with 1 did not differ from that of the apo-protein used as a reference (Fig. S6, ESI†). The experiment results demonstrate that although eIF4E is likely to recognize the Bn7GMP derivatives such as 1,26,36 a SuFEx-type reaction is unfeasible due to the different construction of its binding pocket.
390 Da vs. 34905 Da) revealed a clear shift of + 515 Da, which is consistent with covalent attachment of 1 in a SuFEx reaction with the loss of fluoride. An additional experiment in which the MS spectrum of the cNIIIB and 1 mixture was recorded at 10-minute intervals was next conducted (Fig. S4, ESI†). The resulting data indicated that the reaction fits to first-order kinetics, with an estimated rate constant of k ≈ 0.054 min−1 and a half-life of approximately 13 min (Fig. S5, ESI†). In analogous experiments conducted for ligands 2–4, no changes in MS spectra were observed over time. Similar MS experiments were performed to verify the selectivity of 1 towards cNIIIB. N7-Benzyl derivatives of GMP have been previously shown to bind eukaryotic translation initiation factor 4E (eIF4E).36 Thus, we tested the selectivity of SuFEx ligation of 1 in the context of this protein. The MS spectrum recorded for the sample obtained by 30 min incubation of eIF4E with 1 did not differ from that of the apo-protein used as a reference (Fig. S6, ESI†). The experiment results demonstrate that although eIF4E is likely to recognize the Bn7GMP derivatives such as 1,26,36 a SuFEx-type reaction is unfeasible due to the different construction of its binding pocket.
The results of these experiments corroborate our hypothesis that 1 functions as a covalent inhibitor of cNIIIB, operating through a confined space-induced SuFEx reaction. The covalent targeting of the protein by compound 1, in contrast to the lack of such targeting by other ligands with only subtle structural variations (2, 3), and the substrate selectivity (cNIIIB vs. eIF4E) indicated that the SuFEx reaction takes place exclusively in the binding pocket of cNIIIB and is driven by its defined geometry and microenvironment.
To establish the precise binding mode of 1, we attempted to grow crystals of cNIIIB that were covalently bound to 1. These endeavors were ultimately unsuccessful, and the crystals obtained were of insufficient quality. Subsequently, we conducted LC-MS/MS-based proteomic experiments to identify peptide fragments modified by compound 1. The results obtained for apo-cNIIIB and cNIIIB incubated with 1 revealed no shifts in mass of the peptides obtained after protein digestion. Therefore, these experiments also failed to identify the amino acid in the binding pocket that undergoes SuFEx ligation. However, this result may be consistent with the covalent modifications of histidine, which exhibit instability under acidic conditions commonly employed in peptide analysis.30
Finally, we turned our attention to NMR spectroscopy, which is highly sensitive to any alterations in the chemical structure and does not necessitate crystallization or additional sample treatment that could disrupt the covalent bonding. We anticipated that the sulfonamide modification of the imidazole sidechain of the histidine residue would result in a notable change in the chemical shifts of its hydrogen and nitrogen atoms. To that end, we expressed the uniformly 15N-labeled cNIIIB protein in E. coli cultured in 15N M9 medium supplemented with 15NH4Cl (Fig. S7, ESI†). The incorporation of 15N reached appox. 30% (according to MS, Fig. S8, ESI†), and the MS experiment performed after incubating the labelled enzyme with 1 revealed expected shifts and confirmed covalent binding (Fig. S8, ESI†). Next, we recorded long-range [1H, 15N] HSQC spectra of 15N-labelled cNIIIB preincubated with either 3,4-diFBn7GMP or compound 1 (Fig. 4B). We observed several 15N resonances in the region of 170–230 ppm aligned in a three-peak pattern, characteristic of the ε tautomer of histidine.37 These were virtually identical in both spectra and plausibly correspond to the histidine residues located on the protein surface (Fig. 4A). However, in the case of cNIIIB-1, an additional pair of peaks was observed with δN ≈ 205 and 263 ppm. We assumed this significant shift in the histidine sidechain resonances is related to the covalent N–S linkage. A recently published reports on covalent SuFEx-type ligation of the antiapoptotic Bcl-2 protein hMcl-1 has reported similar observations.34,35 The relative intensity of the observed cross-peaks is similar to the pattern characteristic of the histidine δ tautomer,37 which is consistent with the Nδ substitution suggested by molecular modelling. Interestingly, we were not able to observe the remaining two cross-peaks at δH corresponding to Hε. One possible explanation for this phenomenon is that the proton in question undergoes a faster exchange upon N-sulfonamidation, as it should be significantly more acidic than Hε of unmodified imidazole, analogous to the H8 proton of guanosine methylated at N7.
