
Highly sequence-specific, timing-controllable m6A demethylation by modulating RNA-binding affinity of m6A erasers†
Kenko
Otonari
a,
Yuri
Asami
a,
Kosuke
Ogata
 c,
Yasushi
Ishihama
bc,
Shiroh
Futaki
a and
Miki
Imanishi
*a
c,
Yasushi
Ishihama
bc,
Shiroh
Futaki
a and
Miki
Imanishi
*a
aInstitute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: imiki@scl.kyoto-u.ac.jp
bGraduate School of Pharmaceutical Science, Kyoto University, Kyoto 606-8501, Japan
cNational Institute of Biomedical Innovation, Health and Nutrition, Ibaraki, Osaka 567-0085, Japan
First published on 22nd October 2024
Abstract
Recent advancements in tools using programmable RNA binding proteins and m6A-erasers enable sequence-selective and timing-controllable m6A demethylation. However, off-target effects are still a concern. This study addresses the problem by reducing the RNA-binding ability of m6A-erasers. The modulated m6A-erasers achieved sequence-specific and timing-controllable m6A demethylation with minimal off-target activity.
N 6-Methyladenosine (m6A) is one of the most abundant post-transcriptional modifications in eukaryotic RNA, regulating RNA metabolisms such as alternative splicing1 and mRNA stability.2 It is installed by a methyltransferase complex (m6A-writers) and removed by demethylases (m6A-erasers).3–9 Studies manipulating the expression of the m6A-erasers, fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5), revealed their crucial roles in processes like spermatogenesis,10 tumorigenesis,11 adipogenesis,12 and differentiation.13 However, identifying the m6As responsible for the phenotypes remains difficult because overexpression or knockdown of m6A-erasers impacts global m6A levels. To address this, programmable m6A demethylation tools have been developed utilizing sequence-specific RNA binding proteins to guide FTO or ALKBH5 to target m6As.14–16 Recent advancements in these tools enable temporal control of demethylation by associating the RNA-binding proteins and m6A-erasers in response to stimuli.17,18 These advancements allow for dynamic regulation of individual m6As in sequential or stepwise processes such as differentiation, reprogramming, and circadian clock. However, unintended background demethylation due to overexpressed m6A-erasers remains a concern, because the global decrease in m6A levels by overexpressing m6A-erasers in living cells has been reported.6,19 Therefore, engineered m6A-erasers that target specific sequences at a desired timing are highly needed.
m6A-erasers have intrinsic RNA-binding ability, causing off-target binding of the eraser and non-specific m6A demethylation. Reducing this intrinsic RNA-binding ability is a promising approach to mitigate these effects, allowing the m6A-eraser to act selectively through an RNA-binding protein segment that targets specific RNA sequences. A similar approach has been reported by Liu et al. using an m6A-“writer” consisting of catalytically dead Cas13 (dCas13), for targeting the specific RNA sequences, and the METTL3:METTL14 methyltransferase complex lacking the zinc finger RNA-binding domain of METTL3, to weaken its own RNA-binding ability.20 Unlike the METTL3:METTL14 complex, where RNA-binding domains are separate from the active site,21–24 the RNA-binding and active sites of m6A-erasers are structurally interrelated.4,5,25 While reducing RNA-binding ability is effective in minimizing off-target effects, maintaining catalytic activity while weakening RNA-binding ability in m6A-erasers is challenging.
This study aimed to create m6A-erasers for highly sequence-specific m6A demethylation using two m6A-erasers, FTO and ALKBH5, as templates. Their RNA-binding abilities were modulated based on structural data to suppress nonspecific binding to undesirable off-target RNA sequences, resulting in “modulated” RNA-binding (mod) FTO and modALKBH5. For specific RNA segment recognition, they were fused with the RNA-binding protein PUF; a sequence-specific, monomeric RNA-binding protein that is programmable through arrangement of repeated motifs.14,26,27 Importantly, a switching system using FK-506 binding protein (FKBP) and the FKBP-rapamycin-binding domain of FKBP12-rapamycin associated protein (FRB) was installed in the linker to connect the PUF domain and the mod-m6A-erasers, allowing for time-controllable, sequence-specific m6A demethylation (Scheme 1).
