
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlugging synthetic DNA nanoparticles into the central dogma of life
Kayla
Neyra
 a,
Sara
Desai
b and
Divita
Mathur
*a
a,
Sara
Desai
b and
Divita
Mathur
*a
aDepartment of Chemistry, Case Western Reserve University, Cleveland, OH 44106, USA. E-mail: dxm700@case.edu
bDepartment of Biochemistry, Case Western Reserve University, Cleveland, OH 44106, USA
First published on 29th November 2024
Abstract
Synthetic DNA nanotechnology has emerged as a powerful tool for creating precise nanoscale structures with diverse applications in biotechnology and materials science. Recently, it has evolved to include gene-encoded DNA nanoparticles, which have potentially unique advantages compared to alternative gene delivery platforms. In exciting new developments, we and others have shown how the long single strand within DNA origami nanoparticles, the scaffold strand, can be customized to encode protein-expressing genes and engineer nanoparticles that interface with the transcription–translation machinery for protein production. Remarkably, therefore, DNA nanoparticles – despite their complex three-dimensional shapes – can function as canonical genes. Characteristics such as potentially unlimited gene packing size and low immunogenicity make DNA-based platforms promising for a variety of gene therapy applications. In this review, we first outline various techniques for the isolation of the gene-encoded scaffold strand, a crucial precursor for building protein-expressing DNA nanoparticles. Next, we highlight how features such as sequence design, staple strand optimization, and overall architecture of gene-encoded DNA nanoparticles play a key role in the enhancement of protein expression. Finally, we discuss potential applications of these DNA origami structures to provide a comprehensive overview of the current state of gene-encoded DNA nanoparticles and motivate future directions.
1. Introduction
Synthetic nucleic acids are actively being pursued as a molecular scaffolding to organize biomolecules and inorganic nanoparticles with high spatial precision and efficiency. Through the bottom-up self-assembly of deoxyribonucleic acids (DNA) and ribonucleic acids (RNA), new nanomaterials for diverse applications are being realized, including for plasmonics, photonic wires, multi-enzyme catalytic cascades, spectroscopy, imaging, and targeted drug delivery systems. Such nanoparticles, built under the umbrella of synthetic DNA nanotechnology, harness the base-pairing rules and the physical double-helical shape of DNA/RNA to create self-assembling materials in high quantities, freely dispersed in solution for versatile downstream uses. The design process for application-specific nucleic acid nanoparticles has become highly facile through computational advancements over the past three decades, leading to formalized design principles and software tools that have broadened its accessibility and applicability.1–4 The leading paradigm in DNA-based nanoparticle design is DNA origami, wherein a long single-stranded DNA (ssDNA), referred to as the scaffold strand (typically a few thousand nucleotides long), is folded into a predefined shape using a specific set of short synthetic oligonucleotides called staple strands.5 This technique captivated the interest of scientists and engineers in the early 2000s by folding DNA into a 100-nanometer large smiley face and an “embossed” map of the world.5 By fine-tuning the scaffold – the fundamental building block of DNA origami nanoparticles (potentially approaching 51 kb in length6) – and staple strand architecture,7 remarkable control over structural size as well as dynamic reconfiguration can be achieved.8 Thus, the construction of expansive DNA architectures has opened new avenues for nanoscale platforms that facilitate the organization of proteins, nanoparticles, fluorescent molecules, and other functional entities across a wide range of applications.9Recent advancements in the biomedical front have seen a convergence in DNA nanoparticle shapes towards biomimetic designs, with three-dimensional wireframe polyhedral nanoparticles accelerating the precise display of proteins and ligands for mimicking viral structures while serving as vaccines.10–13 More excitingly, the evolution of DNA nanotechnology has extended beyond structural programmability and into functional programmability. Functional nucleic acid oligonucleotides, such as DNA aptamers and non-coding RNAs, can be seamlessly integrated into DNA nanostructures through standard base pairing or by appending sequences to staple strand ends, imparting additional functionality.14–16 By integrating functional DNA and RNA with DNA-based delivery vehicles, it is possible to enhance the targeting of therapeutic systems and lower non-specific effects.16
We would like to draw attention to another rapidly growing area within functional nucleic acid nanoparticles that merges DNA's biological function as genetic sequences in the central dogma of life with its architectural capabilities. These functional nucleic acids are gene-encoded DNA origami nanoparticles that are created by encoding the scaffold strand with a protein-expressing gene rather than arbitrary or non-compatible sequences. The resultant gene-encoded nanostructures undergo transcription in bacterial and mammalian systems to produce the corresponding proteins. This enabling technology has been demonstrated for prototypical fluorescent protein expression in cells and cell-free systems as well as therapeutic protein (p53) expression in mice (described below). Protein expression via gene-encoded DNA nanoparticles, combined with the ability of DNA nanoparticles to predictably target cells, underscores its importance and significant potential in transforming gene therapies. Herein, we feature the latest progress in creating gene-encoded DNA nanoparticles as well as their prospective applications. Additionally, we provide insight on the developing field by emphasizing how to encode custom genes into DNA nanoparticles, key considerations identified thus far in design and function, as well as future directions that require attention for translational applications.
2. Techniques to produce custom long single stranded DNA
As described above, DNA origami nanoparticles contain a scaffold strand, which forms the basic backbone of the nanoparticle, and a set of short oligonucleotides referred to as staple strands that induce the scaffold into the desired geometry. Generally, naturally occurring ssDNA is employed as the scaffold strand due to easy and reasonable commercial availability. Naturally occurring ssDNA is sourced from bacteriophage genomes such as M13 and pUC. The M13mp18 (7249 nucleotides long) variant is one of the most widely-used scaffold strands, enabling the assembly of roughly 100-nm large DNA nanostructures.17In gene-encoded DNA origami nanoparticles, the scaffold strand is neither an indigenous bacteriophage genomic sequence nor an arbitrary sequence. Rather, the scaffold strand encodes a genetic sequence that is transcribable by RNA polymerases to produce a protein of interest.18 Applying established DNA origami design principles and available computational tools, nanoparticles can be created with a unique scaffold – one that is specific to any desired genetic sequence. Experimental assembly of gene-encoded DNA origami nanoparticles is similar to traditional (M13mp18-encoded) DNA origami nanoparticles; 5- to 10-fold excess staple strands are combined with the gene-encoded scaffold strand in appropriate Mg2+ supplemented buffer and incubated under a thermal annealing program.19 Canonical genetic sequences are encoded into double stranded plasmids or chromosomes; therefore, strategies to extract or produce gene-encoding single DNA strands from double stranded sources are required for the assembly of gene-encoded DNA nanoparticles.
Many therapeutic genes (important for gene delivery purposes) are a few hundred to several thousand bases pairs long. Until de novo DNA synthesis can make technological advancements to produce high-throughput long (>3 kilobases) ssDNA, well-established molecular biology techniques leveraging enzymes and microbial engineering for ssDNA production need to be translated for custom scaffold strands in DNA nanotechnology. There are other comprehensive reviews explaining the different methods of scaffold production of arbitrary sequences, and some of these have led to the production of long single-stranded genes.20,21 For these methods, the precursor DNA template is most often a gene-encoding plasmid. Plasmids encoding prototypical genes can be acquired from non-profit repositories such as Addgene but molecular biology techniques, such as Golden Gate assembly, can also be used to create a plasmid with a custom gene insert.22 The plasmid can be amplified using bacterial culturing and plasmid extraction, after which it is ready for use as a template. Fig. 1 summarizes the various enzymatic methods and Fig. 2 represents bacteriophage-based methods adopted to produce gene-encoded scaffold strands.