 | ||
| Fig. 4 (A) Comparison of 3D models of cNIIIB in complexes with 3,4-diFBn7GMP (X-ray structure)26 and compound 1 (molecular modeling) with a close-up view of the active sites. All histidine residues are shown as blue sticks, while His110 from the active site was highlighted in magenta. (B) Overlay of 1H,15N long range (INEPT delay of 12.5 ms) HSQC NMR spectra of uniformly 15N-labeled cNIIIB in the presence of 3,4-diFBn7GMP (blue contours) and compound 1 (pink contours). | ||
Conclusion
In conclusion, we have designed and tested the first covalent inhibitor of cytosolic nucleotidase IIIB (cNIIIB). By analyzing the crystal structure of the cNIIIB complex with noncovalent inhibitor 3,4-diFBn7GMP, we deduced that the perfect fitting of this compound to the catalytic pocket and the orientation of the side chains responsible for complex formation makes an ideal environment for proximity-induced SuFEx reaction. The structure of this inhibitor was redesigned by introducing reactive sulfonyl fluoride and fluorosulfate groups onto the benzyl group and in silico experiments strongly suggested possibility of S–N covalent bond formation between the SuFEx warhead and His110 present on the periphery of the binding pocket. Among the newly designed and synthesized compounds the one possessing pSO2FBn group (compound 1) appeared to be superior with IC50 = 67 nM under conditions facilitating the covalent binding. The MALDI MS experiments confirmed that 1 acts as a covalent inhibitor that operates according to the SuFEx mechanism. 15N NMR experiments validated the formation of a covalent bond between the inhibitor and His110. Overall, our work represents a rare case of nucleotide-based covalent inhibitor that targets an enzyme other than a kinase.38 Moreover, this is one of the few cases of selective histidine-targeting by SuFEx.Nucleotidases represent a class of nucleotide-processing enzymes that are particularly promiscuous. Unfortunately, the analogous function, structural similarities, low affinity for substrates, and overlapping substrate specificity of these enzymes present a significant challenge to the search for selective inhibitors of individual members of this family. Conversely, these minor structural variations create the space for designing selective covalent inhibitors based on the proximity-driven SuFEx reaction. In such a strategy, the precise geometry of ligands is of pivotal importance and has the potential to identify hitherto unanticipated SuFExable targets inside the binding pockets of nucleotidases. We contend that the findings presented herein offer a promising avenue for enhancing the efficacy of substrate-derived inhibitors of nucleotidases. Moreover, it may prove advantageous in the context of other nucleotide-processing enzymes that exhibit relatively low affinity for their native substrates. The approach provides a versatile research platform for the design of inhibitors of known nucleotidases and the identification of novel proteins involved in nucleotide metabolism. Such studies are underway in our laboratory, and the findings will be duly published.
Author contributions
MCh: conceptualization, synthesis, writing – original draft; MW: conceptualization, NMR measurements, writing – original draft; JP-H: computational studies, writing original draft; MK: biophysical studies; DK: conceptualization, protein sample preparation, preliminary biophysical studies; TS: protein preparation; MZ: additional biophysical studies; JJ: conceptualization, funding acquisition, methodology, project administration, supervision, writing-review and editing; JK: conceptualization, funding acquisition, methodology, project administration, supervision, writing – review and editing.Data availability
The data supporting this article including Fig. S1–S8 and Tables S1, S2 (ESI†), experimental details and compounds characterisation have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre Poland (2023/49/B/ST5/02061 to J. J.). J. P. H. acknowledges support from the Faculty of Physics, University of Warsaw ((PP/BF) 501-D111-01-1110102). Computations and modeling were carried out using infrastructure financed by European Funds: POIG.02.01.00-14-122/09.Notes and references
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC.