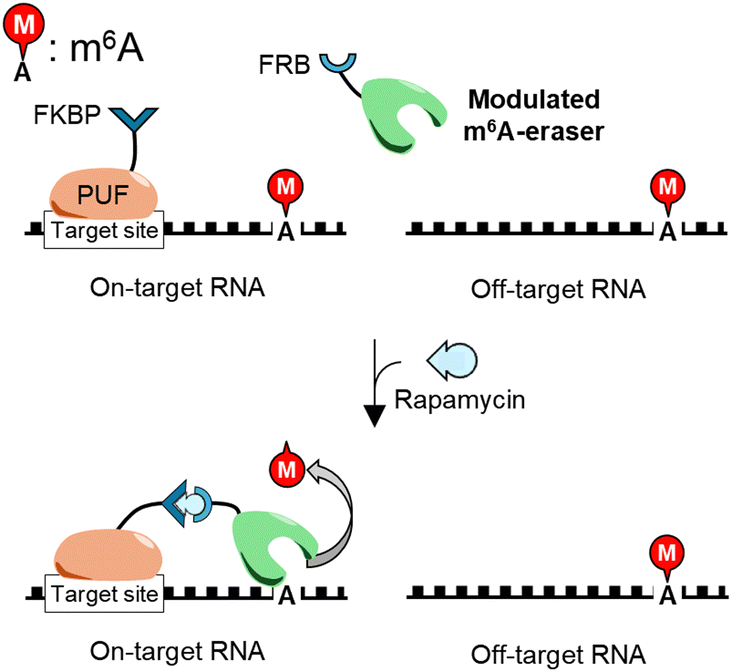 | ||
| Scheme 1 Concept of ligand-inducible sequence-specific m6A demethylation tools with minimal off-target effects. Modulated m6A-erasers do not demethylate in the absence of a ligand but demethylate m6A only nearby the PUF-binding site in the presence of a ligand. | ||
FTO interacts with RNA at a positively charged groove-like region, where basic residues (K88, K213) form a ‘pincer structure’ that holds substrate RNA strands.7,25 To reduce the RNA-binding affinity of FTO itself, alanine substitutions were introduced into FTO at these residues (Fig. 1A). The FTO mutants were fused with PUF, which binds to the UGUAUAUA sequence.14 The m6As near the PUF-binding site and those without a nearby PUF-binding site were designated as on-target and off-target, respectively. The demethylation activity of PUF-fused FTO mutants toward on-target and off-target m6As was examined using the m6A-sensitive endoribonuclease MazF28 (Fig. S1A and Table S1, ESI†). Wild-type FTO-PUF (FP) showed on-target demethylation at concentrations below 500 nM but remarkable off-target demethylation at higher concentrations (Fig. 1B, C and Figure S1B, ESI†). FTOK88A-PUF demethylated both on- and off-target m6As at high concentrations, similar to FP (Fig. 1B and Fig. S1B, C, ESI†), whereas FTOK213A-PUF exhibited slightly reduced off-target demethylation while maintaining on-target RNA demethylation activity (Fig. 1B and Fig. S1B, D, ESI†).