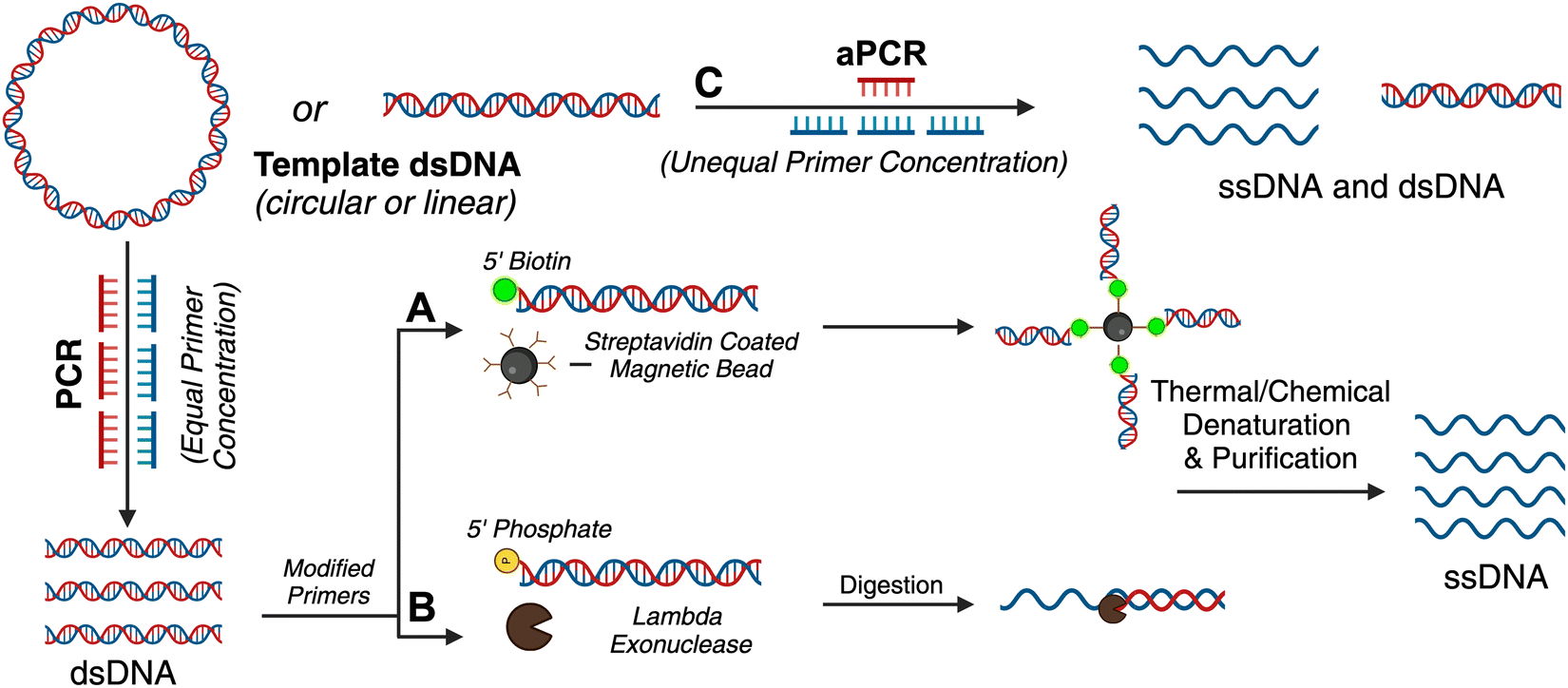 | ||
| Fig. 1 PCR-based methods employed to isolate custom gene encoded scaffolds from dsDNA. Traditional PCR with modified primers enables the isolation of one strand by three different methods. (A) dsDNA labeled with biotin on the 5′ end can be captured by streptavidin coated magnetic beads. The dsDNA is then chemically denatured by NaOH, and the unanchored ssDNA is isolated. (B) dsDNA labeled with a 5′ terminal phosphate group undergoes strand specific digestion by lambda exonuclease to isolate the custom gene encoded scaffold. (C) Asymmetric PCR (aPCR) uses an unequal concentration of primers to selectively amplify the strand of interest. Created in BioRender. Desai, S. (2024), https://BioRender.com/v49e539. | ||
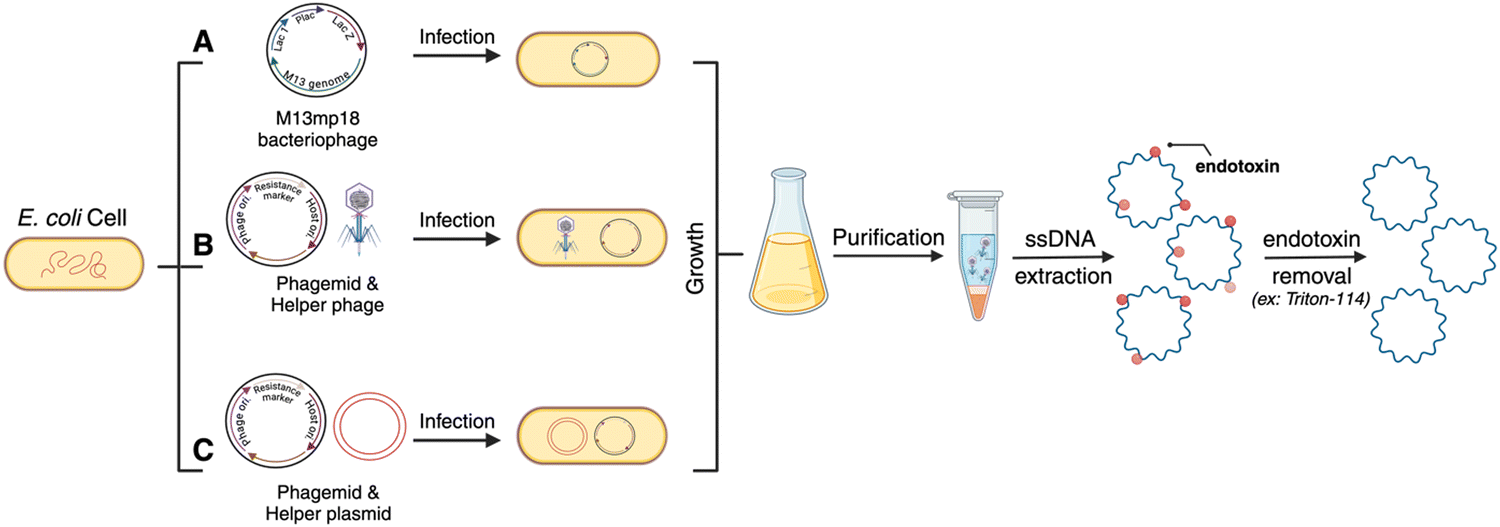 | ||
| Fig. 2 Bacteriophages are used to produce custom gene-encoded scaffolds. (A) The M13mp18 bacteriophage is engineered with the sequence of interest prior to infection of E. coli cells with the phage. The culture is grown and purified to extract the ssDNA scaffolds from the progeny of the bacteriophage. Alternatively, phagemids engineered with the desired sequence can be used alongside a helper phage (B) or plasmid (C) to infect E. coli cells resulting in the similar production of custom ssDNA scaffolds. Created in BioRender. Desai, S. (2024), https://BioRender.com/u58r479. | ||
2.1 Traditional polymerase chain reaction (PCR)
Traditional PCR has been successfully applied for producing gene-encoded scaffold strands. Using a precursor plasmid as the PCR template and equimolar primers, the resultant amplified product can be a double-stranded DNA (dsDNA) gene fragment. If the constituent strands within the dsDNA PCR amplicon (sense and antisense strands) are separated, one can use either or both as the scaffold strand for DNA origami nanoparticles. It is challenging to separate the strands as they lack significant discerning properties – both are equivalent in size, molecular weight, and charge. Typical analytical techniques such as agarose gel electrophoresis and size-exclusion chromatography are unable to separate the two strands unless there is a significant difference in their GC% content; therefore, mechanisms of tagging one of the two strands to impart distinction are required. Some innovative approaches have risen to the challenge and advanced the production of gene-encoded scaffold strands. In 2022, Lin-Shiao et al. carried out traditional PCR to produce a mNeonGreen-encoding scaffold strand from a template plasmid (Fig. 1(A)).18 Forward and reverse primers were designed to enclose this gene, flanking the transcriptional promoter and the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) which falls downstream of the polyadenylation (polyA) signal. In this study, one of the primers was biotinylated on the 5′ end to allow for subsequent isolation of the gene-encoded ssDNA from the dsDNA PCR amplicon. The authors did not explicitly state the specific orientation (sense or antisense) of the template sequence, however, it can be inferred that the biotinylated primer corresponds to the strand that is being removed during the purification process, while the non-biotinylated primer amplifies the strand that is retained for subsequent use as the scaffold strand. After PCR, streptavidin-coated magnetic beads (Dynabeads MyOne Streptavidin C1 beads) were used to precipitate out the biotinylated dsDNA from the PCR reaction mixture, leaving behind the PCR-associated components and salts in solution (Fig. 1(A)). Next, the dsDNA still bound to the beads was chemically dehybridized into its constituent ssDNA strands. This was achieved by resuspending the precipitated beads in a “melt buffer” that consisted of 125 mM sodium hydroxide (NaOH) in ddH2O. Upon precipitating this solution (which contained biotinylated ssDNA still attached to the streptavidin-magnetic beads), the supernatant (containing the desired ssDNA scaffold sequence) was retained. This solution was further purified using SPRI (Solid Phase Reversible Immobilization) beads to clean and concentrate the ssDNA scaffold. SPRI beads work by reversibly binding DNA in the presence of crowding agents (PEG) and high salt concentrations, and variation of these conditions will affect the selectivity of the SPRI beads, allowing for size-specific nucleic acid purification.23 The paramagnetic beads, coated with carboxyl groups, can be easily separated using a magnet and washed to remove contaminants; DNA can be subsequently released by resuspension in a low ionic strength solution.23 The resulting 2716 nt ssDNA sequence was collected and used to anneal an 18-helix bundle DNA origami nanoparticle encoding mNeonGreen (Fig. 3(B)). This construct was subsequently used for visualizing genome integration by CRISPR-mediated homology directed repair (HDR).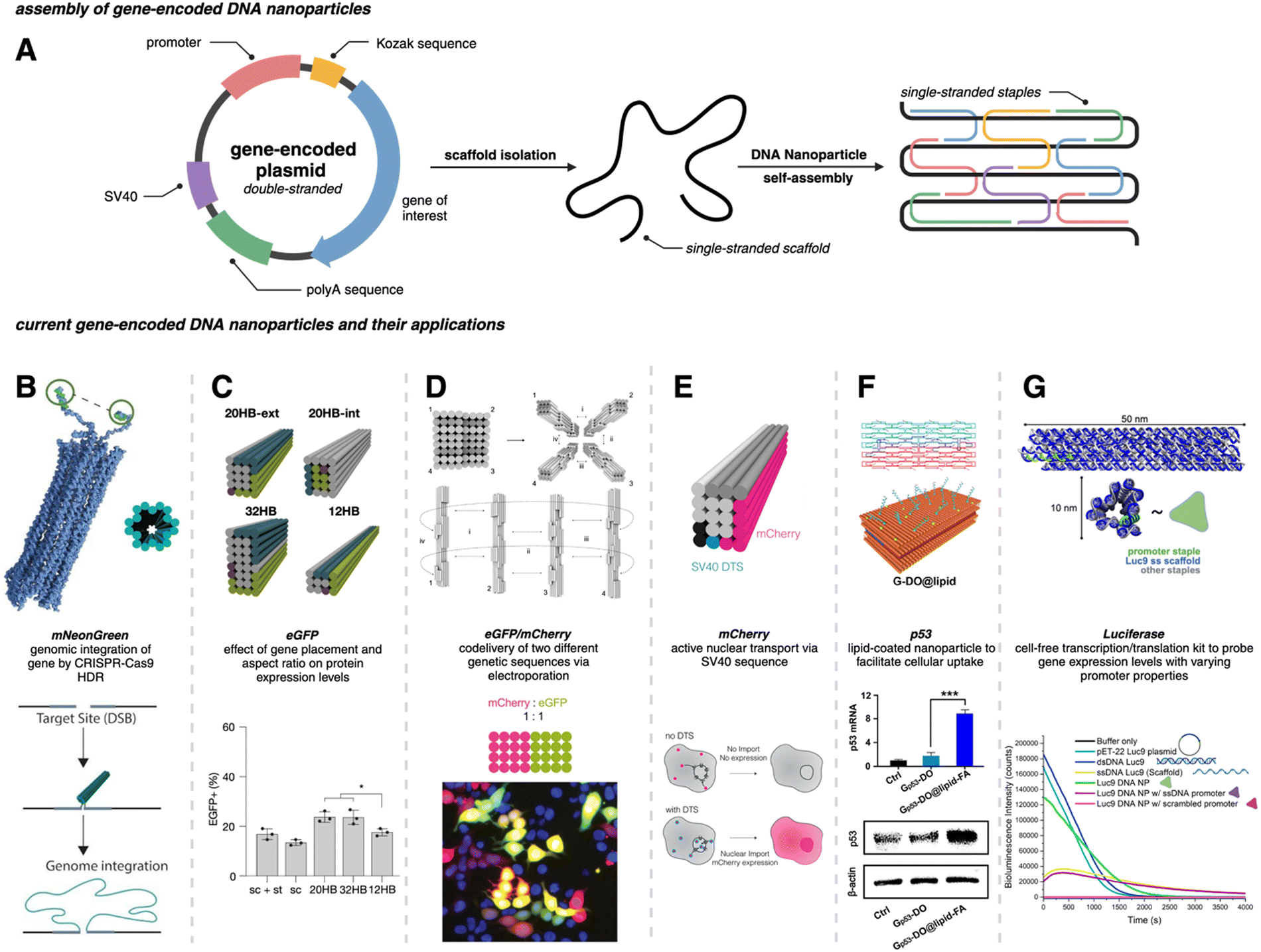 | ||
Fig. 3 Gene-encoded DNA nanoparticles: assembly and current applications. (A) Schematic representation of DNA nanoparticle assembly from a single-stranded gene-encoding scaffold and complementary staple strands. (B) Cylindrical model of 18 hb DNA nanoparticle with extended homology arms for genome targeting. The gene of interest can then be integrated within the genome via CRISPR-Cas9 homology directed repair. Adapted from ref. 18 licensed under CC-BY-NC. (C) Visualization of four DNA nanoparticles with varying aspect ratios. All structures were assembled from the same scaffold strand (sc_EGFP1). eGFP expression levels showed no significant difference between the 20 hb and the 32 hb, but expression as measured by flow cytometry was slightly lower for the 12 hb. Adapted from ref. 22 licensed under CC-BY. (D) Visualization of DNA nanoparticle monomers combining to produce dimer (i), trimer (i and ii), and tetramer (i, ii, iii, and