DOI:
10.1039/D4CC04877F
(Feature Article)
Chem. Commun., 2025,
61, 4483-4494
Recent advancements in synthesis of cyclic oligosaccharides†
Received
20th September 2024
, Accepted 3rd February 2025
First published on 24th February 2025
Abstract
The development of synthetic methods for chemical glycosylation enables the synthesis of various oligosaccharides, including nonnatural cyclic oligosaccharides. Electrochemical glycosylation is an enabling technology not only for automated solution-phase synthesis of linear oligosaccharides but also for the chemical synthesis of cyclic oligosaccharides. In this review, recent syntheses of nonnatural cyclic oligosaccharides are also introduced, and glycosylation methodologies are focused on.

Clockwise from bottom left: Hirofumi Endo, Yu-Cong Sun, Norihiko Sasaki, and Toshiki Nokami
| Hirofumi Endo was born in Tottori, Japan. He obtained his master's degree from Tottori University in 2022. He is continuing his PhD study and spent 4 months in the Baran group at the Scripps Research Institute in La Jolla. His research interests include synthetic organic chemistry, organic electrochemistry, and carbohydrate chemistry. Yu-Cong Sun was born in China and grew up in both China and Japan. He received his bachelor's degree from Tottori University in 2024. He started his master's studies in the same group. His research interests include synthetic organic chemistry, carbohydrate chemistry, and organic electrochemistry. Norihiko Sasaki was born in Okayama, Japan. He received his PhD from Kyushu University under the supervision of Professor Kazunori Sugiyasu in 2021. He spent one year as a postdoc in the same group at the National Institute for Materials Science (NIMS) and rejoined Tottori University as an Assistant Professor in 2022. His research interests include supramolecular chemistry, carbohydrate chemistry, organic electrochemistry, organofluorine chemistry, and ionic liquids. Toshiki Nokami was born in Wakayama, Japan. He obtained his PhD from Kyoto University under the supervision of late Professor Jun-ichi Yoshida in 2004. He spent 11 months at ETH Zurich as a postdoc under the supervision of Professor Peter H. Seeberger. He joined the Yoshida group as an Assistant Professor and moved to the Itoh group at Tottori University as an Associate Professor in 2012. He was promoted to Professor in 2019. His research interests include synthetic organic chemistry, carbohydrate chemistry, and organic electrochemistry. |
Introduction
Macrocyclic molecules including cyclodextrins
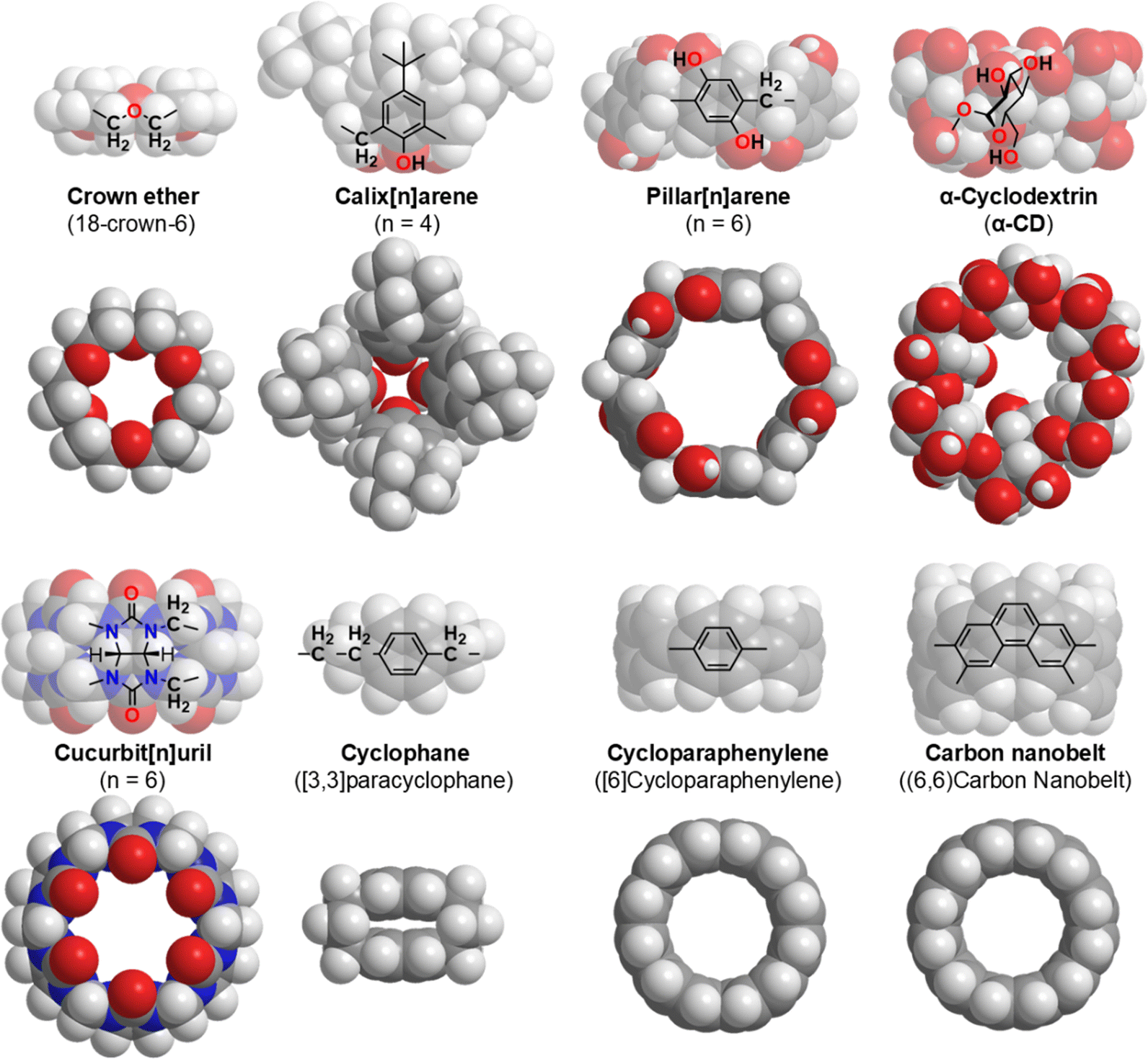
Chemistry is a field of science and technology that enables the manipulation of substances at the atomic and molecular levels. To achieve this goal, chemists have created a variety of macrocyclic molecules, such as crown ethers,1 calixarenes,2 pillar[n]arenes,3 cyclodextrins (CD),4 cucurbit[n]urils,5 cyclophanes,6 cycloparaphenylenes,7 and carbon nanobelts (Fig. 1).8 These macrocyclic molecules are used as host molecules with internal cavities that enclose guest molecules through hydrogen bonding, hydrophobic interactions, electrostatic interactions, and specific molecular shapes or size compatibility. The respective components and their numbers determine the inner and outer diameters of the macrocyclic systems.9 Generally, CDs and cucurbit[n]urils are easy to obtain or synthesize, and the cavity depths are constant at 0.78 and 0.91 nm, respectively. In contrast, the cavity depth of calixarenes ranges from 1.2 to 2.2 nm.
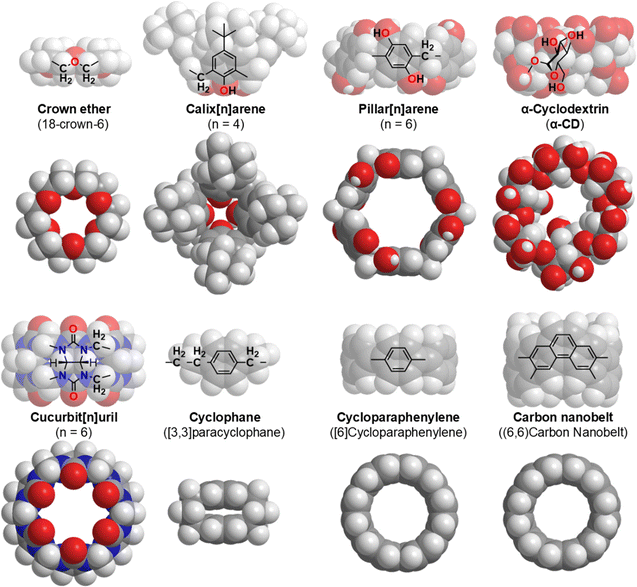 |
| | Fig. 1 Selected macrocyclic molecules.10,11 | |
Sugars are renewable biological resources that exert their structure and function in the form of oligosaccharides and polysaccharides. Oligosaccharides are generally water soluble and flexible; however, CDs are structurally rigid oligosaccharides with a nanosized hydrophobic interior. The reason for this is that, in addition to being a cyclic structure, intramolecular hydrogen bonds reinforce the structure. Such structures have been reproduced by artificial host molecules with advantages such as easy chemical modification; however, they are inferior in terms of biocompatibility and sustainability to CDs.