 | ||
| Fig. 1 Alanine scanning on basic amino acids to lower off-target demethylation of FTO-PUF. (A) The two lysine residues (K88 and K213) in wild-type FTO consist of the pincer structure interacting with the phosphate backbone of the ssRNA (upper; PDB ID: 3LFM) and the corresponding region in modFTO (lower; modified from 3LFM). (B) and (C) The demethylation activities to on-target RNA or off-target RNA of wild-type and alanine-substituted FTO-PUFs. (B) 1 μM or (C) indicated concentration of FTO-PUF or each substitution was incubated with 50 nM of the two m6A-modified RNA oligos (on-target: 5′FAM-AUUGUAUAUAUCUAAG(m6A)CAUUUUA-3′, bold: PUF binding site, off-target: 5′TAMRA-AUAUCUCUUGGGUUCUAUUAG(m6A)CAUUUAG-3′) at 25 °C for 1 h. Values and error bars indicate mean ± SEM (n = 3 or 4, Tukey; ***: p < 0.001). | ||
Interestingly, with the combination of these alanine substitutions, FTOK88A/K213A-PUF successfully reduced off-target demethylation even at high concentrations while maintaining on-target m6A demethylation activity (Fig. 1B and C). K88 and K213 may cooperatively contribute to interactions with the substrate RNA. It is plausible that FTOK88A/K213A-PUF relies on PUF for RNA binding and that FTOK88A/K213A exhibits high sequence selectivity by being guided to its target m6A via PUF. We named the alanine substitution FTOK88A/K213A as ‘modulated-RNA-binding FTO (modFTO)’ (Fig. 1A and Fig. S1E, ESI†).
The two alanine substitutions were introduced to reduce the RNA-binding affinity, and then the Kd values of wild-type FTO and modFTO for ssRNA were measured by fluorescence polarization assay with FAM-labeled ssRNA (Table S1, ESI†). Wild-type FTO and modFTO exhibited Kd values of 0.93 ± 0.39 μM and 2.89 ± 0.64 μM, respectively (Fig. 2A and Fig. S2, ESI†).
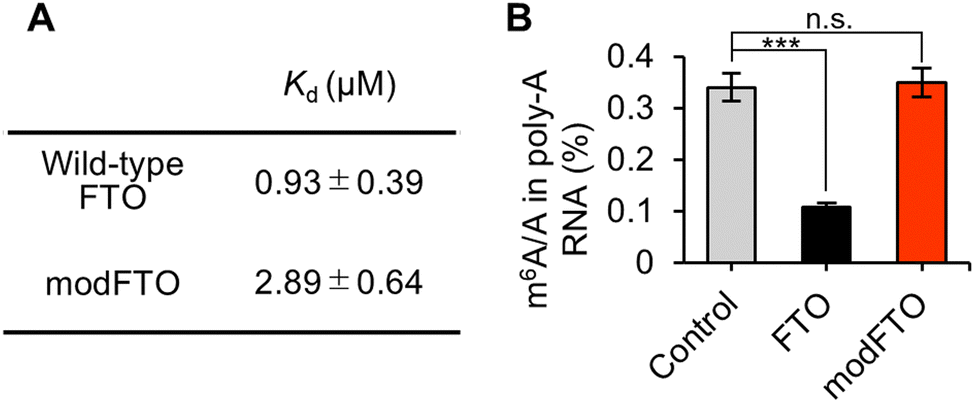 | ||
| Fig. 2 (A) Kd values of wild-type FTO and modFTO analysed by FP assay. The values indicate mean ± SEM (n = 3). (B) Quantification of the m6A/A in poly-A RNA by LC-MS/MS. While wild-type FTO showed a significant decrease of m6A/A compared to the control, modFTO did not show any decrease. Values and error bars indicate mean ± SEM (n = 3, Tukey; n.s.: not significant, ***: p < 0.001). | ||
ModFTO itself, without PUF, showed little demethylation activity towards both on- and off-target m6As, even at high concentrations (Fig. S3, ESI†). We validated the demethylation activity towards mRNA isolated from HEK293T cells. The ratios of m6A to A (m6A/A) were measured using liquid chromatography tandem mass spectrometry (LC-MS/MS). Wild-type FTO demethylated approximately 70% of m6A in mRNA, whereas modFTO did not show any significant demethylation (Fig. 2B and Fig. S4A, ESI†). FP also demethylated m6A at a similar level to wild-type FTO, reflecting PUF-independent demethylation; whereas, modFP showed significantly less demethylation than FP (Fig. S4B, ESI†). Given that modFTO alone rarely demethylates m6A, the observed demethylation by modFP is probably due to PUF–RNA interaction. Although the specificity of the RNA binding protein used should be high enough, the lower background activity of modFTO toward miscellaneous m6A may contribute to the reduced off-target effects of targeted m6A demethylation, even in living cells.