- S. N. Carneiro, S. R. Khasnavis, J. S. Lee, T. W. Butler, J. D. Majmudar, C. A. W. Ende and N. D. Ball, Org. Biomol. Chem., 2023, 21, 1356–1372 RSC.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- R. Shah, E. De Vita, P. Sathyamurthi, D. Conole, X. Zhang, E. Fellows, E. Dickinson, C. Fleites, M. Queisser, J. Harling and E. Tate, J. Med. Chem., 2024, 67, 4641–4654 CrossRef CAS PubMed.
- O. O. Fadeyi, L. R. Hoth, C. Choi, X. D. Feng, A. Gopalsamy, E. C. Hett, R. E. Kyne, R. P. Robinson and L. H. Jones, ACS Chem. Biol., 2017, 12, 2015–2020 CrossRef CAS PubMed.
- C. Dubiella, H. Cui, M. Gersch, A. Brouwer, S. Sieber, A. Krüger, R. Liskamp and M. Groll, Angew. Chem., Int. Ed., 2014, 53, 11969–11973 CrossRef CAS PubMed.
- W. T. Chen, J. J. Dong, L. Plate, D. E. Mortenson, G. J. Brighty, S. H. Li, Y. Liu, A. Galmozzi, P. S. Lee, J. J. Hulce, B. F. Cravatt, E. Saez, E. T. Powers, I. A. Wilson, K. B. Sharpless and J. W. Kelly, J. Am. Chem. Soc., 2016, 138, 7353–7364 CrossRef CAS PubMed.
- J. Ippolito, H. Niu, N. Bertoletti, Z. Carter, S. Jin, K. Spasov, J. Cisneros, M. Valhondo, K. Cutrona, K. Anderson and W. Jorgensen, ACS Med. Chem. Lett., 2021, 12, 249–255 CrossRef CAS PubMed.
- M. Teng, S. Ficarro, H. Yoon, J. Che, J. Zhou, E. Fischer, J. Marto, T. Zhang and N. Gray, ACS Med. Chem. Lett., 2020, 11, 1269–1273 CrossRef CAS PubMed.
- K. Bum-Erdene, D. Liu, G. Gonzalez-Gutierrez, M. Ghozayel, D. Xu and S. Meroueh, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 7131–7139 CrossRef CAS PubMed.
- E. C. Hett, H. Xu, K. F. Geoghegan, A. Gopalsamy, R. E. Kyne, C. A. Menard, A. Narayanan, M. D. Parikh, S. P. Liu, L. Roberts, R. P. Robinson, M. A. Tones and L. H. Jones, ACS Chem. Biol., 2015, 10, 1094–1098 CrossRef CAS PubMed.
- C. Baggio, P. Udompholkul, L. Gambini, A. Salem, J. Jossart, J. Perry and M. Pellecchia, J. Med. Chem., 2019, 62, 9188–9200 CrossRef CAS PubMed.
- X. Wan, T. Yang, A. Cuesta, X. Pang, T. Balius, J. Irwin, B. Shoichet and J. Taunton, J. Am. Chem. Soc., 2020, 142, 4960–4964 CrossRef CAS PubMed.
- L. H. Jones and J. W. Kelly, RSC Med. Chem., 2020, 11, 10–17 RSC.
- S. Hunsucker, B. Mitchell and J. Spychala, Pharmacol. Ther., 2005, 107, 1–30 CrossRef CAS PubMed.
- V. Bianchi and J. Spychala, J. Biol. Chem., 2003, 278, 46195–46198 CrossRef CAS PubMed.