Cyclodextrins as sustainable macrocyclic molecules with various applications
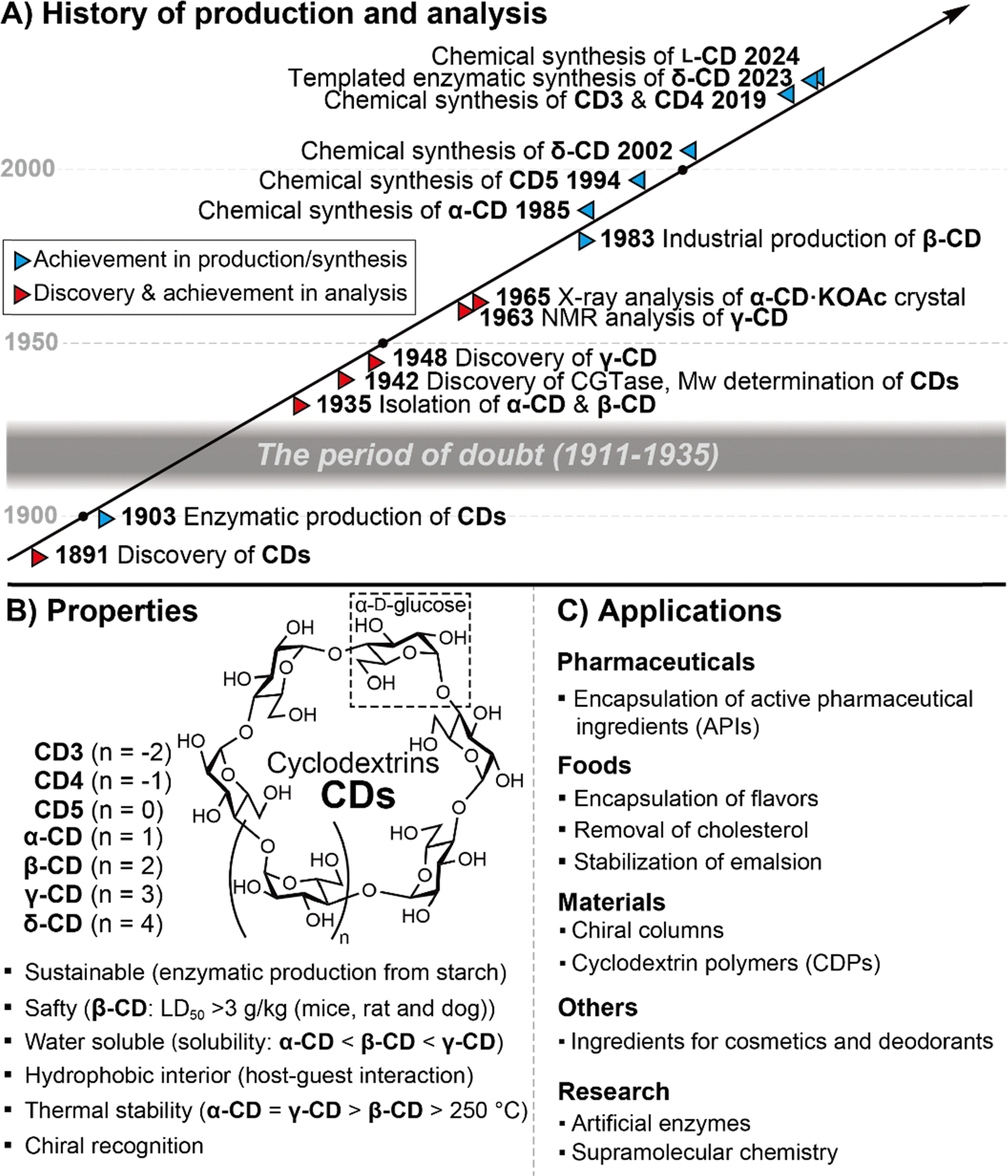
It has been more than 130 years since Villiers discovered CDs,12 however, the large-scale industrial production of β-CD had to wait until the 1980s (Fig. 2A). In the early stages of CD history, researchers experienced impurities and a lack of structural information, the so-called “period of doubt”.4 By the middle of the last century, significant progress had been made in structural determination,13,14 molecular recognition,15 and enzymatic production.16 Many scientists have been attracted to the unique properties of CDs as water-soluble macrocyclic molecules. The first chemical synthesis of CDs had to wait until 1985 because analytical techniques, such as high-resolution NMR and MS, which are crucial for structural determination in modern chemical synthesis, were not common by the 1980s.17 Since then, many cyclic oligosaccharides have been synthesized as macrocyclic compounds with many applications in supramolecular chemistry. Although various types of nanomaterials have been reported so far, CDs are highly appreciated because of their unique properties derived from the D-glucose repeating unit, which is one of the most sustainable natural resources (Fig. 2B). CDs are safe nanomaterials at reasonable prices.18CDs are soluble in water but are equipped with a hydrophobic interior that encapsulates hydrophobic molecules.19 Thermal stability has been an important property of CDs since their discovery, and chiral recognition has also been a distinctive function of CDs.20
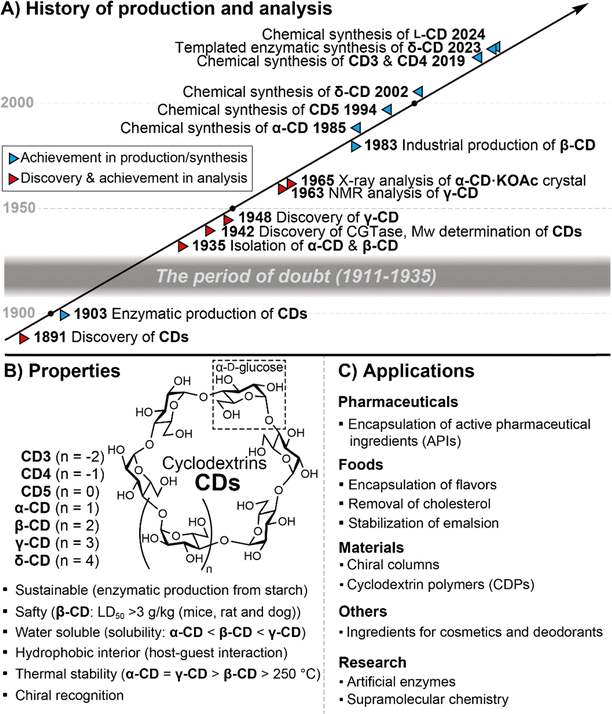 |
| | Fig. 2 History, properties, and applications of cyclodextrins. | |
The applications of CDs and their derivatives have expanded throughout various fields of daily life (Fig. 2C).21 In the pharmaceutical industry, CDs improve the solubility and stability of active pharmaceutical ingredients (APIs).22 By multiplying the function of sustained release, it is possible to adjust the location and concentration of APIs for drug delivery. In the food business, based on the biological safety of CDs, the quality and appearance of food products can be maintained by encapsulating flavour and colouring agents that are vulnerable to light and oxidation.23 In addition, by encapsulating ingredients that are the source of unpleasant odours and bitterness, CDs can mask the odour and bitterness by inhibiting binding to their receptors. This effective anti-odour property allows CDs to be used commercially as deodorants. Using CDs that have been pre-incrusted with fragrance components, it is also possible to release the fragrance components and encapsulate and remove the odour components. CDs are also used for separation.24 For example, β-cyclodextrin-linked silica gels can be used for the optical resolution of organic compounds. Cyclodextrin polymers are new materials that have the properties of CDs but are insoluble in water and organic solvents.25 They can be used as water purification agents because environmental pollutants such as VOCs and dioxins can be encapsulated.
CDs have been important molecules in academic research for many years. The contribution of CDs to supramolecular chemistry is significant, and various derivatives of CDs have been synthesized to date.26 Therefore, the structure and function of cyclic oligosaccharides composed of monosaccharides other than D-glucose are also of interest, and various cyclic oligosaccharides have been synthesized; however, unexplored cyclic oligosaccharides remain due to the underdevelopment of chemical synthesis technology.
Synthetic methods for preparing cyclic oligosaccharides
There are several methods for synthesizing cyclic oligosaccharides, including conventional enzymatic and chemical methods. The enzymatic production of CDs remains one of the most practical methods for producing specific cyclic oligosaccharides.16 For example, cyclodextrin glycosyltransferase (CGtase) produces α-, β-, and γ-CDs from amylose. Due to its selectivity and efficiency, enzymatic synthesis has been industrialized; however, the scope and ratio of cyclic oligosaccharides depend on the enzymes involved. The template-based enzymatic synthesis overcomes the limitations of conventional enzymatic methods. The enzymatic synthesis of larger cyclic oligosaccharides, such as γ- and δ-CDs, is still challenging because of their low yields and difficulty in purification. Zimmerman improved the selectivity for larger cyclic oligosaccharides using engineered CGtases and isolated these products in a single step.27 Beeren reported the selective enzymatic synthesis of δ-CD by introducing a bolaamphiphile template.28
Conventional chemical synthesis greatly expands the variety of cyclic oligosaccharides by assembling intentionally protected mono- or disaccharide units. Since Ogawa's first report on the chemical synthesis of α-CD,17 numbers of artificial cyclic oligosaccharides have been synthesized.29 Although the number of synthetic cyclic oligosaccharides has continued to increase, achieving these syntheses often involves lengthy synthetic routes. Electrochemical glycosylation30–32 is an alternative to conventional chemical glycosylation for synthesizing cyclic oligosaccharides.33 The electrochemical method offers the advantages of controlling reactions and generating highly reactive glycosylation intermediates at low temperatures. Multiple synthetic protocols for producing cyclic oligosaccharides have been developed across diverse research areas; however, each method presents its own advantages and limitations. Therefore, it is crucial to select an appropriate synthetic approach for the desired cyclic oligosaccharides.
Selected synthesis of cyclodextrin analogues
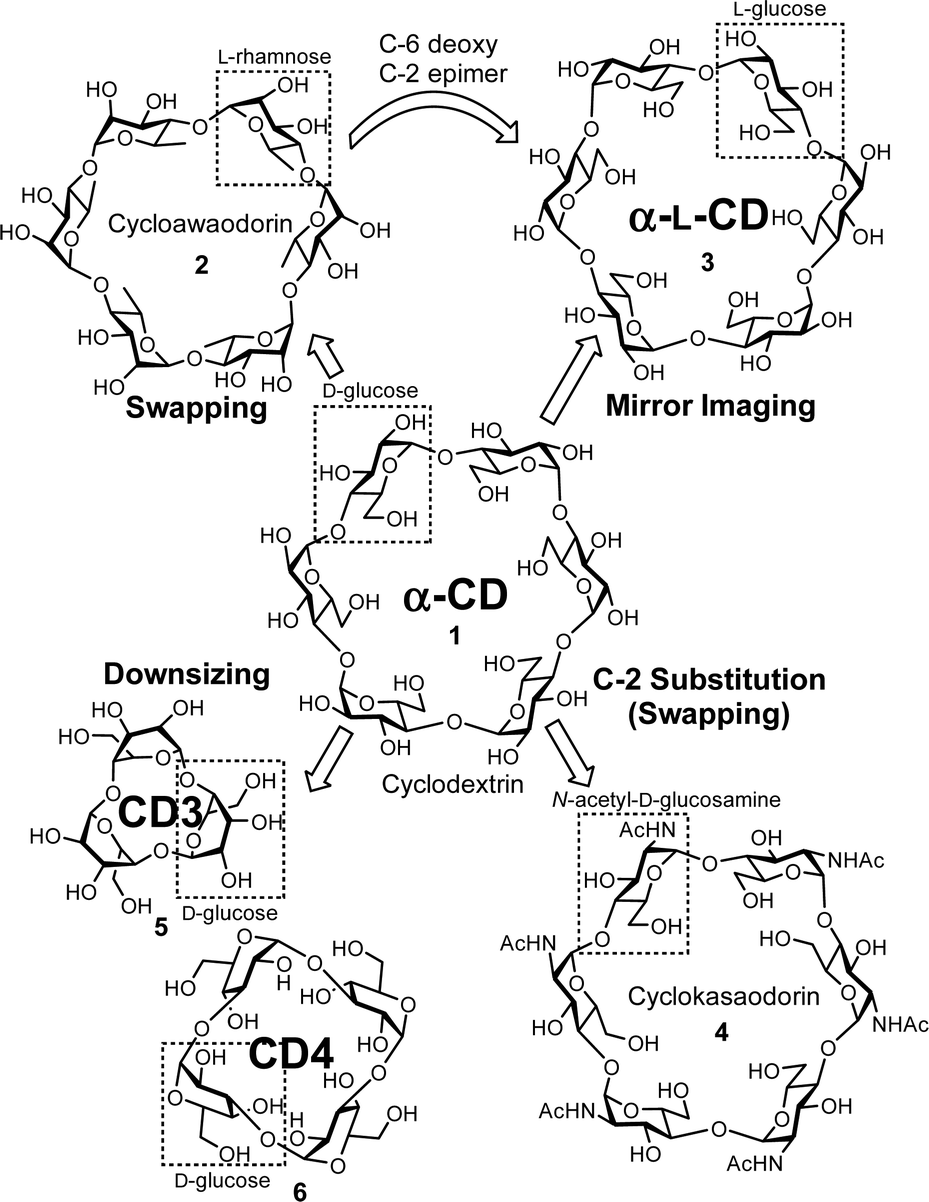
New methodologies are needed for synthesizing cyclodextrin analogues (Fig. 3). After the chemical synthesis of α-CD (1), a cyclic oligosaccharide consisting of L-rhamnose ‘Cycloawaodorin’ (2) was reported by Nishizawa and co-workers, and this is the first example of a cyclic oligosaccharide swapping normal D-sugars for L-sugars.34,35 The mirror-image CD (α-L-CD) (3),36 which is the C-6 deoxy and C-2 epimer of 2, had never been reported, although CDs have received considerable attention for many years. ‘Cyclokasaodorin’ (4) is also a nonnatural cyclic oligosaccharide with N-acetyl-D-glucosamine as a repeating unit.37 Although the structural differences between α-CD (1) and 4 are only the substituents at C-2 positions, it is difficult to convert all the C-2 hydroxyl groups of α-CD (1) into acetamide groups. Therefore, 4 must be prepared from D-glucosamine as a starting material. Recent advances in chemical glycosylation enable the synthesis of unique nonnatural cyclic oligosaccharides. CD3 (5) and CD4 (6) are the smallest CDs.38 In this article, we also focus on recent glycosylation methodologies that enable the synthesis of nonnatural cyclic oligosaccharides. There are some interesting analogues such as thio-linked or glycosidic bond expanded small-ring cyclic oligosaccharides,39,40 but these are beyond the scope of this article.