The generality of this design strategy for highly sequence-specific demethylation was verified using another m6A-eraser, ALKBH5. As expected, the fusion of wild-type ALKBH5 with PUF, ALKBH5-PUF (AP), showed on-target demethylation below 500 nM; however, at higher concentrations, it showed off-target activity owing to its innate recognition ability, similar to FP (Fig. 3A and B). Therefore, we aimed to create a modulated RNA-binding ALKBH5 (modALKBH5) for higher sequence-specific m6A demethylation. Although both ALKBH5 and FTO belong to the AlkB subfamily, the pincer structure that was mutated in modFTO is unique to FTO among AlkB subfamily members25 (Fig. S5, ESI†). In addition, the direction of the RNA phosphoribosyl backbone is reversed in ALKBH5 compared to that of the substrate structure of FTO.5 Therefore, we explored the unique residues that contribute to RNA binding in ALKBH5.
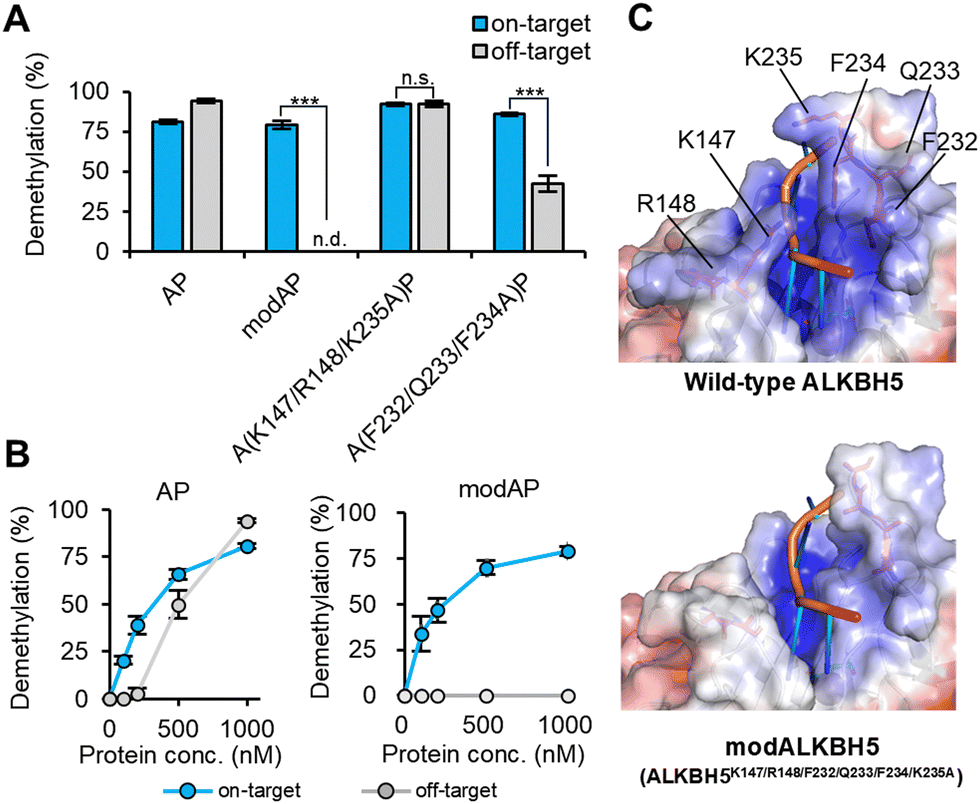 | ||
| Fig. 3 Creation of modAP based on RNA recognition modes. (A) and (B) The sequence-specific demethylation activities of the alanine-substitutions to on- or off-target RNA. (A) 1 μM or (B) indicated concentration of AP, modA(K147/R148/F232/Q233/F234/K235A)P or each substitution were incubated with two m6A-modified RNA oligos at 25 °C for 1 h. (C) The electrostatic surface of wild type ALKBH5 (PDB ID; 7WL0) and modALKBH5 (modified from 7WL0). The six amino acids substituted with alanine in creating modALKBH5 are indicated. Values and error bars indicate mean ± SEM (n = 4, Tukey; n.s., not significant; ***: p < 0.001). | ||
Recent analysis of the crystal structure of ALKBH5 in complex with an m6A-containing ssRNA5 suggested that nucleotide recognition lids (NRL) 2 and βIV-V loop facilitate substrate ssRNA recognition via electrostatic and π–π stacking interactions (Fig. 3C). Among the PUF-fused ALKBH5 mutants with alanine substitutions in these regions, a mutant with six substitutions, ALKBH5K147A/R148A/F232A/Q233A/F234A/K235A-PUF, overcame the off-target activity (Fig. 3A, B and Fig. S6A, ESI†) even at 1 μM and was designated