iv) higher-order assemblies. Codelivery of eGFP and mCherry was carried out with a dimeric assembly with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Epifluorescent microscopy allowed for the visualization of mCherry (red), eGFP (green) and coexpression (yellow) in HEK293T cells. Adapted from ref. 22 licensed under CC-BY. (E) 20 hb DNA nanoparticle encoding for the mCherry protein. The bottom schematic is representative of successful nuclear uptake and subsequent protein expression with a DNA targeting sequence (DTS). Adapted from ref. 24 licensed under CC-BY. (F) Hybridization of two halves of the DNA plasmid template (sense and antisense) annealed with corresponding staple strands to make a singular DNA nanoparticle encoding for the p53 anti-tumor gene. The DNA nanoparticle was coated with lipids (DOPE-FA) to facilitate cellular uptake. Western blot analysis showed significant increase in p53 protein production for Gp53-DO@lipid-FA. Adapted with permission from ref. 25. Copyright (2023) American Chemical Society. (G) Depiction of a 12 hb DNA nanoparticle encoding for the luciferase protein. Delayed addition of the NanoGlo substrate allowed for bioluminescence quantification; modifications made to the promoter region domain exhibited a decrease in bioluminescence intensity. Adapted with permission from ref. 26. Copyright (2024) American Chemical Society. Figure created with https://www.biorender.comhttps://www.biorender.com. :1. Epifluorescent microscopy allowed for the visualization of mCherry (red), eGFP (green) and coexpression (yellow) in HEK293T cells. Adapted from ref. 22 licensed under CC-BY. (E) 20 hb DNA nanoparticle encoding for the mCherry protein. The bottom schematic is representative of successful nuclear uptake and subsequent protein expression with a DNA targeting sequence (DTS). Adapted from ref. 24 licensed under CC-BY. (F) Hybridization of two halves of the DNA plasmid template (sense and antisense) annealed with corresponding staple strands to make a singular DNA nanoparticle encoding for the p53 anti-tumor gene. The DNA nanoparticle was coated with lipids (DOPE-FA) to facilitate cellular uptake. Western blot analysis showed significant increase in p53 protein production for Gp53-DO@lipid-FA. Adapted with permission from ref. 25. Copyright (2023) American Chemical Society. (G) Depiction of a 12 hb DNA nanoparticle encoding for the luciferase protein. Delayed addition of the NanoGlo substrate allowed for bioluminescence quantification; modifications made to the promoter region domain exhibited a decrease in bioluminescence intensity. Adapted with permission from ref. 26. Copyright (2024) American Chemical Society. Figure created with https://www.biorender.comhttps://www.biorender.com. | ||
Wu et al. used a similar approach where one of the primers was modified with a biotin tag for streptavidin magnetic bead-based purification after PCR, but they also added terminal DNA loops to the primer 5′ ends.25 The terminal DNA loop is known to enhance stability of linear dsDNA fragments and protect against nucleases.27 This approach was used to generate both green fluorescent protein (GFP) and p53 encoded scaffold strands that were assembled into two different DNA rectangular nanostructures with varying dimensions (42 nm and 74 nm on one side, respectively). After pulldown of the dsDNA with the magnetic beads, NaOH was again used for separating the two constituent strands. In this work, both antisense and sense strands were recovered from the supernatant (two biotinylated primers were used independently) and subsequently used as scaffold strands. The sense and antisense scaffold strands were each mixed with their respective staple strands in the ratio of 1:10 in 1 × TAE/Mg2+ buffer and thermally annealed. The resulting DNA origami monomers were then hybridized with each other through base pairing to create the gene-encoded DNA origami nanoparticle. Structural integrity was confirmed through agarose gel electrophoretic analysis and AFM images. Aside from biotin tags, the scaffold strand could be separated from its complementary strand using specific nucleases (Fig. 1(B)).
2.2 Asymmetric PCR (aPCR)
As mentioned above, traditional PCR is a well-established technique to amplify dsDNA copies from a template DNA using a DNA polymerase enzyme (Fig. 1(A)). PCR uses equal proportion of forward and reverse primers and exponentially amplifies a DNA template into more dsDNA copies (the sense and antisense pair of strands). Through aPCR, one can disproportionately add an excess of one of the primers (forward or reverse) in the reaction mixture to amplify one of the two strands within the template DNA in higher quantities. As a result, one can achieve two PCR products, one being the amplified dsDNA and the other – which is of interest as a scaffold strand – being the amplified ssDNA (Fig. 1(C)). The precursor DNA template can be either double- or single-stranded for aPCR to work correctly, and the desired ssDNA is typically purified using agarose gel electrophoresis extraction.2Custom scaffold strand generated via aPCR has exponentially increased the impact on DNA nanotechnology. This technique helped realize wireframe polyhedral DNA origami structures in a variety of sizes and shapes, all derived from the same M13mp18 ssDNA template.2 The Veneziano group has demonstrated that use of modified nucleotide triphosphates (dNTPs) in the aPCR reaction in place of canonical dNTPs can introduce different functionalities to the scaffold strand, such as amine-modified dCTP (NH2-dCTP).28 Thus, the programmable landscape of a DNA origami nanostructure is doubled where the staples can be chemically modified as usual but the scaffold can also have programmed functionality such as different densities of thiol groups, fluorescent molecules, and phosphorothioate modifications.