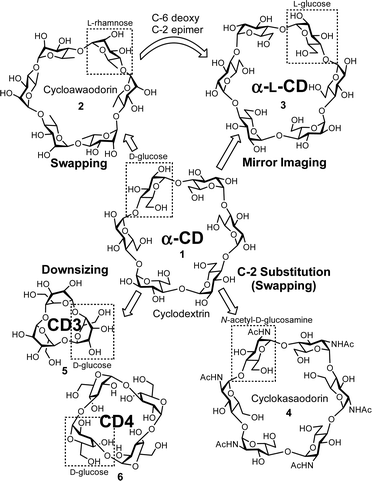 |
| | Fig. 3 Selected analogues of α-CD. | |
Recent examples from other groups
The smallest CDs
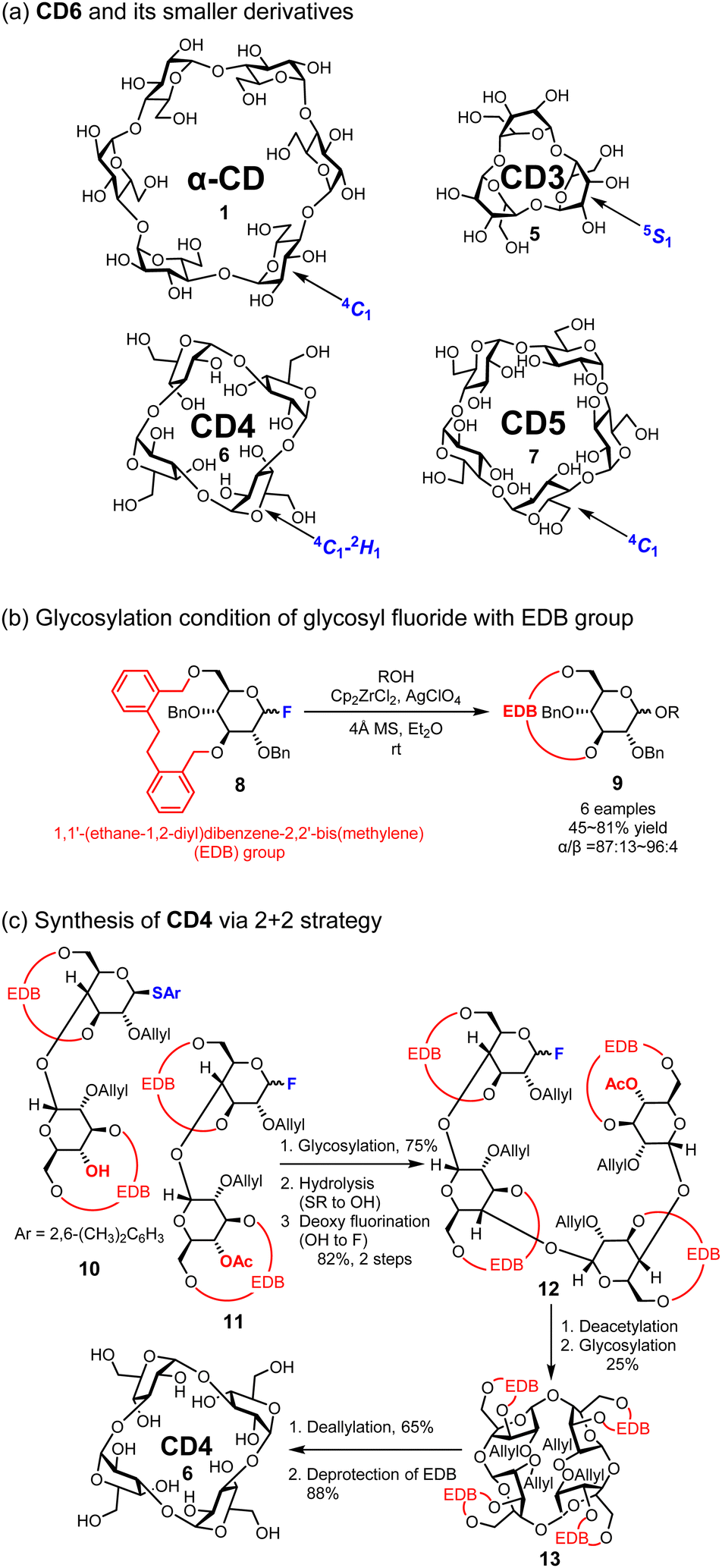
In 1970, Sundararajan and co-workers reported that CDs bearing fewer than six glucose molecules could not be cyclized due to their steric hindrance based on computational analysis.41 After 24 years, Nakagawa and co-workers synthesized a CD composed of five D-glucose units called CD5 (7);42 however, smaller and more strained CDs remained difficult to synthesize for another 25 years. In 2019, Yamada and co-workers reported the first synthesis of smaller CDs, specifically CD3 (5) and CD4 (6), using conformationally supple glucose monomers (Fig. 4).38 These monomers contain a benzyl-type protecting group named, [1,1′-(ethane-1,2-diyl)dibenzene-2,2′-bis(methylene)] (EDB) on 3-OH and 6-OH of D-glucose. The EDB group on the D-glucose monomer enables not only α-selective glycosylation, which is crucial for the synthesis of CDs, but also increases the conformational flexibility of pyran rings, which is also pivotal for synthesizing smaller CDs.43 Both CD3 (5) and CD4 (6) were synthesized via Mukaiyama–Suzuki glycosylation44,45 using the 2 + 1 and 2 + 2 strategies, respectively. CD3 (5) and CD4 (6) were successfully deprotected by removing the allyl groups at 2-OH of the pyranose ring and the EDB groups at 3-OH and 6-OH. The structure of CD3 (5) was also revealed through single-crystal X-ray structural analysis and NMR spectroscopy, and its stable structure of CD3 (6) in D2O is the 5S1 skew-boat conformation. This is an example of how the protecting group influences the conformation of the pyran ring, and the flexible glucose units enable the formation of CD3 (5) and CD4 (6).
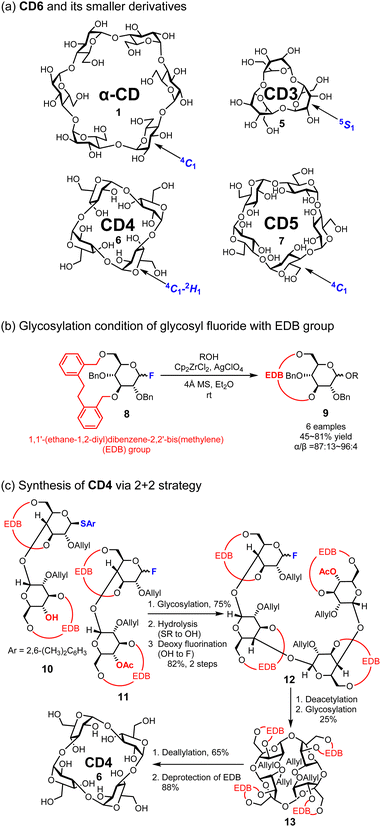 |
| | Fig. 4 Structures of α-CD to CD3 and the synthetic protocol of CD4. | |
The simplest approach towards cyclic oligosaccharides
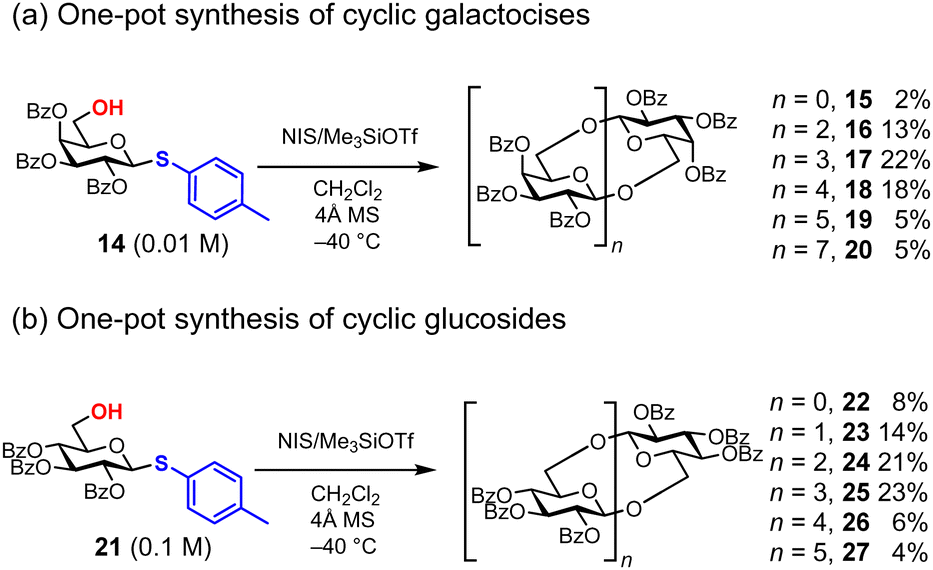
The chemical synthesis of cyclic oligosaccharides often involves a lengthy process, including two glycosylation steps, the elongation of linear oligosaccharides, and their cyclization (cycloglycosylation).46 In 2020, Aoki and co-workers reported the one-pot synthesis of cyclic oligosaccharides 15–20 bearing β-1,6-glycosidic bonds from thiogalactoside 14 as a monomer (Fig. 5a).47 Although the formation of anhydro sugars is a substantial problem to prevent the formation of larger cyclic oligosaccharides, this strategy is the simplest synthesis strategy of cyclic oligosaccharides by integrating the elongation and cyclization of linear oligosaccharides into a single step. Thioglycosides of D-galactose and D-glucose can be used as monomers 14 and 21, each equipped with benzoyl groups at 2, 3, and 4-OH. For this one-pot synthesis, 3.0 equivalents of NIS and 1.5 equivalents of Me3SiOTf in CH2Cl2 provide optimal conditions. In the case of D-glucose, the concentration of the initiating thioglycoside 21 is crucial for successful cyclization; at a low concentration of 21 (0.01 M), linear oligosaccharides, indicating incomplete cyclization, are observed. However, under more concentrated conditions of 21 (0.1 M), complete conversion to cyclic oligosaccharides was achieved (Fig. 5b). In this one-step synthesis, cyclic tetrasaccharides 17 and 25 are the major products of both D-glucose and D-galactose. This polyglycosylation approach works very well because cyclization is slower than intermolecular glycosylation, although it is difficult to predict the relative reaction rate of these competitive glycosylations.
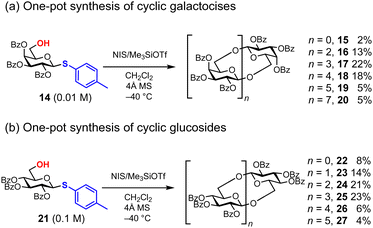 |
| | Fig. 5 Synthesis of cyclic oligosaccharides under polyglycosylation conditions. | |
The largest synthetic cyclic oligosaccharides
The major challenge in the construction of cyclic oligosaccharides is the cycloglycosylation of linear oligosaccharides prepared using multistep elongation sequences. Since the first synthesis of α-CD (1) by Ogawa,17 various cyclic oligosaccharides have been synthesized based on this strategy. The synthesis of ultra-large cyclic polysaccharides remains challenging because of the constrained conformational spaces of the precursors. Recently, Yang and co-workers reported the synthesis of cyclic mannoside of various sizes by promoter-controlled cycloglycosylation using oligosaccharide thioglycosides and (Z)-ynenoates (Fig. 6).48,49 Particularly noteworthy is the synthesis of ultra-large cyclic polymannosides (16-mer 38, 32-mer 39) via cycloglycosylation under high-dilution conditions (0.001 M). Cycloglycosylation is highly dependent on the promoters. In small and middle-sized cyclic oligosaccharides, with the elongation of the linear oligosaccharides (4-mer 28, 8-mer 29), the more effective promoters for the cycloglycosylation of thioglycoside precursors are IBr/AgOTf rather than NIS/Me3SiOTf. Furthermore, excess amounts of promoters are found to dramatically accelerate the cycloglycosylation of the linear oligosaccharides 28 and 29. In contrast, activation of oligosaccharide (Z)-ynenoates by a stoichiometric amount of a gold(I) complex is more effective for the cycloglycosylation of ultra-large linear oligosaccharides 34 and 35 by proper preorganization of the ultra-large cyclic transition state. The stabilizing effect of the oxocarbenium ions also led to higher yields when using toluene as the solvent compared with CH2Cl2. The global deprotection of precursors 34 and 35 afforded the corresponding cyclic oligosaccharides 38 and 39 in reasonable yields. Although the demonstrated monomer is limited to mannoside with β-1,6-glycosidic bonds, the importance of the anomeric leaving groups and their promoters was clearly demonstrated by this study.