modALKBH5. Partial substitutions at K147/R148/K235, which contribute to electrostatic interactions with RNA, still showed off-target demethylation at 1 μM (Fig. 3A and Fig. S6A, B, ESI†). This result is partly in accordance with a previous report demonstrating that ALKBH5K147A retained its demethylation activity.4 ALKBH5F232A/Q233A/F234A-PUF, which has mutations at the π–π stacking interaction sites of ALKBH5, largely reduced off-target demethylation (∼40%) while retaining the demethylation activity against on-target RNA (Fig. 3A and Fig. S6A, B, ESI†). ModALKBH5, with six alanine substitutions, showed minimal m6A demethylation activity on its own (Fig. S3, ESI†), whereas modALKBH5-PUF (modAP) achieved highly sequence-selective demethylation dependent on the PUF binding site. This suggests that ALKBH5 recognizes its substrate RNAs through multiple residues, including basic and aromatic amino acids. Furthermore, the results for modAP, as well as modFP, demonstrated the effectiveness of the strategy to reduce the RNA-binding ability of the m6A-erasers for highly sequence-specific m6A demethylation with low off-target activity.
Ligand-responsive m6A demethylation of a specific sequence is useful for switching of the methylation state of target sites. A rapamycin-inducible modFTO-PUF (imodFP) was designed using modFTO, which does not actively demethylate m6A alone owing to reduced RNA-binding ability (Fig. S3, ESI†). FK-506 binding protein (FKBP) and FKBP-rapamycin-binding domain of FKBP12-rapamycin associated protein (FRB) are known to dimerize rapidly in the presence of rapamycin.29 In imodFP, modFTO and PUF were fused to FRB and FKBP, forming modFTO-FRB and FKBP-PUF, respectively (Scheme 1). As a result, imodFP (modFTO-FRB and FKBP-PUF) successfully demethylated on-target m6A in the presence of rapamycin but not off-target m6A (Fig. 4A and Fig. S7A, ESI†). By contrast, iFP (wild-type FTO-FRB and FKBP-PUF) demethylated both on- and off-target m6As, independent of rapamycin (Fig. S7A and C, ESI†). This may be because the wild-type FTO-FRB can demethylate any m6As.