For gene-encoded DNA nanostructures, aPCR has proven useful for synthesizing a gene-encoding scaffold strand from a precursor plasmid or synthetic dsDNA gene fragment.29 The Henderson group used aPCR-generated scaffold strand to encode GFP into a cylindrical helix bundle DNA origami nanoparticle. This study assessed variations in architectures of their structure by tuning the position of the promoter region of the gene as well as changing the number of staples used to fold the nanostructure. The gene cassette included a T7 promoter, a GFP gene, and a poly A signal. It should be noted that a CMV enhancer and promoter were also included in the scaffold makeup, but these elements were not the focus in this study. aPCR followed by gel extraction purification was used to produce two variants of the ssDNA scaffold (encoding the antisense strand) – one that was inclusive of the T7 promoter and one that was not. In total, five nanostructures were designed, all following the same overall shape with a consistent crossover pattern. The designs were each folded with a varying number of staples and are identified as follows: T7GHL PO (promoter only in duplex form), T7GHL HS (folded with half set of staples), T7GHL FS (folded with full set of staples), T7GHL BP (promoter buried on internal portion of structure), and T7GHL-T7 (folded with scaffold that lacks promoter region). Apart from the T7GHL BP, the promoter of each construct is linear, with no crossovers, and is adjacent to the cylindrical core. To assess gene expression efficiency in each of the constructs, in vitro transcription (IVT) analysis was performed using a commercial T7 polymerase kit. Messenger RNA (mRNA) production of each variant was qualitatively assessed via electrophoretic analysis, and it was found that all DNA nanoparticles yielded RNA products that were identical in size, except for the construct that lacked the T7 promoter sequence in the scaffold. Additionally, there was an inverse correlation between crossover density of the structure and RNA production – meaning that T7 polymerase can transcribe through crossovers, however, this was quantitatively reduced as compactness of the structure increased. This study suggests that T7 RNA polymerase can complete transcription of a gene-encoded DNA nanoparticle despite high crossover density and limited access to the promoter.
In a recent work by us, we created a luciferase-encoding DNA nanostructure where the scaffold strand was synthesized using aPCR.26 We used a pET-22 Luc9 plasmid as the aPCR precursor DNA template in which Renilla luciferase protein (Luc9) was expressed under a bacterial T7 promoter.26 First, the transcription coding (sense) and template (antisense) strands within the plasmid were identified, based on which an excess of forward primer was added for selective amplification of the template strand. Primers were designed to flank the promoter and genetic sequence on the plasmid, but primer design was tuned based on the desired length of the scaffold strand. The Luc-encoding aPCR ssDNA product was purified to remove the dsDNA “byproduct” and other aPCR-related contaminants using gel extraction and applied in assembling a 12-helix bundle DNA origami structure (Fig. 3(G)). This nanostructure was fully compatible with in vitro cell-free transcription translation systems and produced a functional luciferase protein which could catalyze light upon the addition of luciferase substrate (vide infra).
As mentioned above, ssDNA is generally purified via agarose gel electrophoresis after aPCR synthesis. Unfortunately, gel extraction leads to low yield (∼16%) and considerable loss of material in the agarose gel.30 To compound the loss of sample, a second round of purification is a requirement after the DNA origami nanoparticle is assembled to remove excess staple strands. Therefore, custom-scaffolded DNA nanoparticle synthesis is currently a two-step process with significant loss of the scaffold strand in the first step. Notable size-exclusion techniques (other than gel extraction), such as fast protein liquid chromatography, are difficult to employ because they tend to significantly dilute the sample during fraction elution and require additional concentration steps.31 Therefore, new ways to improve the scaffold recovery post aPCR are needed. We have shown that photocleavable biotin tethers strategically placed on staple strands can allow the one-step purification of custom-scaffolded DNA origami nanoparticles.30 A photocleavable linker is a photosensitive (to 365 nm light) single bond between the 5′ terminal O and an aliphatic amino group, to which biotin can be attached via a long spacer.32 This photocleavable biotin tether on a staple strand allows the pulldown of assembled DNA origami particles, leaving behind excess staples as well as any aPCR-related contaminants. While agarose gel extracted custom scaffold strand leads to over 85% loss of the scaffold strand, using photocleavable biotin tethers one can directly use the crude aPCR reaction mix as the scaffold solution and excess staple strands (with one photocleavable biotin tether modification) to assemble a helix bundle or wireframe DNA origami nanoparticle and subsequently acquire purified sample with up to 90% yield.30 Photo-induced nucleotide damage at 365 nm is known to be minimal, making this approach viable (but untested) for gene-encoded DNA nanoparticle purification.
2.3 Bacteriophage-based scaffold production
Bacteriophage culturing can also be harnessed for producing custom gene-encoded scaffold strands. Bacteriophages are specific viruses that infect bacteria to hijack their transcription–translation machinery and multiply in large quantities (Fig. 2). As mentioned before, some bacteriophage genomes – called phagemids – are naturally circular ssDNAs, such as M13. Native M13 genomic sequence can be reengineered by inserting a custom sequence after the origin of replication (Fig. 2(A)). Yet, the permissible length of such phage genomes is limited by the loading capacity of phage capsids. To overcome the length challenge and expand the available space for inserting custom sequences into the phage genome, a “helper phage” plasmid has been engineered that carries the genes necessary for phage production and the origin of phage replication is split between the original phage genome and the helper phage (Fig. 2(B)). As a result, when the bacteria are transformed with the helper phage and the phagemid (encoding the custom sequence of interest), the phage particles are amplified containing the new sequence, up to 10 kb long.33 Genes encoding GFP and mCherry have been synthesized in scaffold forms using custom phagemid culturing.22,24 For ease of use, the Douglas and Dietz labs, who have pioneered and formalized the phage technique for custom scaffold synthesis, have also shared customized helper phage and “empty” phage vectors on the non-profit repository Addgene from where one can purchase the vectors and use cloning techniques to insert a gene of choice (Fig. 2(C)).33,34 More recently, the Douglas group also reengineered the helper phage and the bacterial genome to create a new bacterial strain – called the eScaf – that eliminates the need for a secondary helper phage plasmid.35 Purification of scaffold strand produced via the phagemid method can be sensitive to contamination, however, there is a well-established process wherein the bacteria are lysed and processed via centrifugation at a specific force so as to spin down the bacterial genomic DNA but allow the phage genome (which is the scaffold strand) to remain in the supernatant. One key difference between phage-produced scaffold and enzymatically amplified scaffold (via PCR/aPCR) is that the former contains significant endotoxin contamination. This is essential to remove through downstream purification to avoid triggering an immune response for biomedical applications; endotoxin removal can be performed using commercial kits or by using Triton-114.363. Notable considerations for creating gene-encoded DNA nanoparticles
Protein expression via gene-encoded DNA nanostructures has now been demonstrated in vitro, in vivo, and in cell-free systems.18,22,24–26,29 To facilitate the successful translation towards therapeutic goals, important considerations into designing gene-encoded DNA nanoparticles for efficient protein expression have been studied. Their successful implementation heavily depends on efficient cellular delivery strategies. As research for gene-encoded DNA nanoparticles is still in its early stages, there are only a few methods that have been used to introduce these nanoparticles into mammalian cells. Each approach has its own advantages and limitations, but it is important to note that there is not a consensus on the most translatable strategy, underscoring the importance of continued investigation.3.1 Inclusion of functional non-coding sequences relative to the gene for augmented expression
When a plasmid is transfected into cells, it typically contains non-coding sequences (domains critical for replication and protein expression) in addition to the gene of interest. The extent to which the accessibility of different domains within the encoding scaffold strand affect transcription is being actively addressed. Recently, the Dietz group designed DNA origami nanoparticles encoding eGFP and mCherry to test some sequence-related factors that could affect protein expression from electroporated DNA nanoparticles. They synthesized an eGFP encoding scaffold strand using the phagemid method described above, with which a 20-helix bundle DNA origami structure was assembled and studied in human embryonic kidney (HEK) cells via electroporation (Table 1).22 Notable features in their eGFP-encoded scaffold strand included a Kozak sequence, a WPRE, and Inverted Terminal Repeat (ITR) sequences (Table 1). The Kozak and WPRE sequences are known to be important during translation, thus were expected to have no known effect on DNA nanoparticle transcription.37–39 However, location(s) of the ITRs relative to the gene are known to impact gene expression in adeno-associated viruses (AAVs).40,41 Their inclusion within the scaffold strand is part of a broader strategy to create more versatile gene delivery applications by incorporating functional sequences and structures inspired by viral systems. In this study, ITRs were placed in the scaffold sequence to be either upstream, downstream, or flanking the expression cassette, and it was found that the inclusion of at least one ITR, preferably downstream of the expression cassette, improved gene expression efficiency via the 20-helix bundle as measured by mean fluorescence intensity (MFI).22 It was also found that expression could be further improved by placing the ITR sequences on the DNA nanoparticle in a way that would allow them to assemble into their hairpin secondary structure during the annealing process (rather than only being available upon denaturation of the nanoparticle inside the cell). In adjusting the position of the ITR sequences on the helix bundle structure, expression efficiency increased up to nine-fold.22| Scaffold features | Function | Benefit of incorporation | Ref. |
|---|---|---|---|
| Gene location: adapted from ref. 22 licensed under CC-BY. Internal crosslinking: adapted from ref. 22 licensed under CC-BY. Aspect ratio: reprinted (adapted) with permission from ref. 42. Copyright (2018) American Chemical Society. Scaffold orientation: used with permission from ref. 22 licensed under CC-BY. Promoter domain design: reprinted with permission from ref. 26. Copyright (2024) American Chemical Society. | |||