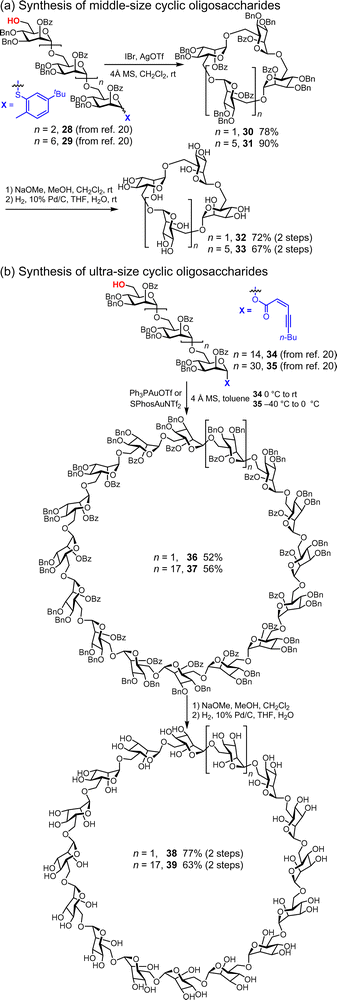 |
| | Fig. 6 Synthesis of cyclic oligomannosides. | |
Synthesis of mirror-image CDs
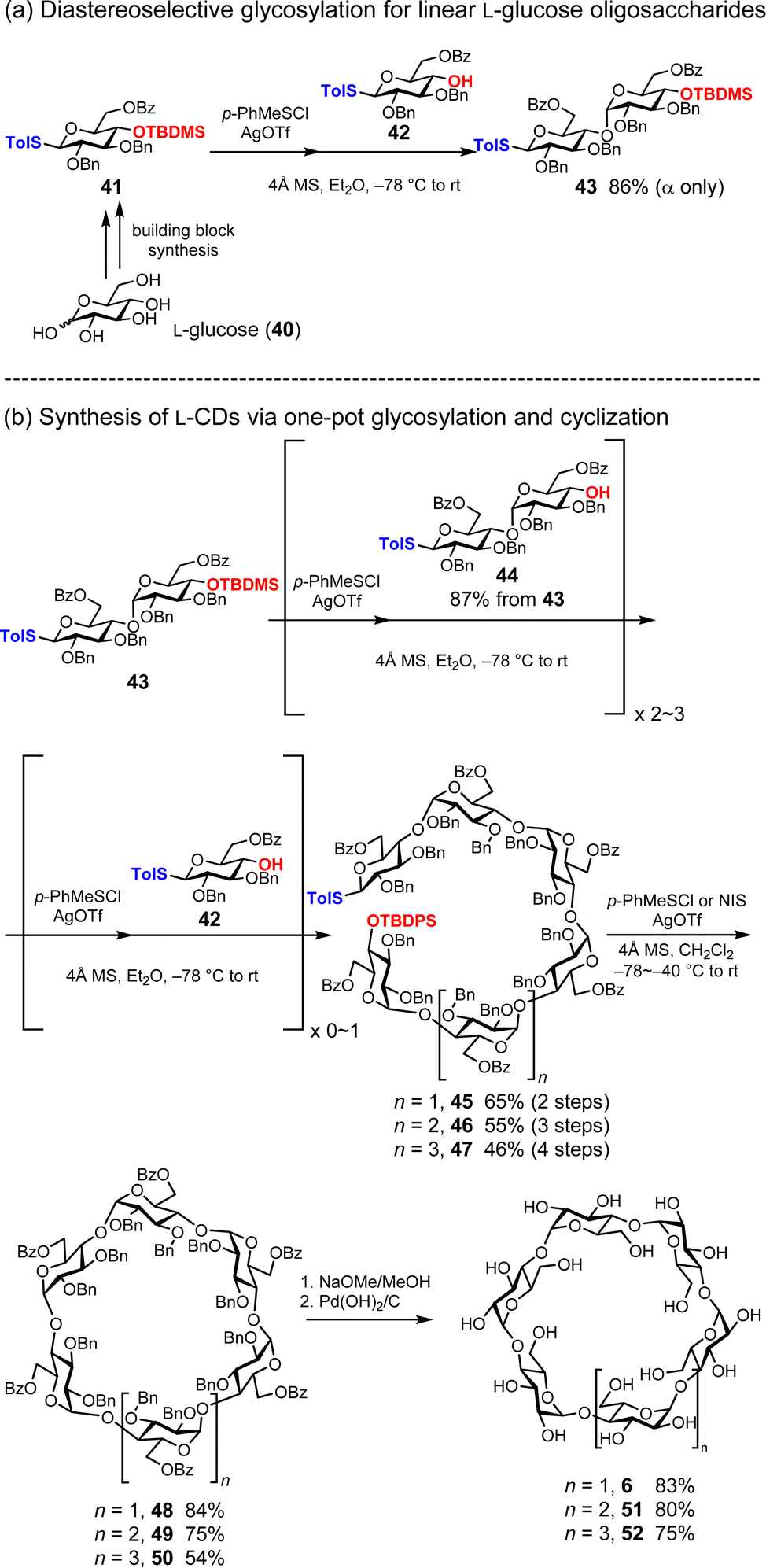
Naturally occurring CDs exhibit chirality originating from their component D-glucose units.50 As mentioned above, Nishizawa and co-workers synthesized nonnatural cyclic oligosaccharides ‘cycloawaodorin’ consisting of L-rhamnose, which is a natural L-sugar. Recently, Stoddard and co-workers reported three mirror images of CDs (α-L-CD) (3), 51, and 52 consisting of nonnatural L-glucose (Fig. 7).37 To this end, they employ L-glucose building blocks 41 and 42 bearing benzyl groups (Bn), benzoyl groups (Bz), tert-butyldimethylsilyl groups (TBDMS), and p-toluene thiol (STol). For the diastereoselective formation of α-1,4-glycosidic bonds with L-glucose, they devised a strategy combining the solvent effect of Et2O51 and remote anchimeric assistance originating from the Bz group at 6-OH.52 As an activator of glycosylation, p-toluenethionyl chloride (p-PhMeSCl) and AgOTf were used, resulting in the desired disaccharide 43 with 86% yield and complete α-selectivity (Fig. 7a). The cyclic hexasaccharide precursor 45 was synthesized in 65% yield by donor preactivation-based one-pot glycosylation,53–57 reducing the need for intermediate purification steps. They extended the linear hexasaccharide 45 to synthesize linear heptasaccharide 46 and octasaccharide 47 in 55% and 45% yields, respectively (Fig. 7b). Cyclization was achieved using the corresponding lengths of linear oligosaccharides 48, 49, and 50, NIS or p-PhMeSCl activators, and CH2Cl2, resulting in yields of 84%, 75%, and 54%, respectively. They investigated the chiral properties of deprotected α-L-CD (3) and found that α-L-CD (3) exhibited opposite chiral properties to those of natural CDs. The preactivation strategy is a powerful method for preparing linear oligosaccharides equipped with reactive anomeric leaving groups and protecting-group-free hydroxyl groups.
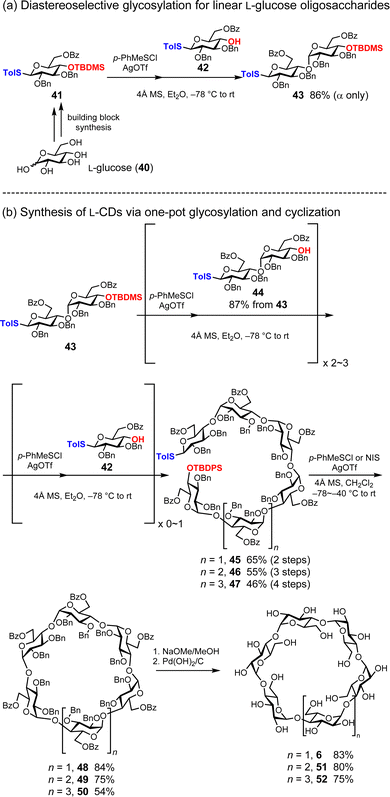 |
| | Fig. 7 Total synthesis of L-CDs. | |
Synthesis of cyclic oligosaccharides via intramolecular electrochemical glycosylation
Automated electrochemical assembly
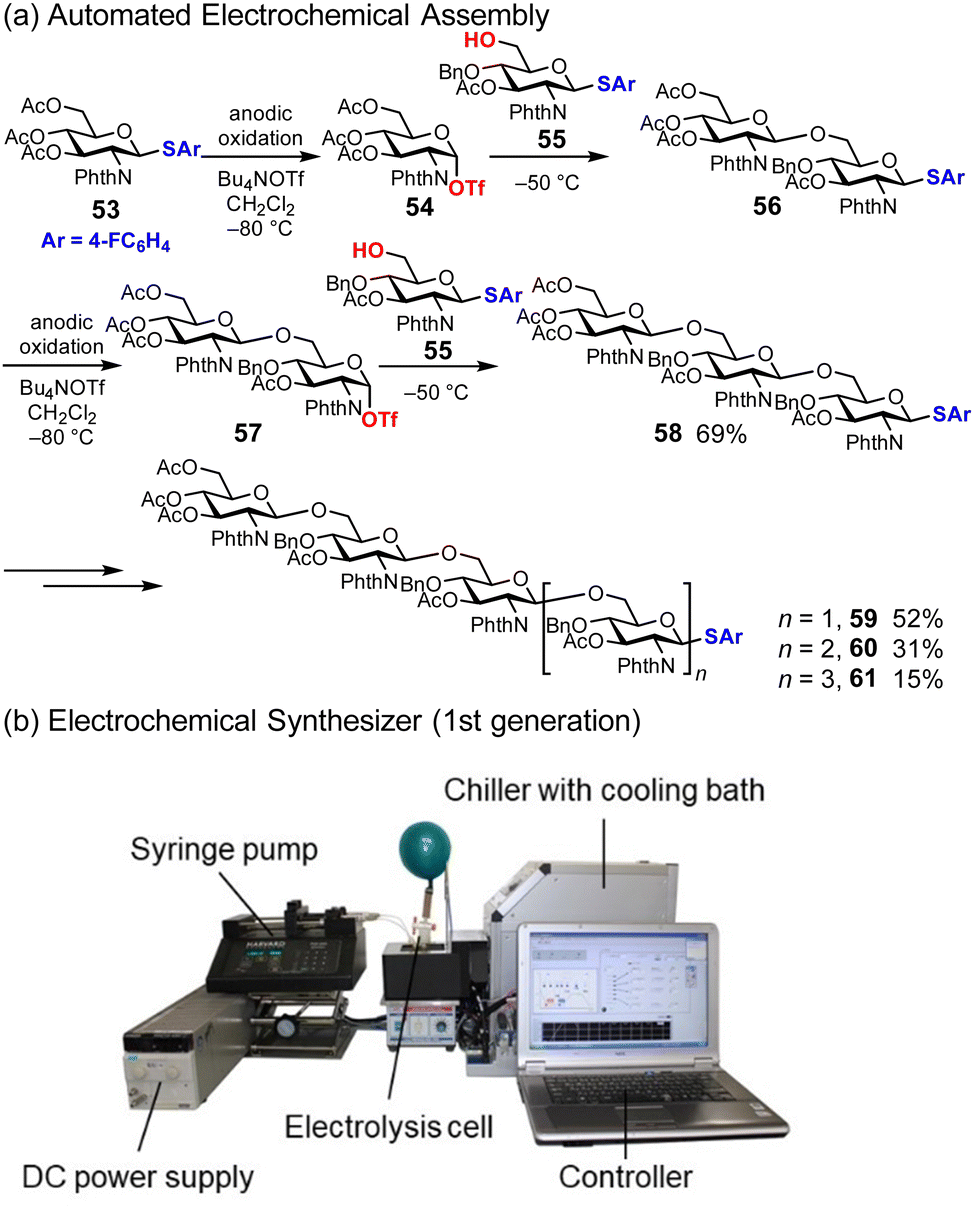
Similar to automated synthesis of oligonucleotides and oligopeptides, automated synthesis of oligosaccharides is developed based on solid-phase synthesis.58,59 In contrast, we have been interested in the solution-phase synthesis of oligosaccharides and inspired by the ‘preactivation method’ and the ‘cation pool method’.60,61 The preactivation method is a two-step process based on the generation of a highly reactive glycosylation intermediate in the absence of nucleophiles; thus the generated intermediate reacts with nucleophiles, which are sugar hydroxyl groups. This method allows the elongation of oligosaccharides multiple times in one pot without the need for deprotection. The cation pool method is based on the electrochemical generation of carbocations in the absence of nucleophiles. Thus, the generated carbocations can react with various nucleophiles with low oxidation potentials because the nucleophiles are not exposed to anodic oxidative conditions. The ‘automated electrochemical assembly’ developed by the authors is an electrochemical solution-phase synthesis to produce oligosaccharides in a one-pot automated manner (Fig. 8a).62,63 For example, thioglycoside 53 is used as a starting material, which is converted to the corresponding glycosyl triflate 54via anodic oxidation at low temperatures. Thus, the generated glycosyl triflate 54 is sufficiently reactive to couple with the sugar hydroxyl group of 55 in one pot, affording disaccharide 56. Oligosaccharides with the desired chain length can be obtained by repeating this two-step process. To conduct this process in an automated manner, we developed an electrochemical synthesizer equipped with an electrolysis cell, DC power supply, chiller, syringe pump, and PC (Fig. 8b). The synthesis of poly-N-acetylglucosamine (PNAG) oligosaccharides was demonstrated using an electrochemical synthesizer, and PNAG oligosaccharides up to hexasaccharide 61 (n = 3) were obtained.62 Automated electrochemical assembly can be used to synthesize other linear oligosaccharides, including TMG-chitotriomycin64,65 and Myc-IV(C16:0, S).66 There are several benefits to automated synthesis using an electrochemical synthesizer, such as reproducibility, observability, and controllability; however, productivity (time, scale, and yield) has to be improved for further applications of this method.