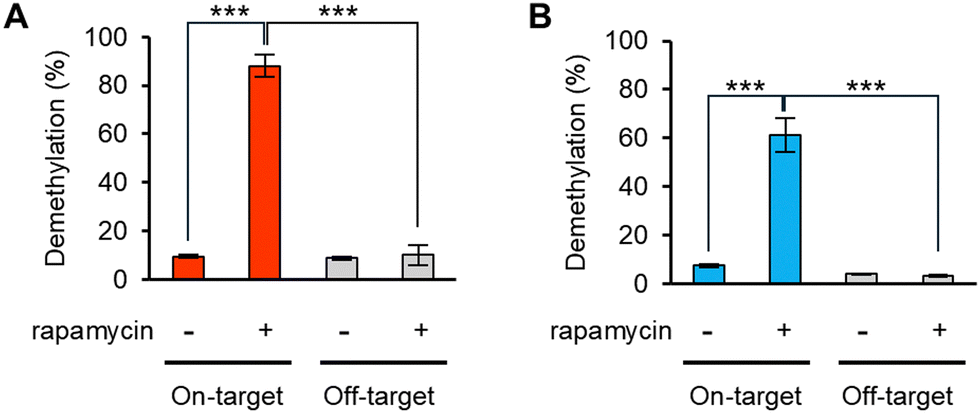 | ||
| Fig. 4 Rapamycin-inducible sequence-specific demethylations of (A) imodFP or (B) imodAP. 1 μM of modFTO-FRB or modALKBH5-FRB and FKBP-PUF were incubated with 50 nM of m6A-modified RNA oligos. Although imodFP and imodAP did not show demethylation activities in the absence of rapamycin, both showed the activities to only on-target RNA in the presence of rapamycin. Values and error bars indicate means ± SEM (n = 3, Tukey's; ***: p < 0.001). | ||
Using the same strategy, we created imodAP (modALKBH5-FRB and FKBP-PUF). imodAP showed on-target demethylation only in the presence of rapamycin (Fig. 4B and Fig. S7B, ESI†), whereas iAP (wild-type ALKBH5-FRB and FKBP-PUF) lacked sequence-specificity or rapamycin-responsiveness (Fig. S7B, D, ESI†). The high sequence- and ligand-dependent demethylation ability of imodFP and imodAP appears to rely on the properties of modFTO and modALKBH5, that do not demethylate on their own unless coupled with PUF (Fig. S3, ESI†). These results suggest that imodFP and imodAP can demethylate a specific m6A at a particular time with high accuracy.
In summary, we created a novel system that regulates the timing of sequence-specific m6A demethylation with minimal off-target effects by fine-tuning the RNA-binding affinity of m6A-erasers. To reduce RNA-binding affinity, residues on the RNA recognition surface, but not in its active site, were substituted with alanine so that they exerted their demethylation ability only when in proximity to RNA. Ultimately, the ligand-inducible imodFP and imodAP were designed using modFTO and modALKBH5, which were activated by rapamycin and demonstrated highly sequence-specific demethylation.
To date, approaches to reduce non-specific substrate recognition of enzymes have been employed in the target RNA methylation system using an m6A-writer lacking the zinc finger domain20 and in highly target-specific genome-editing systems using Cas9 mutants with substituted residues in the substrate recognition or Rec3 domains.30–32 In contrast to these approaches, which use enzymes with independent substrate-recognition and catalytic domains, our strategy could be applied to other epitranscriptomic and epigenetic enzymes; however, optimization is required for each enzyme.
For timing control, targeted m6A demethylation systems that respond to stimuli, such as abscisic acid and light irradiation, have been reported but they utilize wild-type m6A-erasers and do not consider background demethylation.17,18 Our mod-m6A-erasers with reduced RNA-binding ability can be integrated with various systems that respond to stimuli, such as light and chemicals, along with various RNA binding domains, including CRISPR/dCas13, to achieve more accurate timing- and sequence-specific m6A demethylation than wild-type m6A-erasers. Thus, they will be powerful tools to reveal the period-dependent roles of individual m6A modifications in dynamic physiological processes. In cellulo experiments for biological application are underway.