| Kozak sequence | Initiates translation in eukaryotic cells; affects the probability of a ribosome recognizing the start codon | Ensures that the transcribed mRNA is efficiently translated into protein | 22 and 37 |
| Woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) | Enhances mRNA stability and protein expression | Increases transgene expression when placed downstream of the gene and proximal to the polyA signal | 38 and 39 |
| Inverted terminal repeats (ITRs) | Single-stranded sequence of nucleotides directly followed by its reverse complement | Improves expression efficiency when placed upstream of expression cassette (mimics viral expression mechanism of AAV) | 22 and 41 |
| Simian virus 40 (SV40) | Multiple transcription factors bind to SV40 DTS sequence within the cytoplasm; recruit transcription factors and bind with them in the cytoplasm | Direct transportation of DNA nanostructure to nucleus | 24 |
| polyA sequence | Important for stability of mRNA upon nuclear export – prevents degredation | Ensures that the transcribed mRNA is stable enough to undergo translation | 22 |
| Origami design feature/assessed variable | Definition | Summarized findings | Ref. | |
|---|---|---|---|---|
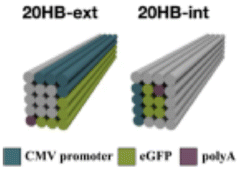
| Gene location |
|
Internal versus external placement of gene on origami structure | Placement of the gene on a certain region of the origami structure does not have an effect on gene expression – it should be noted that in both cases, the promoter sequence was on the exterior of the structure | 22 |
| Internal crosslinking |
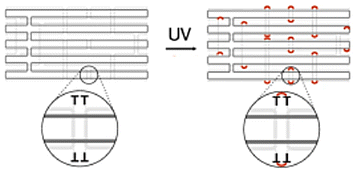
|
Extra thymine residues were included on the staple strands to induce crosslinking via UV point welding – when structure is exposed to UV light, thymine dimers are created preventing the dissociation of scaffold and staples | These variants had almost complete suppression of eGFP signal – indicating that the unfolding of the origami must occur in order for the desired gene to be transcribed | 22 |
| Aspect ratio (AR) |

|
Value comparing the length to width dimensions on origami structure | Tested Structures: 20 hb (AR = 5), 32 hb (AR = 2), 12 hb (AR= 15) | 22 and 42 |
| No difference in levels of expression between 20 hb and 32 hb; levels were slightly lower for 12 hb, but should be noted that cell density was also lower for this treatment group | ||||
| Scaffold orientation |
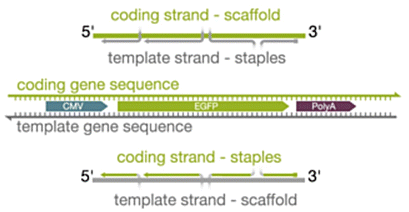
|
Varying the scaffold sequence to correspond with the coding (sense) or template (antisense) strand | Use of the template strand exhibited higher electroporation efficiencies when the scaffold was delivered alone and when the scaffold and staples were codelivered (non-annealed) – when either scaffold was folded into an origami structure, there was no significant difference between expression values | 22 |
| Promoter domain design |
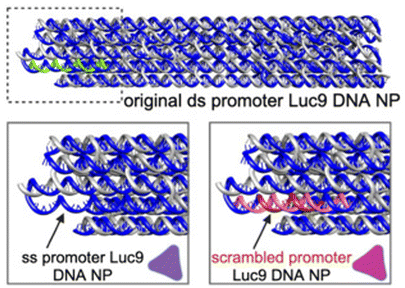
|
Analyzed three promoter variants: native double-stranded, scrambled (arbitratry DNA sequence replacing promoter) double-stranded, and single-stranded (no staple bound) | Single-stranded promoter regions significantly decrease gene expression and those with arbitrary sequences completely turn off expression | 26 |
To maximize efficiency of gene delivery, the gene-encoded nanoparticle would require efficient transport to the nucleus since it is the site of transcription, but transport is often a challenge due to the selective permeability of the nuclear membrane. The nuclear membrane consists of a lipid bilayer that is perforated by various nuclear pore complexes; these complexes regulate the passage of molecules between the cytoplasm and the nucleus. Larger molecules (such as DNA nanoparticles) can enter the nucleus more easily through active transport, but often require specific nuclear localization sequences (NLS) or DNA nuclear targeting sequences (DTS) that aid in nuclear import.43 To assist with the nuclear import of their mCherry-encoded DNA nanoparticle, Liedl et al. included multiple Simian virus 40 (SV40) derived DTS.44,45 The mCherry-encoded scaffold strand was bacteriophage-produced and included a CMV promoter, mCherry reporter gene, and a bovine growth hormone polyadenylation (bGH polyA) signal (coding/sense strand). Additionally, zero, one, three, or six repeats of the 72 bp SV40 DTS were inserted in the scaffold, however, the location of these repeats varied between each of the DNA nanoparticle constructs. In all cases, the coding domain (mCherry gene) was always on the outer helix of the nanoparticles (Fig. 3(E)). 24 hours after electroporating 0.75 μg (∼0.545 pmol) of these nanostructures into HEK293T cells, it was found that three repeats of the SV40 DTS downstream of the polyA signal maximized mCherry expression, as quantified by MFI and flow cytometry.
3.2 Aspect ratio and coding domain position within helix bundle DNA nanoparticles may not tune overall protein expression
While a plasmid is a simple circular or linear dsDNA molecule wherein the sequence can be easily parsed by different transcription proteins and factors, a gene-encoded DNA origami nanoparticle is complicated, with the gene-encoded scaffold strand rasterized through a 3D shape via single and double crossover sites. Part of the scaffold can be buried inside the DNA nanoparticle depending on the overall geometry. Therefore, it becomes important to consider the architecture and sequence design since complex molecular coordination of several transcription factors and proteins (in addition to RNA polymerase II) culminates in successful DNA transcription. Following successful scaffold isolation via the bacterial phage technique, Kretzmann et al. designed a series of eGFP encoded DNA nanoparticles of different aspect ratio helix bundle (hb) geometries, as geometric design is known to affect cellular uptake (Fig. 3(C)).42 Initially, they varied the placement of the coding domain within the scaffold (the gene sequence) to route to the outer versus inner helices of a 20-helix bundle structure (Table 1). Subsequently, the number of helices per structure was varied to give aspect ratios of 15 (12 hb), 5 (20 hb), and 2 (32 hb). The CMV promoter (essential for transcription initiation) was on one of the outer helices in each helix bundle design. They characterized total eGFP expression by electroporating 0.5 μg (∼0.20 pmoles) of DNA nanoparticles into HEK293T cells and measured fluorescence by flow cytometry after 48 hrs. However, coding domain position and aspect ratio did not seem to significantly affect eGFP expression levels.To account for the fact that gene placement did not have a significant impact on expression, it was hypothesized that the DNA nanostructure unfolded for the gene to be successfully transcribed by RNA polymerase in the nucleus. To test this, extra thymine residues were included on the staple strands to induce internal crosslinking via UV point welding (Table 1). When the nanostructures were exposed to UV light (310 nm, 2 hours), thymine dimers were created between neighboring staple strands, preventing the dissociation of the scaffold and staple strands upon delivery. In electroporating these crosslinked variants into the HEK293T cell line, there was almost complete suppression of the eGFP signal, leading to the interpretation that DNA nanoparticle unfolding into constituent staple and scaffold strands occurs during transcription. This is an exciting observation on the processing of DNA nanoparticles through transcription, but it is unclear whether there was any loss in biological activity in the gene-encoded DNA nanoparticle owing nucleic acid damage from prolonged UV exposure.
3.3 Design of the promoter domain can control protein expression
The promoter is a 20–200 nucleotide long conserved sequence domain upstream of the coding gene sequence necessary for transcription initiation. It is also crucial to ensure the right strand in the dsDNA gene is recognized as the coding strand (which is homologous to the transcribed mRNA) and the noncoding strand (which serves as the template that is transcribed by RNA polymerase to produce the mRNA). Consequently, transcription can be significantly impeded in a gene (or gene-encoded DNA nanoparticle) if the promoter region is not designed optimally. Oftentimes DNA origami nanoparticle design includes ssDNA domains in the form of scaffold loops or staple extensions for reducing nanoparticle aggregation or for other specific downstream functionality.46 However, the extent to which DNA domains affected transcription accuracy is unknown. We designed a Renilla luciferase encoding DNA helix bundle with a bacterial T7 promoter driving gene expression to probe this.26 Luciferase-encoding scaffold was synthesized using aPCR from a precursor plasmid strand, with the scaffold containing the T7 promoter, luciferase gene, and the T7 terminator. Three variants of the helix bundle were assembled, one with a dsDNA promoter, one missing the staple complementary to the T7 promoter, and one where the T7 promoter was replaced with an arbitrary sequence (Table 1). The DNA nanoparticle variant missing the T7 promoter complementary staple thus displayed a promoter in ssDNA form. All nanoparticle constructs were tested for luciferase expression in a bacterial cell-free transcription–translation system that allowed facile characterization of protein expression directly on a plate reader, avoiding the need to grow cell cultures. Results showed a significant reduction of luciferase expression (measured as total bioluminescence signal) from the ssDNA promoter DNA nanoparticle compared to the dsDNA promoter nanoparticle. The construct with an arbitrary sequence in place of the conserved T7 sequence showed nominal protein expression (Fig. 3(G)). Thus, nature of how the promoter domain is designed in a gene-encoded DNA nanoparticle is critical.4. Potential applications of gene-encoded DNA nanoparticles
Encodement of the scaffold with a functional genetic sequence allows for harnessing every component of DNA origami nanostructures to its full potential. Traditionally, staple strands have been the primary site of chemical modifications within DNA nanostructures to organize other molecules, attach fluorescent reporters, or display moieties for cellular interfacing. However, with gene-encodement of the scaffold strand, gene delivery can now be added to the repertoire of functionality.The next translation, thus, could be combining the spatial control afforded by modified staple strands to display targeting proteins and ligands with a gene-encoded scaffold strand to create gene therapy platforms. Recently, Wu et al. investigated the targeted delivery of a p53 gene in HeLa (human cervical epithelial) cells and in tumor xenograft BALB/c (Bagg Albino immunodeficient) nude mice through a gene-encoded DNA nanoparticle (Fig. 3(F)).25 The p53 gene is known to be an important tumor suppressing gene as it induces apoptosis and cell-cycle arrests.47 The DNA nanoparticle delivery system was created by joining two gene-encoded DNA origami monomer nanoparticles. Each monomer consisted of a custom scaffold strand generated via PCR. Interestingly, both scaffold strands were produced from the same precursor p53 encoding plasmid, but one scaffold encoded the sense strand while the other scaffold encoded the antisense strand. Each strand was folded into a pre-designed shape (using complementary staples) to form DNA origami monomers. The two monomers hybridized with each other to form the gene-encoded DNA origami (DO) construct.