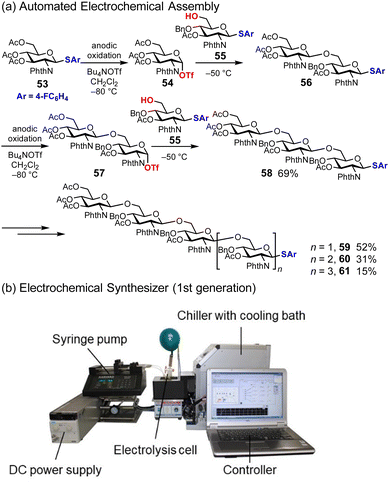 |
| | Fig. 8 Automated electrochemical assembly for the synthesis of oligosaccharides. | |
Electrochemical synthesis of cyclic oligoglucosamines
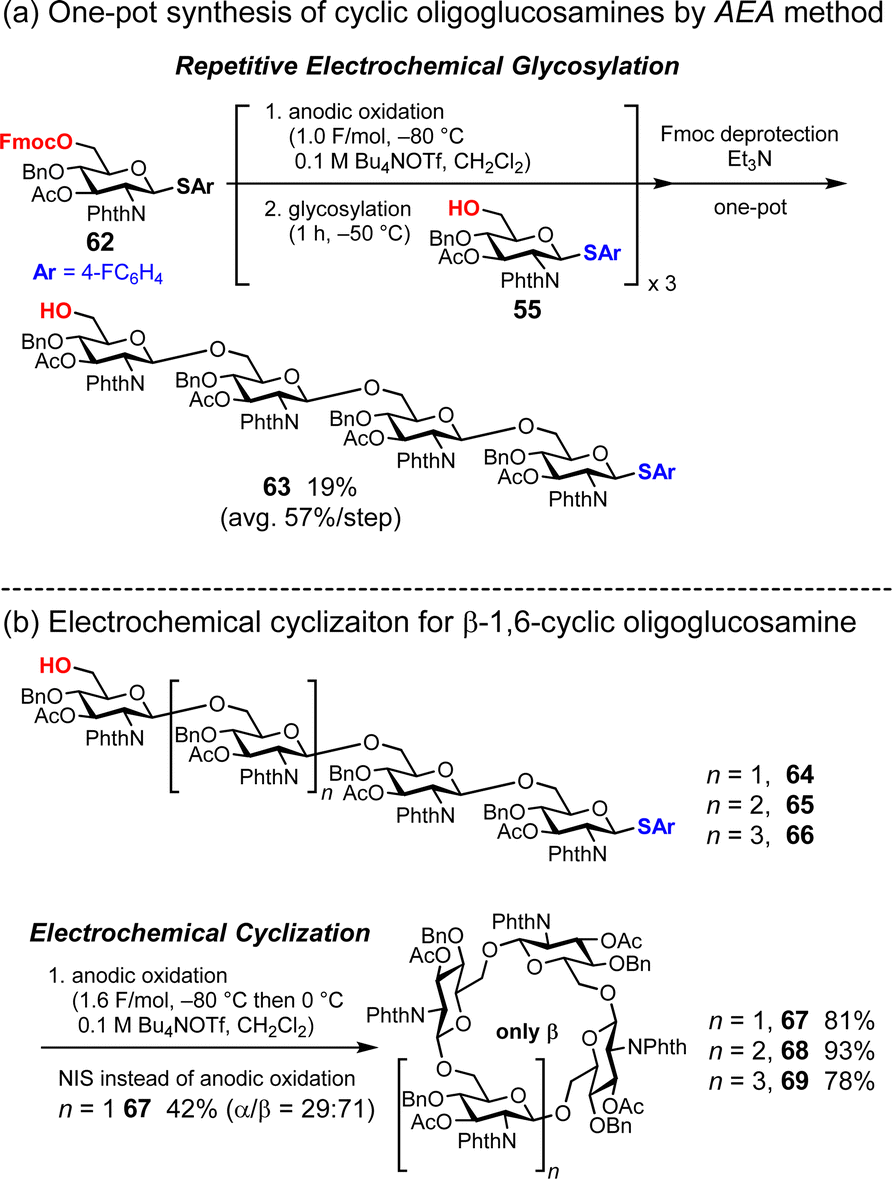
Cyclic oligoglucosamines are nonnatural cyclic oligosaccharides. Nifantiev and co-workers have achieved the synthesis of cyclic oligo-1,6-β-D-glucosamines using thioglycosides as building blocks and N-iodosuccinimide (NIS) and triflic acid (TfOH) as the promoter system.67,68 However, in the case of linear oligosaccharides containing four, six, and seven glucosamine units, the control of stereochemistry is not perfect, even in the presence of a strongly participating 2-N-phthaloyl group to control β-glycosylation. In contrast, the electrochemical method enabled the stereoselective synthesis of cyclic oligoglucosamines.33 Linear oligoglucosamines (the precursors of the cyclic oligoglucosamines) are readily prepared using an automated electrochemical synthesizer (Fig. 9a). In the cyclization process, stereoselective electrochemical glycosylation afforded the corresponding cyclic oligoglucosamines in high yields (cyclic tetrasaccharide 67: 81%; cyclic pentasaccharide 68: 93%; and cyclic hexasaccharide 69: 78%) (Fig. 9b). We also performed the cyclization of linear tetrasaccharide 63 under conventional chemical glycosylation conditions using the NIS/TfOH system. The reaction afforded both α- and β-isomers of cyclic oligoglucosamine 67. Therefore, electrochemical glycosylation is a powerful tool for synthesizing cyclic oligosaccharides, allowing for complete stereoselectivity by optimizing the electrochemical glycosylation conditions, such as temperature, electrolyte, and reaction time.