This work was supported by JSPS KAKENHI (Grant No. JP21K19046, 21H05110, 22H02210) to M. I.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- W. Xiao, S. Adhikari, U. Dahal, Y.-S. Chen, Y.-J. Hao, B.-F. Sun, H.-Y. Sun, A. Li, X.-L. Ping, W.-Y. Lai, X. Wang, H.-L. Ma, C.-M. Huang, Y. Yang, N. Huang, G.-B. Jiang, H.-L. Wang, Q. Zhou, X.-J. Wang, Y.-L. Zhao and Y.-G. Yang, Mol. Cell, 2016, 61, 507–519 CrossRef CAS PubMed.
- H. Du, Y. Zhao, J. He, Y. Zhang, H. Xi, M. Liu, J. Ma and L. Wu, Nat. Commun., 2016, 7, 12626 CrossRef CAS PubMed.
- J. Liu, Y. Yue, D. Han, X. Wang, Y. Fu, L. Zhang, G. Jia, M. Yu, Z. Lu, X. Deng, Q. Dai, W. Chen and C. He, Nat. Chem. Biol., 2014, 10, 93–95 CrossRef CAS PubMed.
- C. Feng, Y. Liu, G. Wang, Z. Deng, Q. Zhang, W. Wu, Y. Tong, C. Cheng and Z. Chen, J. Biol. Chem., 2014, 289, 11571–11583 CrossRef CAS PubMed.
- S. Kaur, N. Y. Tam, M. A. McDonough, C. J. Schofield and W. S. Aik, Nucleic Acids Res., 2022, 50, 4148–4160 CrossRef CAS PubMed.
- G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y.-G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS PubMed.
- Z. Han, T. Niu, J. Chang, X. Lei, M. Zhao, Q. Wang, W. Cheng, J. Wang, Y. Feng and J. Chai, Nature, 2010, 464, 1205–1209 CrossRef CAS PubMed.
- K. D. Meyer, Y. Saletore, P. Zumbo, O. Elemento, C. E. Mason and S. R. Jaffrey, Cell, 2012, 149, 1635–1646 CrossRef CAS PubMed.
- D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, M. Salmon-Divon, L. Ungar, S. Osenberg, K. Cesarkas, J. Jacob-Hirsch, N. Amariglio, M. Kupiec, R. Sorek and G. Rechavi, Nature, 2012, 485, 201–206 CrossRef CAS PubMed.
- G. Zheng, J. A. Dahl, Y. Niu, P. Fedorcsak, C.-M. Huang, C. J. Li, C. B. Vågbø, Y. Shi, W.-L. Wang, S.-H. Song, Z. Lu, R. P. G. Bosmans, Q. Dai, Y.-J. Hao, X. Yang, W.-M. Zhao, W.-M. Tong, X.-J. Wang, F. Bogdan, K. Furu, Y. Fu, G. Jia, X. Zhao, J. Liu, H. E. Krokan, A. Klungland, Y.-G. Yang and C. He, Mol. Cell, 2013, 49, 18–29 CrossRef CAS PubMed.
- S. Yang, J. Wei, Y.-H. Cui, G. Park, P. Shah, Y. Deng, A. E. Aplin, Z. Lu, S. Hwang, C. He and Y.-Y. He, Nat. Commun., 2019, 10, 2782 CrossRef PubMed.
- W. Wu, J. Feng, D. Jiang, X. Zhou, Q. Jiang, M. Cai, X. Wang, T. Shan and Y. Wang, Sci. Rep., 2017, 7, 41606 CrossRef CAS PubMed.
- P. J. Batista, B. Molinie, J. Wang, K. Qu, J. Zhang, L. Li, D. M. Bouley, E. Lujan, B. Haddad, K. Daneshvar, A. C. Carter, R. A. Flynn, C. Zhou, K.-S. Lim, P. Dedon, M. Wernig, A. C. Mullen, Y. Xing, C. C. Giallourakis and H. Y. Chang, Cell Stem Cell, 2014, 15, 707–719 CrossRef CAS PubMed.
- K. Shinoda, A. Suda, K. Otonari, S. Futaki and M. Imanishi, Chem. Commun., 2020, 56, 1365–1368 RSC.
- K. Rau, L. Rösner and A. Rentmeister, RNA, 2019, 25, 1311–1323 CrossRef CAS PubMed.
- J. Li, Z. Chen, F. Chen, G. Xie, Y. Ling, Y. Peng, Y. Lin, N. Luo, C.-M. Chiang and H. Wang, Nucleic Acids Res., 2020, 48, 5684–5694 CrossRef CAS PubMed.
- J. Zhao, B. Li, J. Ma, W. Jin and X. Ma, Small, 2020, 16, 1907301 CrossRef CAS PubMed.
- H. Shi, Y. Xu, N. Tian, M. Yang and F.-S. Liang, Nat. Commun., 2022, 13, 1958 CrossRef CAS PubMed.
- J. Wei, F. Liu, Z. Lu, Q. Fei, Y. Ai, P. C. He, H. Shi, X. Cui, R. Su, A. Klungland, G. Jia, J. Chen and C. He, Mol. Cell, 2018, 71, 973–985 CrossRef CAS PubMed.
- C. Wilson, P. J. Chen, Z. Miao and D. R. Liu, Nat. Biotechnol., 2020, 38, 1431–1440 CrossRef CAS PubMed.
- X. Wang, J. Feng, Y. Xue, Z. Guan, D. Zhang, Z. Liu, Z. Gong, Q. Wang, J. Huang, C. Tang, T. Zou and P. Yin, Nature, 2016, 534, 575–578 CrossRef CAS PubMed.
- P. Wang, K. A. Doxtader and Y. Nam, Mol. Cell, 2016, 63, 306–317 CrossRef CAS PubMed.
- P. Śledź and M. Jinek, eLife, 2016, 5, e18434 CrossRef.
- A. Yoshida, T. Oyoshi, A. Suda, S. Futaki and M. Imanishi, Nucleic Acids Res., 2022, 50, 449–457 CrossRef CAS.
- X. Zhang, L.-H. Wei, Y. Wang, Y. Xiao, J. Liu, W. Zhang, N. Yan, G. Amu, X. Tang, L. Zhang and G. Jia, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 2919–2924 CrossRef CAS PubMed.
- K. Shinoda, S. Tsuji, S. Futaki and M. Imanishi, ChemBioChem, 2018, 19, 171–176 CrossRef CAS.
- C.-G. Cheong and T. M. T. Hall, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13635–13639 CrossRef CAS PubMed.
- M. Imanishi, S. Tsuji, A. Suda and S. Futaki, Chem. Commun., 2017, 53, 12930–12933 RSC.
- L. A. Banaszynski, C. W. Liu and T. J. Wandless, J. Am. Chem. Soc., 2005, 127, 4715–4721 CrossRef CAS PubMed.
- I. M. Slaymaker, L. Gao, B. Zetsche, D. A. Scott, W. X. Yan and F. Zhang, Science, 2016, 351, 84–88 CrossRef CAS PubMed.
- B. P. Kleinstiver, V. Pattanayak, M. S. Prew, S. Q. Tsai, N. T. Nguyen, Z. Zheng and J. K. Joung, Nature, 2016, 529, 490–495 CrossRef CAS PubMed.
- J. S. Chen, Y. S. Dagdas, B. P. Kleinstiver, M. M. Welch, A. A. Sousa, L. B. Harrington, S. H. Sternberg, J. K. Joung, A. Yildiz and J. A. Doudna, Nature, 2017, 550, 407–410 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04070h |
| This journal is © The Royal Society of Chemistry 2025 |