The final DO construct (made of the two monomers) was structurally designed with short 8 nucleotide oligo extensions as tethers. These oligo extensions were incorporated to accelerate the disassembly of the gene-encoded DO construct during the transcription process, supporting efficient gene expression. To improve cellular uptake, the DO was coated with lipid via a templated growth method. A lipid–DNA conjugate was synthesized and precisely organized onto the DO's surface. Phospholipid with folate (DOPE-FA) was included in the lipid growth around the DO to enhance the targeted cellular uptake of the nanoparticles since folate receptors are often overexpressed in most tumor cells.48 The lipid-coated gene-encoded DO (FA-DO) was characterized by transmission electron microscopy. The DO and FA-DO were labeled with Cyanine5 (Cy5) and incubated with HeLa cells for 6 hours. Cy5 allowed for successful tracking of the DOs, and it was found that FA-DO had a stronger signal when compared to DO. This was confirmed by the MFI recorded through flow cytometry. Quantitative real time PCR (qRT-PCR) determined that the FA-DO had nearly 8 times higher p53 mRNA levels than DO only, and western blot analysis confirmed protein expression. Flow cytometry showed that 81.4% of HeLa cell apoptosis was induced by FA-DO. A cell-viability assay established a dose-dependent inhibition of viability. Remarkably, 80% of tumor inhibition was achieved under the dosage of 3.2 nM with no noticeable cytotoxicity.
The tumor inhibition effects of the FA-DO were investigated in vivo as well through a HeLa tumor xenograft model in BALB/c nude mice. The HeLa tumor bearing mice were treated with equal doses of DO or FA-DO (DNA scaffolds: 1.2 mg kg−1) via tail vein injections every three days for four treatments. By monitoring the tumor volumes and weight, as well as protein, mRNA expression and apoptosis levels, it was deduced that the FA-DO group was the best platform for tumor inhibition. The mice organs also did not demonstrate any observable system toxicity. These results demonstrate the validity of using DNA nanoparticles as a platform for gene delivery, particularly in gene therapy-based tumor inhibition. Wu et al. established that the complementary DNA strands of a functional gene can be directly used to form genetically-encoded DNA origami nanoparticle which can act as a template for lipid growth. The lipid-coating enables the DNA nanoparticle to penetrate the cell membrane in doses that do not elicit dangerous cytotoxicity levels.
One of the challenges of current gene delivery systems made from lipid and viral nanoparticles is the limited DNA loading capacity. For genes larger than 5 kilobases, it becomes necessary to cleave the gene into two parts and co-deliver using multiple delivery vehicles.49,50 DNA nanoparticles are advantageous in overcoming the loading capacity limits since one can engineer DNA nanoparticles of any size. Moreover, it is possible to co-deliver multiple genes simultaneously. To that end, the Dietz group developed multiplexed assemblies for the codelivery of eGFP and mCherry (each being 4816 nt long) genes. DNA nanoparticles encoding for these respective genes into the scaffold strands were created in stoichiometric ratios of 1:1, 1:2, and 1:3 mCherry to eGFP (Fig. 3(D)). The overall design concept involved creating individual origami blocks for each genetic sequence (mCherry and eGFP), which could then be linked together to form a single multifunctional origami structure. This linkage was achieved through complementary base pairing of extended staple sequences, and their modular approach allowed for precise control over the ratio of delivered genes within a single nanostructure. Here, they found that the expression level of eGFP was directly proportional to the number of monomers present within the nanostructure as confirmed by fluorescence microscopy and flow cytometry.22
5. Future directions
Despite being in the early stages of research and development, the potential of gene-encoded DNA nanoparticles is vast as a basic science tool and a translational device. The leading paradigm in gene delivery is plasmid transfections wherein the protein of interest is encoded into bacterial vectors and delivered in vitro and in vivo via lipid and viral formulations. Therefore, any clinical application of gene-encoded DNA nanoparticles warrants comparatively evaluating its performance against plasmid DNA. Looking at cellular uptake of DNA nanoparticles (designed in previous works for pharmacochemical drug delivery) and stability in cellular environments (media, cell lysates, nuclease enzymes, and live cell cytosol), it is known to be improved compared to plasmid or linear dsDNA.51 However, transfection efficiency from the point of view of total protein expression from gene-encoded DNA nanoparticles versus plasmids is an important question. To the best of our knowledge, one study has quantified this; the cell-free transcription–translation of Luc9 protein was 91% and 70% efficient through corresponding plasmid and gene-encoded DNA nanoparticle, respectively, when the protein output from the gene-encoded dsDNA was normalized to 100%.26 Based on this study, DNA nanoparticle was 75% as efficient as the plasmid in protein expression. How the efficiency translates to mammalian cells is still unknown and could be considered a significant uncertainty needing priority attention.On the design level, we also now have a preliminary understanding that the secondary and tertiary structure of DNA nanoparticles (crossover density and promoter location) can influence the overall protein expression efficiency. Surveying the studies that have contributed to this understanding, some studies were designed in in vitro bacterial IVT systems while others used mammalian cell electroporation setup. It is important to note that T7 polymerases are not present in mammalian cells, and therefore the behavior of these constructs may vary when comparing efficiencies in IVT systems versus a mammalian system. T7 polymerase originates from the T7 bacteriophage, a virus known for infecting bacteria; this polymerase is very specific to its promoter, and because of that, T7 systems can be tightly controlled in an experimental setting.52 For gene expression in mammalian cells, RNA polymerase II recognizes a CMV promoter. This promoter interacts with the general eukaryotic transcription machinery, allowing for expression in a wide range of mammalian cell types, but expression can ultimately be affected by discrepancies between cellular environments. Additionally, the complex nuclear environment of mammalian cells as well as accompanying post-transcriptional processes are not replicated in IVT systems. Despite these fundamental differences, DNA nanoparticles that retain a T7 promoter for expression in cell-free systems are able to provide insight on design features that affect gene expression in a more time efficient manner. Similarly, electroporation of gene-encoded DNA nanoparticles were among the first experiments demonstrating transgene expression in mammalian cells. The translational potential of electroporation is limited, as it is primarily suitable for ex vivo cell therapies or transdermal applications. Looking forward, the invasive nature and unpredictability of potential cell damage may need to be addressed for its use in many clinical scenarios. Thus, while both experimental systems (IVT and electroporation) are excellent tools for fundamental research, there is a noticeable need for investigations focused on mammalian cell transfections via gene-encoded DNA nanoparticles. Furthermore, several traditional methods used to augment translation of DNA in vivo are yet to be explored and implemented, such as codon usage and additional regulatory elements.
Despite significant advances in ssDNA production, strategies to produce gene-encoded scaffold strands are key to broadening the scope of the lengths and multiplexity of genes that can be encoded into these nanoparticles and accelerating their use in clinical applications. The bacteriophage-based scaffold synthesis has delivered milligram-scale quantities of ssDNA, and with proper standardization of quality control (and removal of endotoxins) it could be translated for production of gene-encoded scaffolds.53
Studies using DNA origami nanocarriers have consistently demonstrated low cytotoxicity in different cell lines.54–56 Several studies have also established the basic biosafety of DNA nanoparticles in vivo through body weight measurements and histopathological examinations of major organs (heart, liver, kidney, lung, etc.) after treatment with the DNA nanoparticle.57–59 Compared to carriers containing inorganic materials that the body cannot decompose or clear, DNA nanoparticles are a promising alternative that consist of organic monomers that can be processed by the body, eliminating the risk of unwanted accumulation. There remain a few aspects that must be thoroughly investigated prior to using DNA nanoparticles in gene therapy, but in most studies, DNA nanoparticles have not stimulated an immune response.57,59–61 In 2022, an M13mp18-encoded DNA nanoparticle was administered to ICR mice at a dosage of 12 mg kg−1 to maximize the potential immunostimulatory response. Here, the CD11b+ cell levels were monitored, as high cell populations tend to indicate inflammation or tissue damage, however, there was no indication that these DNA nanoparticles were the cause for adverse effects.60 Similarly in 2023, the Bathe group administered DNA nanoparticles (M13mp18 scaffold) intravenously to BALB/c mice at a dosage of 4 mg kg−1 to stay within the standard dose range for nucleic acid therapeutics (1–10 mg kg−1).62 Toxicity readouts were assessed, and it was found that there was no evident liver or kidney damage as indicated by histology.62 Despite these findings, Perrault et al. found that their DNA nano-octahedron induced an inflammatory cytokine response similar to that produced by foreign bacterial or viral nucleic acids.63 They combated this by encapsulating the nano-octahedron in lipid bilayers resembling viral membranes. The immune response and the pharmacokinetics of DNA nanoparticles are not yet fully understood, and continued research is crucial for the successful translation of these structures as a biosafe delivery platform.
The above-mentioned studies utilized DNA nanoparticles as a nanocarrier as opposed to a gene-encoded system. Gene-encoded DNA nanoparticles are yet to be scrutinized for concerns associated with currently applied gene therapy vectors since gene therapy research is heavily regulated due to safety concerns. Predominant gene therapy vectors such as AAVs have shown success in different trials but have also been associated with immune responses and off-target effects.64,65 In some cases, the immune responses can result in effects as severe as death.66 Wild types of Adenoviruses can also infect humans and so there remains a possibility that antibodies targeting these viruses can reduce the efficiency of AAV vectors. Additionally, the risk of unintended genomic integration can result in the inactivation and dysregulation of genes.67 Each of these risks must be fully investigated in gene-encoded DNA nanoparticles before they can be extensively used in gene therapy.
As a basic science tool, gene-encoded DNA nanoparticles have the potential to increase resolution of characterization of DNA nanoparticles to the base-pair level. They could be combined with an optical readout for probing at the base pair level the stability of DNA nanoparticles under different physical and chemical stressors since transcription (and concomitant protein expression) depends on having a contiguous gene sequence and a double stranded promoter region. Any damage to the scaffold strand would result in loss of protein expression, something that is not resolved by traditional means of studying DNA nanoparticle stability.17 For example, using a cell-free transcription–translation system we evaluated how promoter domain configuration within a gene-encoded DNA nanostructure affected its protein expression, as described above. In this case, protein luminescence was the optical readout which allowed time-resolved determination of the physical state of the DNA nanoparticle. UV crosslinking of thymine dimers is emerging as a way to increase DNA nanoparticle stability.68–70 In case of linear gene cassettes, considerable loss in biological activity can be seen post UV exposure. Perhaps a quick in vitro protein expression study can confirm the extent of photo-induced loss in biological activity in a DNA nanoparticle. The optical readout can be made multi-faceted by integrating chromophores engaged in multistep Förster resonance energy transfer (FRET) into gene-encoded nanoparticles. Previously, a strong correlation between the shape of small DNA nanoparticles and its cytosolic stability was observed using multi-step FRET and single cell manipulation, suggesting that future gene-encoded DNA nanoparticles would require additional modifications for resistance to degradation.71 Notwithstanding all the challenges that need to be addressed through further research,72 the promise of gene-encoded nanoparticles is increasingly positive and an area worth keeping an eye on.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. D. was supported by a summer research scholarship provided by CWRU Undergraduate Research Office, Case Alumni Association (CAA) and Flora Stone Mather Center for Women. This work was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health (R00EB030013). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- S. M. Douglas, A. H. Marblestone, S. Teerapittayanon, A. Vazquez, G. M. Church and W. M. Shih, Nucleic Acids Res., 2009, 37, 5001–5006 CrossRef CAS PubMed.
- R. Veneziano, S. Ratanalert, K. Zhang, F. Zhang, H. Yan, W. Chiu and M. Bathe, Science, 2016, 352, 1534 CrossRef CAS.
- H. Jun, X. Wang, M. F. Parsons, W. P. Bricker, T. John, S. Li, S. Jackson, W. Chiu and M. Bathe, Nucleic Acids Res., 2021, 49, 10265–10274 CrossRef CAS PubMed.
- W. G. Pfeifer, C. M. Huang, M. G. Poirier, G. Arya and C. E. Castro, Sci. Adv., 2023, 9, eadi0697 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS.
- A. N. Marchi, I. Saaem, B. N. Vogen, S. Brown and T. H. LaBean, Nano Lett., 2014, 14, 5740–5747 CrossRef CAS PubMed.
- C. J. Kearney, C. R. Lucas, F. J. O’Brien and C. E. Castro, Adv. Mater., 2016, 28, 5509–5524 CrossRef CAS PubMed.
- P. E. Beshay, J. A. Johson, J. V. Le and C. E. Castro, Methods Mol. Biol., 2023, 2639, 21–49 CrossRef CAS.
- D. Mathur, S. A. Díaz, N. Hildebrandt, R. D. Pensack, B. Yurke, A. Biaggne, L. Li, J. S. Melinger, M. G. Ancona, W. B. Knowlton and I. L. Medintz, Chem. Soc. Rev., 2023, 52, 7848–7948 RSC.
- R. Veneziano, T. J. Moyer, M. B. Stone, E.-C. Wamhoff, B. J. Read, S. Mukherjee, T. R. Shepherd, J. Das, W. R. Schief, D. J. Irvine and M. Bathe, Nat. Nanotechnol., 2020, 15, 716–723 CrossRef CAS PubMed.
- Y. C. Zeng, O. J. Young, C. M. Wintersinger, F. M. Anastassacos, J. I. MacDonald, G. Isinelli, M. O. Dellacherie, M. Sobral, H. Bai, A. R. Graveline, A. Vernet, M. Sanchez, K. Mulligan, Y. Choi, T. C. Ferrante, D. B. Keskin, G. G. Fell, D. Neuberg, C. J. Wu, D. J. Mooney, I. C. Kwon, J. H. Ryu and W. M. Shih, Nat. Nanotechnol., 2024, 19, 1055–1065 CrossRef CAS.
- R. R. Du, E. Cedrone, A. Romanov, R. Falkovich, M. A. Dobrovolskaia and M. Bathe, ACS Nano, 2022, 16, 20340–20352 CrossRef CAS PubMed.
- M. A. Dobrovolskaia and M. Bathe, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2021, 13, e1657 CAS.
- Y. Sakai, M. S. Islam, M. Adamiak, S. C. Shiu, J. A. Tanner and J. G. Heddle, Genes, 2018, 9, 571 CrossRef PubMed.
- Y. Wu, L. Luo, Z. Hao and D. Liu, Med. Rev., 2024, 4, 207–224 CrossRef.
- S. Zhao, R. Tian, J. Wu, S. Liu, Y. Wang, M. Wen, Y. Shang, Q. Liu, Y. Li, Y. Guo, Z. Wang, T. Wang, Y. Zhao, H. Zhao, H. Cao, Y. Su, J. Sun, Q. Jiang and B. Ding, Nat. Commun., 2021, 12, 358 CrossRef CAS.
- K. Neyra, H. R. Everson and D. Mathur, Anal. Chem., 2024, 96, 3687–3697 CrossRef CAS.
- E. Lin-Shiao, W. G. Pfeifer, B. R. Shy, M. Saffari Doost, E. Chen, V. S. Vykunta, J. R. Hamilton, E. C. Stahl, D. M. Lopez, C. R. Sandoval Espinoza, A. E. Deyanov, R. J. Lew, M. G. Poirer, A. Marson, C. E. Castro and J. A. Doudna, Nucleic Acids Res., 2022, 50, 1256–1268 CrossRef.
- S. Dey, C. Fan, K. V. Gothelf, J. Li, C. Lin, L. Liu, N. Liu, M. A. D. Nijenhuis, B. Saccà, F. C. Simmel, H. Yan and P. Zhan, Nat. Rev. Methods Primers, 2021, 1, 13 CrossRef CAS.
- C.-Y. Oh and E. R. Henderson, DNA, 2022, 2, 56–67 CrossRef.
- J. Bush, S. Singh, M. Vargas, E. Oktay, C. H. Hu and R. Veneziano, Molecules, 2020, 25(15), 3386 CrossRef CAS.
- J. A. Kretzmann, A. Liedl, A. Monferrer, V. Mykhailiuk, S. Beerkens and H. Dietz, Nat. Commun., 2023, 14, 1017 CrossRef CAS.
- C. Chau, G. Mohanan, I. Macaulay, P. Actis and C. Wälti, Small, 2024, 20, 2308776 CrossRef CAS PubMed.
- A. Liedl, J. Grießing, J. A. Kretzmann and H. Dietz, J. Am. Chem. Soc., 2023, 145, 4946–4950 CrossRef CAS PubMed.
- X. Wu, C. Yang, H. Wang, X. Lu, Y. Shang, Q. Liu, J. Fan, J. Liu and B. Ding, J. Am. Chem. Soc., 2023, 145, 9343–9353 CrossRef CAS PubMed.
- A. R. Galvan, C. M. Green, S. L. Hooe, E. Oktay, M. Thakur, S. A. Díaz, R. Veneziano, I. L. Medintz and D. Mathur, ACS Appl. Nano Mater., 2024, 7, 12891–12902 CrossRef CAS.
- M. A. Zanta, P. Belguise-Valladier and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 91–96 CrossRef CAS.
- E. Oktay, J. Bush, M. Vargas, D. V. Scarton, B. O’Shea, A. Hartman, C. M. Green, K. Neyra, C. M. Gomes, I. L. Medintz, D. Mathur and R. Veneziano, ACS Appl. Mater. Interfaces, 2023, 15, 27759–27773 CrossRef CAS PubMed.
- C. Y. Oh and E. R. Henderson, Sci. Rep., 2023, 13, 12961 CrossRef CAS.
- H. R. Everson, K. Neyra, D. V. Scarton, S. Chandrasekhar, C. M. Green, T. L. Schmidt, I. L. Medintz, R. Veneziano and D. Mathur, ACS Appl. Mater. Interfaces, 2024, 16, 22334–22343 CrossRef CAS PubMed.
- A. Shaw, E. Benson and B. Högberg, ACS Nano, 2015, 9, 4968–4975 CrossRef CAS PubMed.
- J. Olejnik, E. Krzymanska-Olejnik and K. J. Rothschild, Nucleic Acids Res., 1998, 26, 3572–3576 CrossRef CAS PubMed.
- P. M. Nafisi, T. Aksel and S. M. Douglas, Synth. Biol., 2018, 3(1), ysy015 CrossRef CAS PubMed.
- B. Kick, F. Praetorius, H. Dietz and D. Weuster-Botz, Nano Lett., 2015, 15, 4672–4676 CrossRef CAS PubMed.
- K. Shen, J. J. Flood, Z. Zhang, A. Ha, B. R. Shy, J. E. Dueber and S. M. Douglas, Nucleic Acids Res., 2024, 52, 4098–4107 CrossRef CAS PubMed.
- M. M. Koga, A. Comberlato, H. J. Rodríguez-Franco and M. M. C. Bastings, Biomacromolecules, 2022, 23, 2586–2594 CrossRef CAS PubMed.
- J. M. Acevedo, B. Hoermann, T. Schlimbach and A. A. Teleman, Sci. Rep., 2018, 8, 4018 CrossRef PubMed.
- T. Higashimoto, F. Urbinati, A. Perumbeti, G. Jiang, A. Zarzuela, L. J. Chang, D. B. Kohn and P. Malik, Gene Ther., 2007, 14, 1298–1304 CrossRef CAS PubMed.
- S. Brun, N. Faucon-Biguet and J. Mallet, Mol. Ther., 2003, 7, 782–789 CrossRef CAS.
- J. T. Bulcha, Y. Wang, H. Ma, P. W. L. Tai and G. Gao, Signal Transduction Targeted Ther., 2021, 6, 53 CrossRef CAS.
- L. F. Earley, L. M. Conatser, V. M. Lue, A. L. Dobbins, C. Li, M. L. Hirsch and R. J. Samulski, Hum. Gene Ther., 2020, 31, 151–162 CrossRef CAS PubMed.
- M. M. C. Bastings, F. M. Anastassacos, N. Ponnuswamy, F. G. Leifer, G. Cuneo, C. Lin, D. E. Ingber, J. H. Ryu and W. M. Shih, Nano Lett., 2018, 18, 3557–3564 Search PubMed.
- J. Lu, T. Wu, B. Zhang, S. Liu, W. Song, J. Qiao and H. Ruan, Cell Commun. Signaling, 2021, 19, 60 CrossRef.
- E. V. van Gaal, R. S. Oosting, R. van Eijk, M. Bakowska, D. Feyen, R. J. Kok, W. E. Hennink, D. J. Crommelin and E. Mastrobattista, Pharm. Res., 2011, 28, 1707–1722 CrossRef CAS.
- Y. T. Le Guen, C. Pichon, P. Guégan, K. Pluchon, T. Haute, S. Quemener, J. Ropars, P. Midoux, T. Le Gall and T. Montier, Mol. Ther.–Nucleic Acids, 2021, 24, 477–486 CrossRef CAS.
- L. Piantanida, J. A. Liddle, W. L. Hughes and J. M. Majikes, Nanotechnol., 2024, 35(27), 273001 CrossRef CAS PubMed.
- R. V. Sionov and Y. Haupt, Oncogene, 1999, 18, 6145–6157 CrossRef CAS.
- C. Chen, J. Ke, X. E. Zhou, W. Yi, J. S. Brunzelle, J. Li, E. L. Yong, H. E. Xu and K. Melcher, Nature, 2013, 500, 486–489 CrossRef CAS PubMed.
- Z. Wu, H. Yang and P. Colosi, Mol. Ther., 2010, 18, 80–86 CrossRef CAS PubMed.
- H. Zhang, N. Bamidele, P. Liu, O. Ojelabi, X. D. Gao, T. Rodriguez, H. Cheng, K. Kelly, J. K. Watts, J. Xie, G. Gao, S. A. Wolfe, W. Xue and E. J. Sontheimer, GEN Biotechnol., 2022, 1, 285–299 CrossRef CAS.
- D. Mathur and I. L. Medintz, Adv. Healthcare Mater., 2019, 8, e1801546 CrossRef PubMed.
- A. Dousis, K. Ravichandran, E. M. Hobert, M. J. Moore and A. E. Rabideau, Nat. Biotechnol., 2023, 41, 560–568 CrossRef CAS.
- F. Praetorius, B. Kick, K. L. Behler, M. N. Honemann, D. Weuster-Botz and H. Dietz, Nature, 2017, 552, 84–87 CrossRef CAS.
- M. I. Setyawati, R. V. Kutty and D. T. Leong, Small, 2016, 12, 5601–5611 CrossRef CAS.
- P. D. Halley, C. R. Lucas, E. M. McWilliams, M. J. Webber, R. A. Patton, C. Kural, D. M. Lucas, J. C. Byrd and C. E. Castro, Small, 2016, 12, 308–320 CrossRef CAS.
- Q. Pan, C. Nie, Y. Hu, J. Yi, C. Liu, J. Zhang, M. He, M. He, T. Chen and X. Chu, ACS Appl. Mater. Interfaces, 2020, 12, 400–409 CrossRef CAS PubMed.
- Q. Zhang, Q. Jiang, N. Li, L. Dai, Q. Liu, L. Song, J. Wang, Y. Li, J. Tian, B. Ding and Y. Du, ACS Nano, 2014, 8, 6633–6643 CrossRef CAS PubMed.
- Y. Huang, W. Huang, L. Chan, B. Zhou and T. Chen, Biomaterials, 2016, 103, 183–196 CrossRef CAS.
- W. Tang, T. Tong, H. Wang, X. Lu, C. Yang, Y. Wu, Y. Wang, J. Liu and B. Ding, Angew. Chem., Int. Ed., 2023, 62, e202315093 CrossRef CAS PubMed.
- C. R. Lucas, P. D. Halley, A. A. Chowdury, B. K. Harrington, L. Beaver, R. Lapalombella, A. J. Johnson, E. K. Hertlein, M. A. Phelps, J. C. Byrd and C. E. Castro, Small, 2022, 18, e2108063 CrossRef PubMed.
- D. Jiang, Z. Ge, H. J. Im, C. G. England, D. Ni, J. Hou, L. Zhang, C. J. Kutyreff, Y. Yan, Y. Liu, S. Y. Cho, J. W. Engle, J. Shi, P. Huang, C. Fan, H. Yan and W. Cai, Nat. Biomed. Eng., 2018, 2, 865–877 CrossRef CAS.
- E. C. Wamhoff, G. A. Knappe, A. A. Burds, R. R. Du, B. W. Neun, S. Difilippantonio, C. Sanders, E. F. Edmondson, J. L. Matta, M. A. Dobrovolskaia and M. Bathe, ACS Appl. Bio Mater., 2023, 6, 1960–1969 CrossRef CAS PubMed.
- S. D. Perrault and W. M. Shih, ACS Nano, 2014, 8, 5132–5140 CrossRef CAS.
- D. B. Kohn, Y. Y. Chen and M. J. Spencer, Gene Ther., 2023, 30, 738–746 CrossRef CAS PubMed.
- M. Chavez, X. Chen, P. B. Finn and L. S. Qi, Nat. Rev. Nephrol., 2023, 19, 9–22 CrossRef CAS PubMed.
- A. P. Cotrim and B. J. Baum, Toxicol. Pathol., 2008, 36, 97–103 CrossRef CAS PubMed.
- D. R. Deyle and D. W. Russell, Curr. Opin. Mol. Ther., 2009, 11, 442–447 CAS.
- T. Gerling, M. Kube, B. Kick and H. Dietz, Sci. Adv., 2018, 4, eaau1157 CrossRef CAS.
- A. Rajendran, M. Endo, Y. Katsuda, K. Hidaka and H. Sugiyama, J. Am. Chem. Soc., 2011, 133, 14488–14491 CrossRef CAS.
- H. Bila, E. E. Kurisinkal and M. M. C. Bastings, Biomater. Sci., 2019, 7, 532–541 RSC.
- D. Mathur, K. E. Rogers, S. A. Díaz, M. E. Muroski, W. P. Klein, O. K. Nag, K. Lee, L. D. Field, J. B. Delehanty and I. L. Medintz, Nano Lett., 2022, 22, 5037–5045 CrossRef CAS.
- X. Wu, Q. Liu, F. Liu, T. Wu, Y. Shang, J. Liu and B. Ding, Nanoscale, 2021, 13, 12848–12853 RSC.
| This journal is © The Royal Society of Chemistry 2025 |