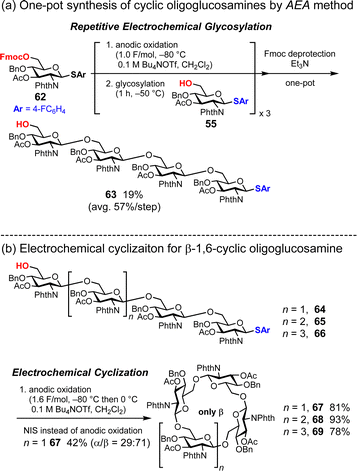 |
| | Fig. 9 Synthesis of cyclic oligoglucosamines. | |
Electrochemical synthesis of cyclic β-glucans
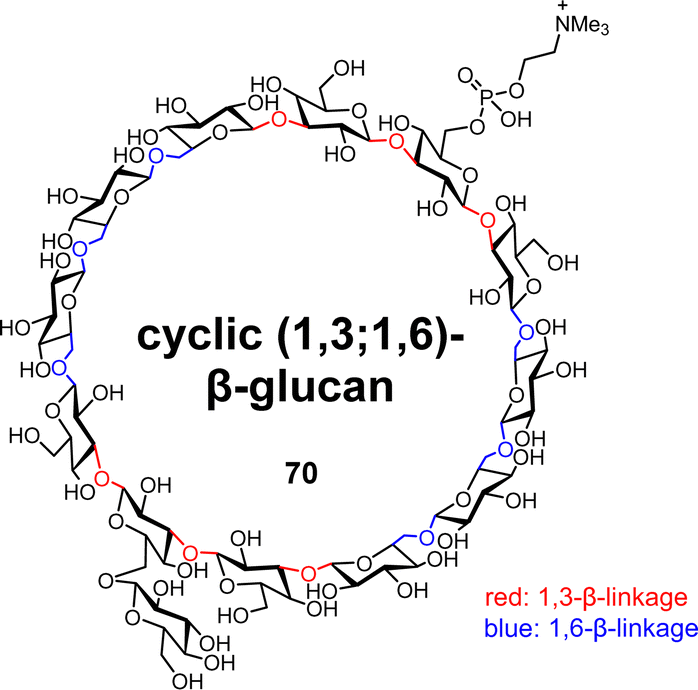
β-glucans are polysaccharides consisting of glucose repeating units linked by β-glucosidic bonds. Among them, cyclic β-glucans play various roles. For example, they exhibit biological activities such as osmotic regulation,69 rhizogenesis,70 and antioxidant activity,71 and the ability to encapsulate target compounds.72 Cyclic β-glucans have a variety of different structures, and we are particularly interested in the chemical synthesis of naturally occurring cyclic (1,3;1.6)-β-glucans 70 with two different glycosidic linkages (Fig. 10).73,74
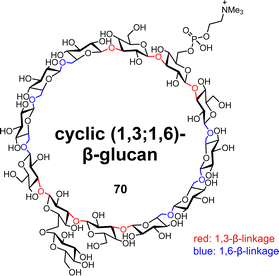 |
| | Fig. 10 Structure of the cyclic (1,3;1,6)-β-glucan tridecasaccharide. | |
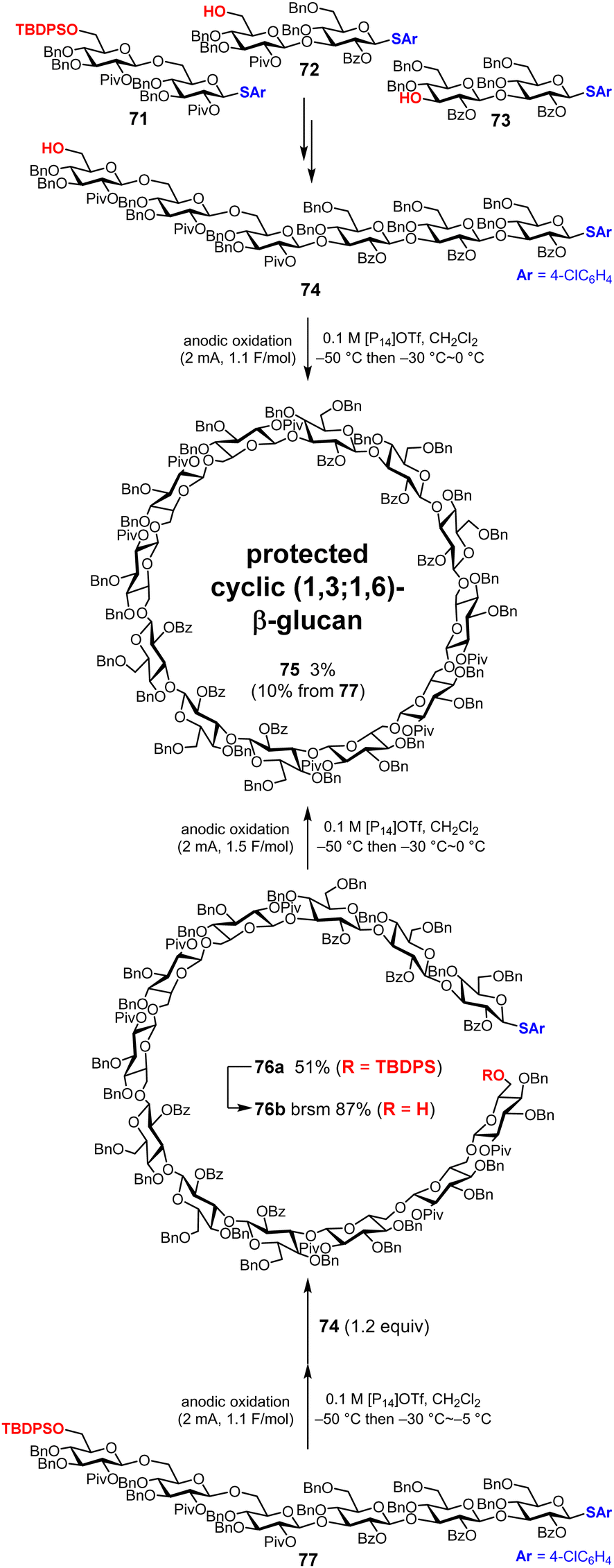
Initially, we synthesized a linear hexasaccharide 74 as the target cyclic oligosaccharide (Fig. 11). We synthesized three disaccharide building blocks 71–73 by electrochemical glycosylation, and the linear hexasaccharide 74 was synthesized by assembling disaccharide 71 and the tetrasaccharide obtained from the remaining two disaccharide building blocks 72 and 73. During these processes, ionic liquid 1-butyl-1-methylpyrrolidinium ([P14]OTf) instead of tetrabutylammonium triflate (Bu4NOTf) was used as an electrolyte to improve the yields of oligosaccharide building blocks. Thus, [P14]OTf is used for dimerization and cyclization as well. Next, we synthesized the protected cyclic (1,3;1,6)-β-glucan dodecasaccharide 75via a one-pot synthesis under electrochemical dimerization–cyclization conditions;48 however, the yield of 75 was only 3%. This low yield was due to the intramolecular cyclization of linear hexasaccharide 74 and excessive reactions of hexasaccharide 74 with each other to form smaller and larger cyclic oligosaccharides, which are not desired compounds. To suppress these side reactions, stepwise synthesis via intramolecular glycosylation of linear dodecasaccharide 76b was also performed. As a result, the yield of 75 increased from 3% (1 step) to 10% (3 steps).75 Further optimization of the reaction conditions and global deprotection of cyclic oligosaccharide 75 are currently underway.
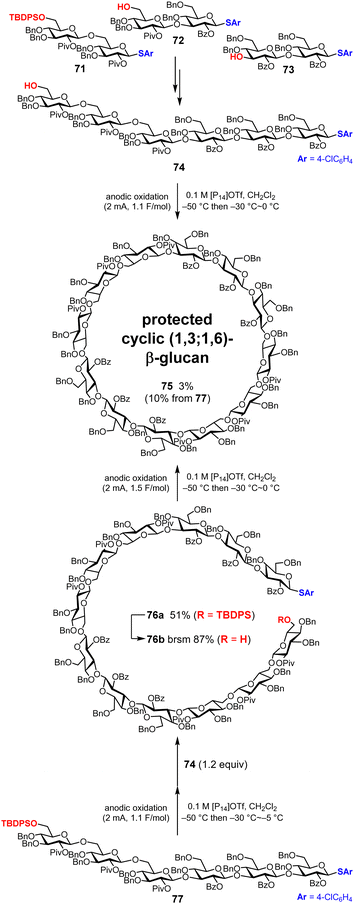 |
| | Fig. 11 Synthesis of the protected precursor cyclic (1,3;1,6)-β-glucan dodecasaccharide. | |
Synthesis of cyclic oligosaccharides via electrochemical polyglycosylation
Electrochemical polyglycosylation of linear oligosaccharides
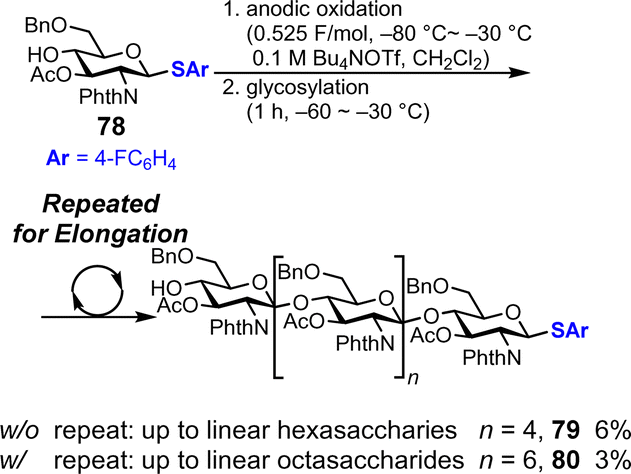
Although polyglycosylation is not very useful for synthesizing oligosaccharides with specific chain lengths, it is a practical method for simultaneously preparing oligosaccharides with different chain lengths. The first report on polyglycosylation was published by Yoshimura in 1988.76 After this work, protocols for the polyglycosylation were explored; however, controlling the degree of polymerization of products remained difficult with conventional chemical reagents,77 which often caused insufficient reactivity for polymerization or overreaction. In 2022, our group reported the electrochemical polyglycosylation of linear oligoglucosamine with β-1,4-glycosidic bonds consisting of D-glucosamine monomers, and this method enabled the control of the degree of polymerization of the product by simply changing the reaction conditions for the reaction (Fig. 12).78 We investigated various conditions such as the temperature of anodic oxidation and glycosylation, leaving groups, and the number of electrolysis cycles. By optimizing the reaction conditions for elongation, we successfully synthesized linear octasaccharide 80 (n = 6) in 3% yield, which was previously too long to be chemically synthesized.
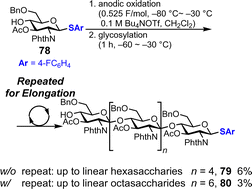 |
| | Fig. 12 Synthesis of linear oligosaccharides under polyglycosylation conditions. | |
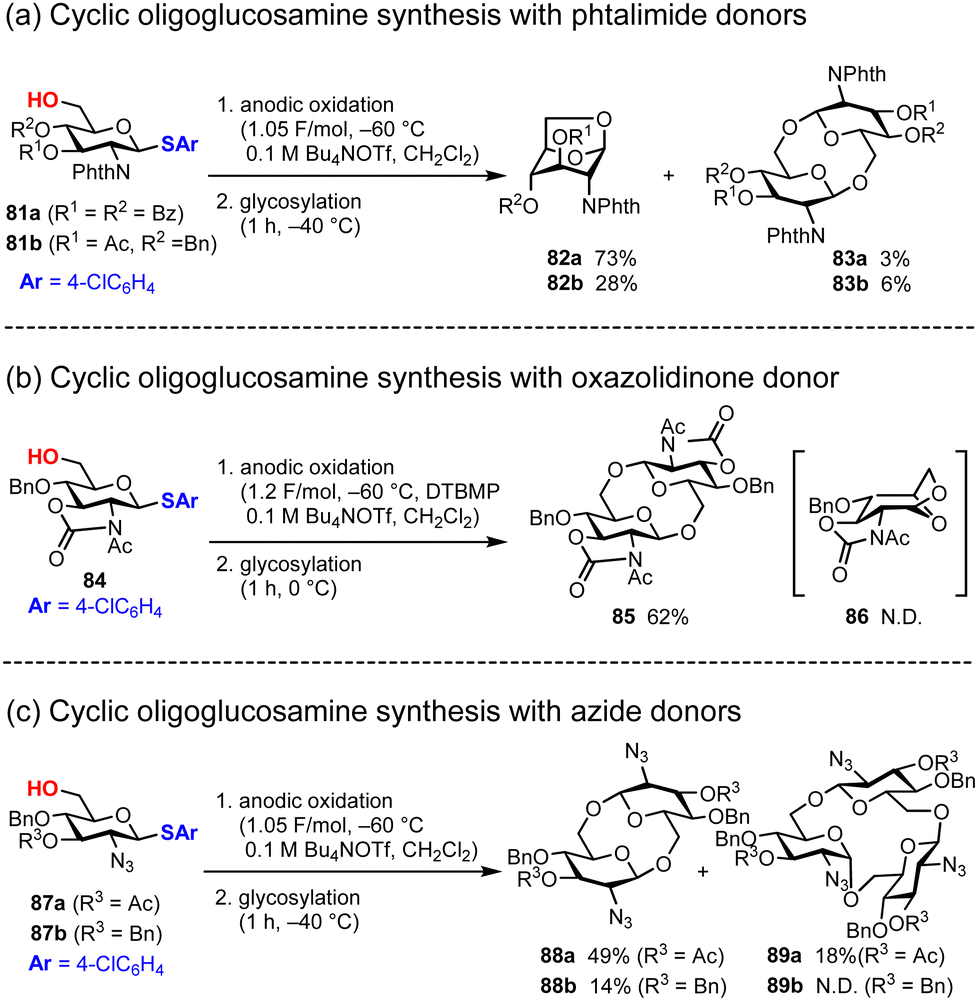
Based on these results, we reported the electrochemical polyglycosylation of cyclic oligoglucosamine with several D-glucosamine monomers in 2024.79 Although polyglycosylation for cyclic oligosaccharides with D-glucose and D-galactose has already been reported by Aoki, the polyglycosylation with D-glucosamine has remained untouched due to the poor reactivity of D-glucosamine monomers, which often have electron-withdrawing protecting groups for the reactive amine. Consistent with this hypothesis, we report that the D-glucosamine building block bearing a phthalimide protecting group 81, one of the most common electron-withdrawing protecting groups for amines, does not yield more than cyclic trisaccharides; instead, we obtained only anhydrosugar 82 exclusively (Fig. 13a). For the synthesis of cyclic oligoglucosamine, we used D-glucosamine monomer 84 equipped with a 2,3-oxazolidinone protecting group that suppressed inversion of the pyranose ring, which is the major cause of the generation of anhydrosugar 86 (Fig. 13b). By employing D-glucosamine bearing 2,3-oxazolidinone groups, we exclusively obtained cyclic disaccharide 85 in 62% yield. We also tried a D-glucosamine monomer protected with azide groups (Fig. 13c). In the case of the azide group, the stereoselectivity of glycosylation could be a problem because it does not have any acryl groups, which are crucial for neighboring group participation; however, we obtained cyclic disaccharide 88a in 49% with perfect β-selectivity. In the same reaction, cyclic trisaccharide 89a was also obtained; however, it contains both α- and β-glycosidic linkages. Although the mechanistic details of the formation of cyclic trisaccharide 89a was not clear, the formation of an α-glycosidic linkage at the first glycosylation may be crucial to obtain 89a. Therefore, a better strategy for the synthesis of a larger cyclic oligosaccharide with α-selective glycosylation has to be developed with β-1,6-glycosidic linkages of glucosamine via the polyglycosylation approach.