- M. Meurillon, Z. Marton, A. Hospital, L. Jordheim, J. Béjaud, C. Lionne, C. Dumontet, C. Périgaud, L. Chaloin and S. Peyrottes, Eur. J. Med. Chem., 2014, 77, 18–37 CrossRef CAS PubMed.
- T. Van, A. Hospital, C. Lionne, L. Jordheim, C. Dumontet, C. Périgaud, L. Chaloin and S. Peyrottes, Beilstein J. Org. Chem., 2016, 12, 1476–1486 CrossRef CAS PubMed.
- E. Garvey, G. Lowen and M. Almond, Biochemistry, 1998, 37, 9043–9051 CrossRef CAS PubMed.
- E. Garvey and K. Prus, Arch. Biochem. Biophys., 1999, 364, 235–240 CrossRef CAS PubMed.
- M. Camici, M. Garcia-Gil, R. Pesi, S. Allegrini and M. Tozzi, Cancers, 2019, 11 Search PubMed.
- L. Jordheim and L. Chaloin, Curr. Med. Chem., 2013, 20, 4292–4303 CrossRef CAS PubMed.
- J. Buschmann, B. Moritz, M. Jeske, H. Lilie, A. Schierhorn and E. Wahle, J. Biol. Chem., 2013, 288, 2441–2451 CrossRef CAS PubMed.
- T. Monecke, J. Buschmann, P. Neumann, E. Wahle and R. Ficner, PLoS One, 2014, 9(3), e90915 CrossRef PubMed.
- D. Kubacka, M. Kozarski, M. R. Baranowski, R. Wojcik, J. Panecka-Hofman, D. Strzelecka, J. Basquin, J. Jemielity and J. Kowalska, Pharmaceuticals, 2022, 15, 27 CrossRef PubMed.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- N. Fischer, M. Oliveira and F. Diness, Biomater. Sci., 2023, 11, 719–748 RSC.
- L. Hillebrand, X. Liang, R. Serafim and M. Gehringer, J. Med. Chem., 2024, 67, 7668–7758 CrossRef CAS PubMed.
- J. Che and L. Jones, RSC Med. Chem., 2022, 13, 1121–1126 RSC.
- D. Bullough and W. Allison, J. Biol. Chem., 1986, 261, 5722–5730 CrossRef CAS PubMed.
- K. Harlow and R. Switzer, Biochemistry, 1985, 24, 3360–3361 Search PubMed.
- J. Cruite, G. Dann, J. Che, K. Donovan, S. Ferrao, S. Ficarro, E. Fischer, N. Gray, F. Huerta, N. Kong, H. Liu, J. Marto, R. Metivier, B. Zerfas and L. Jones, RSC Chem. Biol., 2022, 3, 1105–1110 RSC.
- G. Alboreggia, P. Udompholkul, C. Baggio, K. Muzzarelli, Z. Assar and M. Pellecchia, J. Med. Chem., 2024, 67, 8172–8185 CrossRef CAS PubMed.
- G. Alboreggia, P. Udompholkul, E. Atienza, K. Muzzarelli, Z. Assar and M. Pellecchia, J. Med. Chem., 2024, 67, 20214–20223 CrossRef CAS PubMed.
- X. Chen, D. Kopecky, J. Mihalic, S. Jeffries, X. Min, J. Heath, J. Deignan, S. Lai, Z. Fu, C. Guimaraes, S. Shen, S. Li, S. Johnstone, S. Thibault, H. Xu, M. Cardozo, W. Shen, N. Walker, F. Kayser and Z. Wang, J. Med. Chem., 2012, 55, 3837–3851 CrossRef CAS PubMed.
- V. Simplaceanu, J. Lukin, T. Fang, M. Zou, N. Ho and C. Ho, Biophys. J., 2000, 79, 1146–1154 CrossRef CAS PubMed.
- R. Beltman, A. Herppich, H. Bremer and M. Pflum, Bioconjugate Chem., 2023, 34, 1054–1060 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, computational methods, supporting figures and tables, compounds characterisation. See DOI: https://doi.org/10.1039/d5cb00005j |
| This journal is © The Royal Society of Chemistry 2025 |