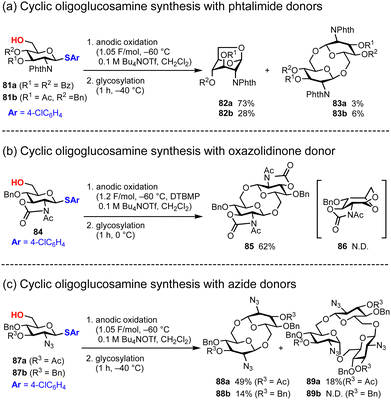 |
| | Fig. 13 Synthesis of cyclic oligosaccharides under polyglycosylation conditions. | |
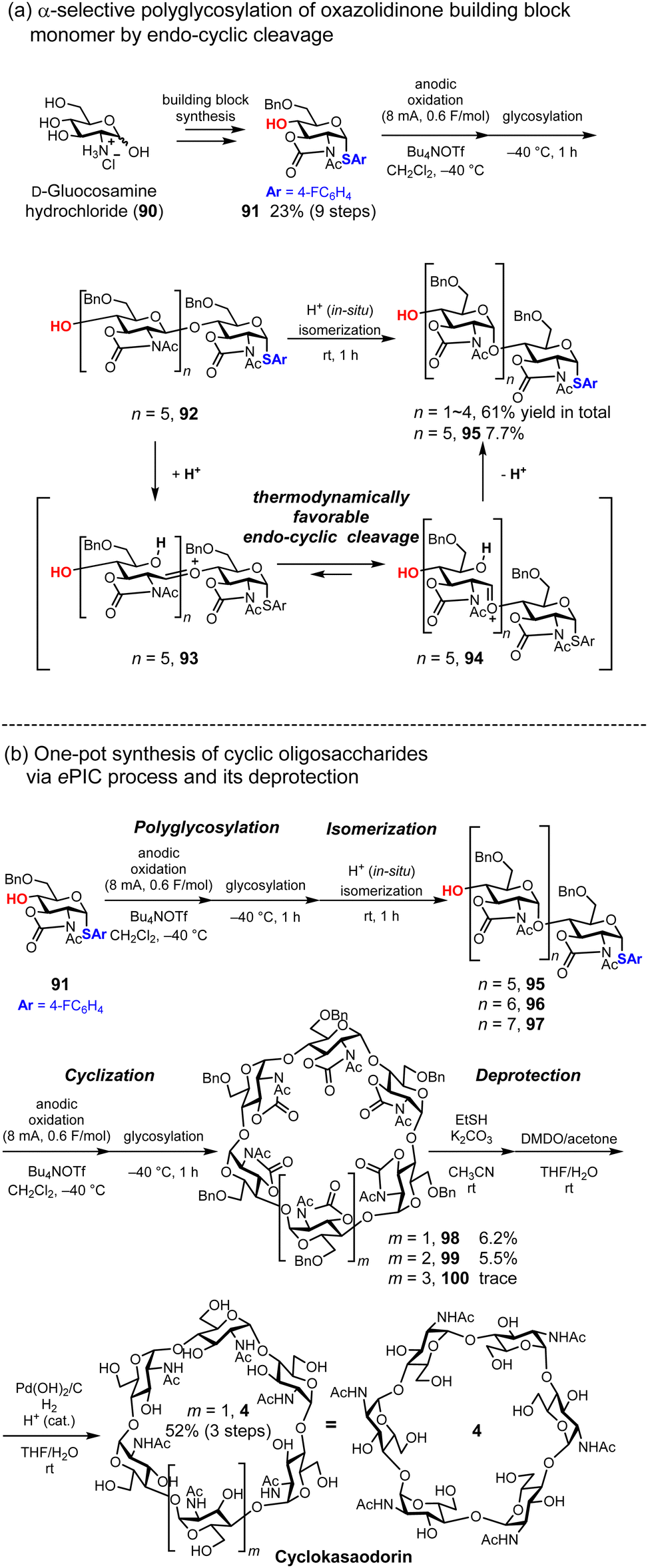
Synthesis of cyclokasaodorin
Although D-glucose exhibits structural variety, including amylose (linear α-1,4-glycosidic bonds), cellulose (linear β-1,4-glycosidic bonds), and CD (cyclic α-1,4-glycosidic bonds), D-glucosamine, which is an aminated form of D-glucose at the 2-position of the pyranose ring, occurs in nature exclusively as chitin and chitosan, which have linear β-1,4-glycosidic bonds. In 2022, we achieved the first total synthesis of unnatural cyclic oligosaccharides composed of N-acetylglucosamine with α-1,4-glycosidic bonds, naming it cyclokasaoddorin (4),36 inspired by the synthesis of cycloawaodorin (2) reported by Nishizawa.34,35 Synthesis of cyclodextrin-shaped cyclic oligoglucosamine requires exclusive 1,2-cis selectivity at the anomeric position and 2,3-oxazolidinone protection, which enables α-selective glycosylation via endo-cyclic cleavage induced by the internal strain of the oxazolidinone ring.80–84 We designed and prepared a D-glucosamine-based thioglycoside donor 91 bearing 2,3-oxazolidinone and Bn groups. Then, we established a polyglycosylation protocol to convert monosaccharides into oligosaccharides via anodic oxidation. Electrochemical glycosylation provided control over the degree of polymerization, and we determined that 0.6 F mol−1 of electricity was optimal for polymerization of the monomer (Fig. 14a). After electrolysis, the reaction temperature is increased with isomerization from β-glycosidic bonds to α-glycosidic bonds, driven by in situ generated acid. Subsequently, we demonstrate the cyclization of a linear α-1,4-hexasaccharide 95 (n = 5) into the corresponding cyclic oligosaccharide, achieving a 44% yield of the desired cyclic hexasaccharide 98 (m = 1). By integrating polyglycosylation and cyclization into a one-pot process, we developed a synthetic protocol for cyclic hexasaccharides, heptasaccharides, and octasaccharides. We named this process ePIC, which stands for ‘electrochemical polyglycosylation, isomerization, and cyclization’. After the initial electrochemical polyglycosylation and acid-induced thermodynamic isomerization, electrolysis was repeated with 1.0 F mol−1 of electricity to promote cyclization. This process enables the one-pot synthesis of cyclic oligoglucosamines, yielding cyclic hexasaccharide 98, heptasaccharide 99, and octasaccharide 100 in 6.2%, 5.5%, and trace amounts, respectively. Deprotection of hexasaccharide 5 was accomplished through the site-selective cleavage of carbonyl groups using ethanethiol,85 oxidative removal of the thioester with dimethyl dioxirane, and deprotection of the benzyl groups (Fig. 14b). Although the desired cyclic oligosaccharide 5 was obtained, the yield of its linear oligosaccharide precursor 95 was moderate via the ePIC process. Therefore, a stepwise process using a disaccharide building block, as shown in the L-CD synthesis (Fig. 7b), might be a useful alternative for preparing linear oligosaccharide precursors 95–97.
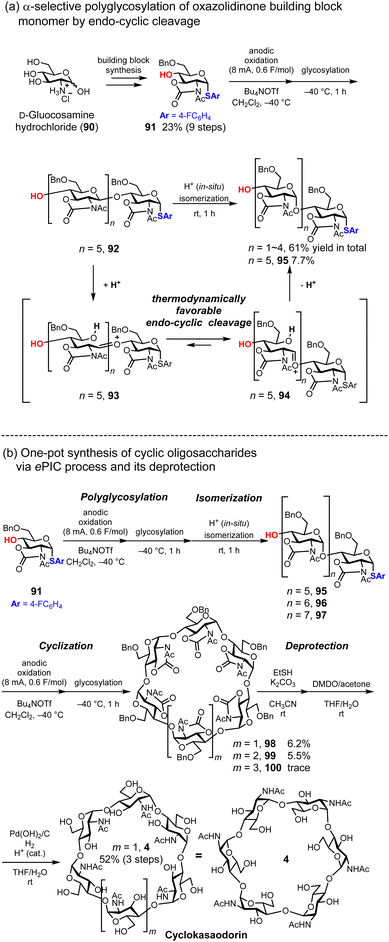 |
| | Fig. 14 Synthesis of ‘cyclokasaodorin’ via electrochemical polyglycosylation. | |
Conclusions
In this feature article, we introduce electrochemical approaches to cyclic oligosaccharides, with recent reports from other groups. Although it is still not clear why the electrochemical method is useful for converting linear oligosaccharides into cyclic oligosaccharides, the generation of reactive intermediates under low-temperature conditions may play a pivotal role. The development of synthetic protocols, protecting groups, and leaving groups with their activation system have led to the synthesis of novel cyclic oligosaccharides. Thus, further advancements in the synthetic methods of complex oligosaccharides, including cyclic oligosaccharides, will allow the development of synthetic approaches toward unique oligosaccharide-based structures and materials.
Data availability
No primary research results, software, or code have been included and no new data were generated or analyzed as part of this feature article.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge financial support from the JSPS (grant no. JP25410115, JP19K05714, and JP23K26654) and the MEXT (grant no. JP15H05844). We thank Koganei Corporation for developing the second-generation electrochemical synthesizer and their continuous support.
References
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021 Search PubMed.
- C. D. Gutsche, Acc. Chem. Res., 1983, 16, 161 Search PubMed.
- T. Ogoshi, T. Yamagishi and Y. Nakamoto, Chem. Rev., 2016, 116, 7937 Search PubMed.
- G. Crini, Chem. Rev., 2014, 114, 10940 Search PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320 Search PubMed.
-
R. Gleiter and H. Hopf, Modern Cyclophane Chemistry, Wiley-VCH Verlag, 2004 Search PubMed.
- S. E. Lewis, Chem. Soc. Rev., 2015, 44, 2221 Search PubMed.
- D. Imoto, A. Yagi and K. Itami, Precis. Chem., 2023, 1, 516 Search PubMed.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794 Search PubMed.
- Molecular structures except α-CD were obtained using CONFLEX 9.
- R. Granero-García, F. J. Lahoz, C. Paulmann, S. Saouane and F. P. A. Fabbiani, CrystEngComm, 2012, 14, 8664 Search PubMed.
- A. Villiers, Bull. Soc. Chim. Paris, 1891, 45, 468 Search PubMed.
- V. S. R. Rao and J. F. Foster, J. Phys. Chem., 1963, 67, 951 Search PubMed.
- A. Hybl, R. E. Rundle and D. E. Williams, J. Am. Chem. Soc., 1965, 87, 2779 Search PubMed.
-
M. L. Bender and M. Komiyama, Cyclodextrin Chemistry, Springer Verlag, Berlin, 1978 Search PubMed.
- A. Biwer, G. Antranikian and E. Heinzle, Appl. Microbiol. Biotechnol., 2002, 59, 609 Search PubMed.
- Y. Takahashi and T. Ogawa, Carbohydr. Res., 1985, 138, C5 Search PubMed.
- A. Mortensen, F. Aguilar, R. Crebelli, A. Di Domenico, B. Dusemund, M. Jose Frutos, P. Galtier, D. Gott, U. Gundert-Remy, J.-C. Leblanc, O. Lindtner, P. Moldeus, P. Mosesso, D. Parent-Massin, A. Oskarsson, I. Stankovic, I. Waalkens-Berendsen, R. A. Woutersen, M. Wright, M. Younes, P. Boon, D. Chrysafidis, R. Gürtler, P. Tobback, D. Arcella, A. M. Rincon and C. Lambré, EFSA J., 2016, 14, 4628 Search PubMed.
-
A. Hedges, Starch: Chemistry and Technology, Academic Press, Cambridge, 3rd edn, 2009, p. 833 Search PubMed.
- A. K. Gataitulin, I. A. Grishin, A. V. Buzyurov, T. A. Mukhametzyanov, M. A. Ziganshin and V. V. Gorbatchuk, Int. J. Mol. Sci., 2022, 23, 13120 Search PubMed.
- A. R. Hedges, Chem. Rev., 1998, 98, 2035 Search PubMed.
- M. Davis and M. Brewster, Nat. Rev. Drug Discovery, 2004, 3, 1023 Search PubMed.
- Z. Zhang, J. Niu, J. Wang, Q. Zheng, W. Miao, Q. Lin, X. Li, Z. Lin, C. Qiu, S. Sang and H. Ji, Food Res. Int., 2024, 195,
114952 Search PubMed.
- E. Schneiderman and A. M. Stalcup, J. Chromatogr. B: Biomed. Sci. Appl., 2000, 745, 83 Search PubMed.
- Z. Liu, L. Ye, J. Xi, J. Wang and Z.-G. Feng, Prog. Polym. Sci., 2021, 118,
101408 Search PubMed.
- G. Gattuso, S. A. Nepogodiev and J. F. Stoddart, Chem. Rev., 1998, 98, 1919 Search PubMed.
- C. Sonnendecker, S. Melzer and W. Zimmermann, MicrobiologyOpen, 2019, 8, e00757 Search PubMed.
- A. Erichsen, G. H. J. Peters and S. R. Beeren, J. Am. Chem. Soc., 2023, 145, 4882 Search PubMed.
-
M. Krishnagopal, G. K. Samanta, G. C. Daskhan and N. Jayaraman, Carbohydrate Chemistry, RSC, 2017, vol. 42, p. 165 Search PubMed.
- R. Noyori and I. Kurimoto, J. Org. Chem., 1986, 51, 4320 Search PubMed.
- C. Amatore, A. Jutand, J.-M. Mallet, G. Meyer and P. Sinäy, J. Chem. Soc., Chem. Commun., 1990, 718 Search PubMed.
- G. Balavoine, A. Gref, J.-c Fischer and A. Lubineau, Tetrahedron Lett., 1990, 31, 5761 Search PubMed.
- S. Manmode, S. Tanabe, T. Yamamoto, N. Sasaki, T. Nokami and T. Itoh, ChemistryOpen, 2019, 8, 869 Search PubMed.
- M. Nishizawa, H. Imagawa, Y. Kan and H. Yamada, Tetrahedron Lett., 1991, 32, 5551 Search PubMed.
- M. Nishizawa, H. Imagawa, K. Kubo, Y. Kan and H. Yamada, Synlett, 1992, 447 Search PubMed.
- H. Endo, M. Ochi, A. M. Rahman, T. Hamada, T. Kawano and T. Nokami, Chem. Commun., 2022, 58, 7948 Search PubMed.
- Y. Wu, S. Aslani, H. Han, C. Tang, G. Wu, X. Li, H. Wu, C. L. Stern, Q. Guo, Y. Qiu, A. X.-Y. Chen, Y. Jiao, B. Zhang, A. H. G. David, D. W. Amstrong and J. F. Stoddart, Nat. Synth., 2024, 3, 698 Search PubMed.
- D. Ikuta, Y. Hirata, S. Wakamori, H. Shimada, Y. Tomabechi, Y. Kawasaki, K. Ikeuchi, T. Hagimori, S. Matsumoto and H. Yamada, Science, 2019, 364, 674 Search PubMed.
- L. Fan and O. Hindsgaul, Org. Lett., 2002, 4, 4503 Search PubMed.
- K. Maiti, G. C. Samanta and N. Jayaraman, ARKIVOC, 2021, iv, 113 Search PubMed.
- P. R. Sundarajan and V. S. R. Rao, Carbohydr. Res., 1970, 13, 351 Search PubMed.
- T. Nakagawa, K. Ueno, M. Kashiwa and J. Watanabe, Tetrahedron Lett., 1994, 35, 1921 Search PubMed.
- A. Motoyama, T. Arai, K. Ikeuchi, K. Aki, S. Wakamori and H. Yamada, Synthesis, 2018, 282 Search PubMed.
- T. Mukaiyama, Y. Murai and S. Shoda, Chem. Lett., 1981, 431 Search PubMed.
- T. Matsumoto, H. Maeta and K. Suzuki, Tetrahedron Lett., 1988, 29, 3567–3570 Search PubMed.
- D. V. Titov, M. L. Gening, A. G. Gerbst, A. O. Chizhov, Y. E. Tsvetkov and N. E. Nifantiev, Carbohydr. Res., 2013, 381, 161 Search PubMed.
- H. Someya, T. Seki, G. Ishigami, T. Itoh, Y. Saga, Y. Yamada and S. Aoki, Carbohydr.
Res., 2020, 487,
107888 Search PubMed.
- X. Li, C. Li, R. Liu, J. Wang, Z. Wang, Y. Chen and Y. Yang, Org. Lett., 2019, 21, 9693 Search PubMed.
- X. Li, C. D. Carluccio, H. Miao, L. Zhang, J. Shang, A. Molinaro, P. Xu, A. Silipo, B. Yu and Y. Yang, Angew. Chem., Int. Ed., 2023, 62, e202307851 Search PubMed.
- F. Cramer and W. Dietsche, Chem. Ber., 1959, 92, 378 Search PubMed.
- H. Satoh, H. Hansen, S. Manabe, W. F. van Gunsteren and P. H. Huneberger, J. Chem. Theory Comput., 2010, 6, 1783 Search PubMed.
- A. A. Hettikankanamalage, R. Lassfolk, F. S. Ekholm, R. Leino and D. Crich, Chem. Rev., 2020, 120, 7104 Search PubMed.
- D. Crich and S. Sun, J. Org. Chem., 1996, 61, 4506 Search PubMed.
- S. Yamago, T. Yamada, O. Hara, Y. Ito and J. Yoshida, Org. Lett., 2001, 3, 3867 Search PubMed.
- J. D. C. Codée, L. J. van den Bos, R. E. J. N. Litjens, H. S. Overkleeft, J. H. van Boom and G. A. van der Marel, Org. Lett., 2003, 5, 1947 Search PubMed.
- S. Yamago, T. Yamada, T. Maruyama and J. Yoshida, Angew. Chem., Int. Ed., 2004, 43, 2145 Search PubMed.
- X. Huang, L. Huang, H. Wang and X.-S. Ye, Angew. Chem., Int. Ed., 2004, 43, 5221 Search PubMed.
- O. J. Plante, E. R. Palmacci and P. H. Seeberger, Science, 2001, 291, 1523 Search PubMed.
- N. V. Ganesh, K. Fujikawa, Y. H. Tan, K. J. Stine and A. V. Demchenko, Org. Lett., 2012, 14, 3036 Search PubMed.
- J. Yoshida, A. Shimizu, Y. Ashikari, T. Morofuji, R. Hayashi, T. Nokami and A. Nagaki, Bull. Chem. Soc. Jpn., 2015, 88, 763 Search PubMed.
- J. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702 Search PubMed.
- T. Nokami, R. Hayashi, Y. Saigusa, A. Shimizu, C.-Y. Liu, K.-K. T. Mong and J. Yoshida, Org. Lett., 2013, 15, 4520 Search PubMed.
- A. Shibuya and T. Nokami, Chem. Rec., 2021, 21, 2389 Search PubMed.
- T. Nokami, Y. Isoda, N. Sasaki, A. Takaiso, S. Hayase, T. Itoh, A. Shimizu, R. Hayashi and J. Yoshida, Org. Lett., 2015, 17, 1525 Search PubMed.
- Y. Isoda, N. Sasaki, K. Kitamura, S. Takahashi, S. Manmode, N. Takeda-Okuda, J. Tamura, T. Nokami and T. Itoh, Beilstein J. Org. Chem., 2017, 13, 919 Search PubMed.
- K. Yano, T. Itoh and T. Nokami, Carbohydr. Res., 2020, 492,
108018 Search PubMed.
- M. L. Gening, D. V. Titov, A. A. Grachev, A. G. Gerbst, O. N. Yudina, A. S. Shashkov, A. O. Chizhov, Y. E. Tsvetkov and N. E. Nifantiev, Eur. J. Org. Chem., 2010, 2465 Search PubMed.
- D. V. Titov, M. L. Gening, A. G. Gerbst, A. O. Chizhov, Y. E. Tsvetkov and N. E. Nifantiev, Carbohydr. Res., 2013, 381, 161 Search PubMed.
- N. Tanaka, R. Saito, K. Kobayashi, H. Nakai, S. Kamo, K. Kuramochi, H. Taguchi, M. Nakajima and T. Masaike, Appl. Microbiol. Biotechnol., 2024, 108, 187 Search PubMed.
- A. A. Bhagwat, A. Mithöfer, P. E. Prefer, C. Kraus, N. Spickers, A. Hotchkiss, J. Ebel and D. L. Keister, Plant Physiol., 1999, 119, 1057 Search PubMed.
- A. V. Nair, S. N. Gummadi and M. Doble, Biotechnol. Lett., 2016, 38, 1519 Search PubMed.
- N. S. V. Kambhampati, S. Kar, S. S. K. Pinnepalli, J. Chelli and M. Doble, Spectrochim. Acta, Part A, 2018, 203, 494 Search PubMed.
- S. Manmode, M. Kato, T. Ichiyanagi, T. Nokami and T. Itoh, Asian J. Org. Chem., 2018, 7, 1802 Search PubMed.
- A. Shibuya, M. Kato, A. Saito, S. Manmode, N. Nishikori, T. Itoh, A. Nagaki and T. Nokami, Eur. J. Org. Chem., 2022, e202200135 Search PubMed.
- A. Shibuya, Y. Ishisaka, A. Saito, M. Kato, S. Manmode, H. Komatsu, M. A. Rahman, N. Sasaki, T. Itoh and T. Nokami, Faraday Discuss., 2023, 247, 59 Search PubMed.
- H. Hashimoto, Y. Abe, H. Shigemori and J. Yoshimura, J. Carbohydr. Chem., 1988, 8, 307 Search PubMed.
- H. J. Schuster, B. Vijayakrishnan and B. G. Davis, Carbohydr. Res., 2015, 403, 135 Search PubMed.
- A. M. Rahman, K. Kuroda, H. Endo, N. Sasaki, T. Hamada, H. Sakai and T. Nokami, Beilstein J. Org. Chem., 2022, 18, 1133 Search PubMed.
- A. M. Rahman, H. Endo, T. Yamamoto, S. Okushiba, N. Sasaki and T. Nokami, Beilstein J. Org. Chem., 2024, 20, 1421 Search PubMed.
- K. Benakli, C. Zha and R. J. Kerns, J. Am. Chem. Soc., 2001, 123, 9461 Search PubMed.
- S. Manabe, K. Ishii and Y. Ito, J. Am. Chem. Soc., 2006, 128, 10666 Search PubMed.
- S. Manabe, K. Ishii and Y. Ito, J. Org. Chem., 2007, 72, 6107 Search PubMed.
- H. Satoh, S. Manabe, Y. Ito, H. P. Luthi, T. Laino and J. Hutter, J. Am. Chem. Soc., 2011, 133, 5610 Search PubMed.
- T. Nokami, A. Shibuya, Y. Saigusa, S. Manabe, Y. Ito and J. Yoshida, Beilstein J. Org. Chem., 2012, 8, 456 Search PubMed.
- R. Koinuma, K. Tohda, T. Aoyagi and H. Tanaka, Chem. Commun., 2020, 56, 12981 Search PubMed.
Footnote |
| † This article is dedicated to late Professor Sir Fraser Stoddart. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab