
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Implications of non-native metal substitution in carbonic anhydrase – engineered enzymes and models
Dyuti
Bhandary
 a,
Sam P.
de Visser
*b and
Gourab
Mukherjee
*a
a,
Sam P.
de Visser
*b and
Gourab
Mukherjee
*a
aDepartment of Catalysis & Fine Chemicals, CSIR – Indian Institute of Chemical Technology, Uppal Road, Hyderabad 500007, India. E-mail: gmchem99@gmail.com
bManchester Institute of Biotechnology and Department of Chemical Engineering, The University of Manchester, 131 Princess Street, Manchester M1 7DN, UK. E-mail: sam.devisser@manchester.ac.uk
First published on 4th December 2024
Abstract
The enzyme carbonic anhydrase has been intensely studied over decades as a means to understand the role of zinc in hydrating CO2. The naturally occurring enzyme has also been immobilized on distinct heterogeneous platforms, which results in a different hybrid class of catalysts that are useful for the adsorption and hydration of CO2. However, the reusability and robustness of such natural and immobilized systems are substantially affected when tested under industrial conditions, such as high temperature and high flow rate. This led to the generation of model systems in the form of metal-coordination complexes, metal–organic frameworks, metallo-peptide self-assembled supramolecules and nanomaterials that mimic the primary, and, to some extent, secondary coordination sphere of the active site of the natural carbonic anhydrase enzymes. Furthermore, the effects of zinc-substitution by other relevant transition metals in both the naturally occurring enzymes and model systems has been reported. It has been observed that some other transition metal ions in the active site of carbonic anhydrase and its models can also accomplish similar activity, established by various reaction probes and ideas. Herein, we present a comprehensive highlight about substituting zinc in the active site of the modified enzymes and its biomimetic model systems with non-native metal ions and review how they affect the structural orientation and reactivity towards CO2 hydration. In addition, the utility of artificially engineered carbonic anhydrases towards a number of non-natural reactions is also discussed.
Introduction
Metalloproteins are a major class of biomolecules with widespread utilities ranging from oxygen and/or metal transport to biocatalytic functions.1–12 They are indispensable in biological systems through their catalytic properties that involve the biosynthesis of important biomolecules for signaling and biofunction, but also as part of the biodegradation and defense of the organism. Due to their high reaction selectivity and ability to operate at room temperature and ambient pressure, these metalloenzymes have gained significant applications in bio-inspired chemical reactions.13–16 From an environmental and biotechnological perspective, an important industrial reaction involves the adsorption and use of carbon dioxide from the air. Specifically, research is focused on large scale adsorption of CO2 in storage media, including materials and solvents.17–23 In addition, ongoing research is applied to converting CO2 into valuable materials by reacting it to either CO, formaldehyde, formate or even methanol products.24–31 These reactions are particularly relevant due to the increasing concentrations of CO2 in the atmosphere. Consequently, processes involving carbon dioxide as a substrate are being investigated extensively.32,33 As the research on CO2 activation is widespread, we will start with a summary of knowledge on the actual enzyme carbonic anhydrase and specifically on its catalytic mechanism for CO2 activation. We then will review engineered carbonic anhydrase systems and look into biomimetic models and complexes that enable CO2 activation.Carbonic anhydrase, structure and mechanism
In biology, carbonic anhydrase (CA) is an important zinc-containing metalloenzyme that plays a premier role in reversible CO2 transport and sequestration in biological systems that maintains the acid–base balance and intracellular pH homeostasis by equilibrating the CO2 and bicarbonate concentrations in the body. The enzyme in its active site holds a Zn(II) ion in a tetrahedral geometry that facilitates hydration of CO2 at pH 9. In its much-established mechanistic cycle, the hydration process is initiated by coordination of water molecules to the bivalent Zn(II) centre. This results in lowering of the pKa of bound water molecules from 15.7 to about 6.8–7.3. The lowering of the pKa of the water molecule in the zinc–aqua complex in CA is consistent with the fact that under physiological conditions the zinc-bound water exists in its deprotonated form. The newly formed nucleophilic centre reacts with CO2 leading to the formation of a bicarbonate–CA complex (Scheme 1). The bicarbonate undergoes prototropic rearrangement and eventually gets replaced by a water molecule thereby completing the catalytic cycle.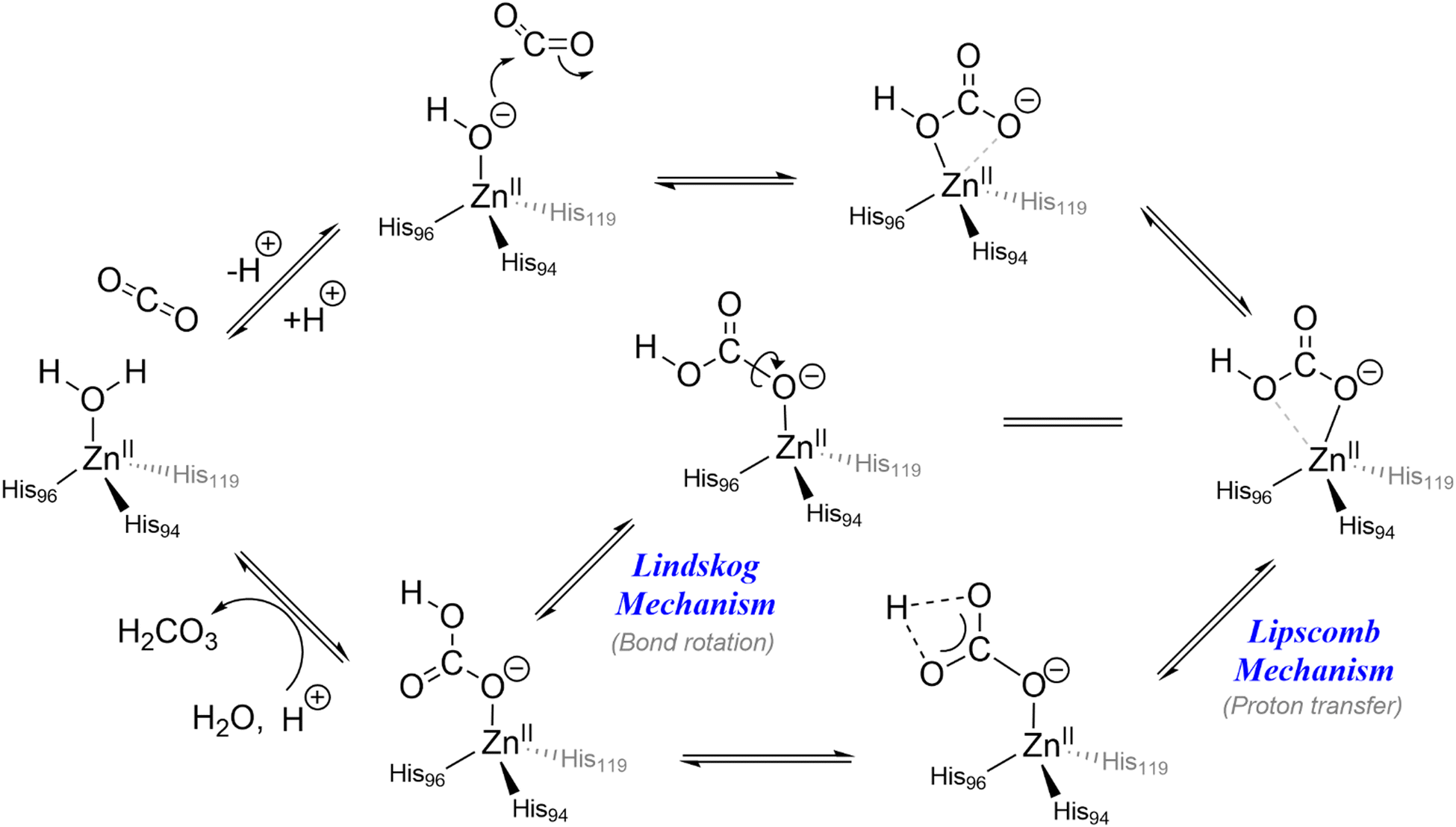 | ||
| Scheme 1 Catalytic cycle of human carbonic anhydrase II. | ||
The mechanistic cycle of CO2 hydration by carbonic anhydrases is controversial and two possible pathways have been proposed (shown in Scheme 1), namely the Lipscomb and the Lindskog mechanisms.34 The active site of CA holds a Zn(II) centre that is coordinated to the side chains of three histidine residues. The binding pocket is highly polar and allows for the flow of water molecules and the relay of protons. As a consequence, the enzyme is pH-dependent and functions within the pH range of 4.5–9.5 with an optimal pH of 7.5.35 The secondary coordination sphere around the active site is believed to play a hydrophobic zone around the CO2 binding pocket. Moreover, the Thr199 and Glu106 residues in the entrance conduit are believed to be responsible for water and product exchange and His64 provides a hydrophilic exit channel for bicarbonate. In spite of having a high catalytic activity and reaction rate in nature, CA enzymes are found unsuitable for industrial post-combustion CO2 fixation under elevated temperature due to factors such as thermal intolerance and chemical instability.
There are various CA isozymes that are structurally different and have dramatically different biochemical and biophysical properties. In mammals, there exist 16 different forms of CAs that differ in their catalytic activity, subcellular location and susceptibility towards inhibitors.36 These isozymes are designated α, β, γ, δ, ζ and η isoforms and have different amino acid residues in the primary coordination sphere.37 In α, γ and δ isoforms, Zn(II) is structurally coordinated by three histidine residues, whereas in β and ζ isoforms, two of those histidine residues have been replaced by cysteine residues.38,39 The η isoform has two histidine and one glutamine residue as ligand. The metal ion in the active centre for all the naturally occurring isoforms of CA is Zn(II), however the CA γ isoform from M. thermophila contains Fe(II) and the ζ-class CA from the marine diatom Thalassiosira weissflogii contains Cd(II).38,40–45
In biomimetic chemistry, catalysts are designed that bear similar active site coordination of the metal but lack the protein environment that regulate substrate, oxidant and proton access.13–15,46–56 These biomimetic models overcome the shortcomings of the naturally available enzyme (such as thermostability, substrate scope, etc.), and can be engineered easily to achieve specific desirable properties and are easier to apply in an industrial setting. In particular, with biomimetic model complexes the axial and equatorial ligand effects of the coordination environment of the metal centre can be tested. In addition, these biomimetic models as compared to enzymatic structures give insight into the second coordination sphere effects of the protein matrix.57 In the case of CA, synthetic biomimetic models (in the forms of coordination complexes, including metal organic frameworks (MOFs), self-assembled peptides, etc.) were designed to enhance their thermal and chemical stability, structural integrity, catalytic efficacy and prevent the denaturation due to shearing and stress.58–62 The active site of the CA mimicking catalysts should emulate two vital structural features viz. (i) presence of a Zn(II)-bound OH group at physiological pH derived from a Zn(II)-bound water molecule with a pKa ∼ 7, and (ii) the presence of electron donating ligands mimicking the imidazole group of the histidine residue. Since the catalytic cycle is initiated upon approach of CO2 substrate onto the zinc(II)–hydroxide centre, the catalytic activity of biomimetic models will also need a ZnII–OH centre. Research has shown, however, that the best performance is obtained with zinc(II) in tetrahedral (Td) geometry.63–65
The metal centre in CA plays a significant role in determining the catalytic activity of the enzyme. In particular, the metal coordination environment determines the physicochemical properties of the complex, namely its acidic or basic character in solution, and controls the activation energy of the catalytic reaction mechanism. Studies have shown that the first-row transition metals and lanthanides could also be used as non-native metals in the CA mimetics.66 Other than metals, alterations in the metal-binding scaffold can alter the catalytic activity of the enzyme mimics. Different types of scaffolds have been tested, including coordination complexes, MOFs, micelles, and molecular cages.
Role of non-native metal substitution in engineered carbonic anhydrase enzymes
One of the first reports on the replacement of the naturally present Zn(II) in CA enzymes was by Coleman.67 Acetazolamide is a well-known CA inhibitor binding to the Zn(II) ion that limits its activity (Scheme 2).68 Embarking on the previous reports of Lindskog et al., the binding affinity of acetazolamide on various metallocarbonic anhydrases was studied with Zn(II), Co(II), Cu(II), Mn(II), Ni(II), Cd(II) and Hg(II). It was observed that the apocarbonic anhydrase had negligible affinity towards acetazolamide, thus the presence of the metal ion was crucial for its inhibition activity. Firm binding of the inhibitor was observed for Zn(II) and Co(II), while its binding to other ions decreased in the order Mn(II) > Cu(II) > Cd(II) > Hg(II) > Ni(II).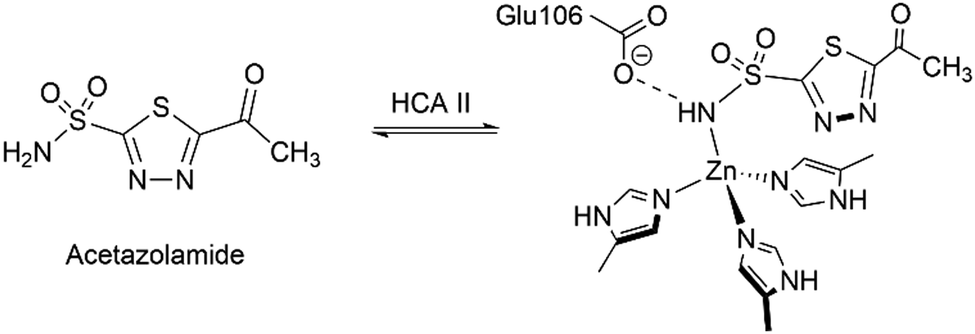 | ||
| Scheme 2 Binding of acetazolamide with human carbonic anhydrase II. | ||
In the γ class of CA enzymes, found mainly in prokaryotes, Ferry, Krebs and their coworkers have reconstituted the Zn(II) ion by unfolding the protein using a denaturant in the presence of a metal chelator and subsequent refolding by removal of the denaturant. This was the process through which variants of CA with Mn(II), Fe(II), Co(II), Ni(II), Cu(II), Zn(II), and Cd(II) were generated.69 It was observed that the catalytic activity of the Fe(II)-reconstructed enzyme had surpassed that of the Zn(II)- and Co(II)-reconstructed enzymes, when it was purified under anaerobic conditions from E. coli or overproduced in M. acetivorans and purified anaerobically. However, the existence of Fe in its ferrous state is crucial for the enhanced activity. In the presence of air, Fe(II) is oxidized to Fe(III) very quickly leading to its loss from the active site, which is subsequently replaced by Zn(II) during the purification process. This phenomenon of oxidation of Fe(II) to Fe(III) was observed by exposure to H2O2 and subsequent Mössbauer studies.70
trans-Metalation experiments along with in vitro studies showed that the catalytic activity of Mn(II) substituted CA is about 7%, while that of the Cd(II) variant is only 2%.71 Furthermore, the use of Cu(II) and Hg(II) led to the complete loss of any catalytic activity.63,72 It was observed that among all the transition metals tested, Co–CA is a highly functional alternative with respect to Zn(II)–CA with an efficacy of kcat/KM = 8.8 × 107 M−1 s−1.73 With the help of differential scanning calorimetry (DSC) and density functional theory (DFT) studies Lisi et al. determined the various stabilities as well as the catalytic efficacies of the metal-substituted CAs. In general, the stability of the transition metal complexes follows the Irving–Williams series which refers to the stability of a complex by exchange of an aqua ligand with another ligand being exclusively independent of the incoming ligand.74 It was established that the metal substitution often does not follow the Irving–Williams series and hence, Co(II), Cd(II) and Cu(II) readily shows catalytic activity.75
Kim et al. studied the behavioral patterns of CA, when substituted with non-native, but biologically relevant, metal ions like Cu(II), Ni(II), Co(II), Cd(II) and Mn(II).76 Using the cryocooling technique of protein crystallization, structural studies were conducted which helped in inferring that the change in geometry of the active site of CA due to a change in the metal ion present is directly proportional to the catalytic activity of the enzyme. Changing the geometry (see Fig. 1) from tetrahedral (ZnII) to octahedral (CoII and NiII) and trigonal bipyramidal (CuII), a drastic change in the catalytic activity was observed. The catalytic activity was reduced by ∼50% (for CoII); ∼2% (for NiII) and ∼0% (for CuII). Hence, the catalytic activity of the modified CA enzymes were of the order Zn(II) ⋙ Co(II) > Ni(II) > Cu(II).
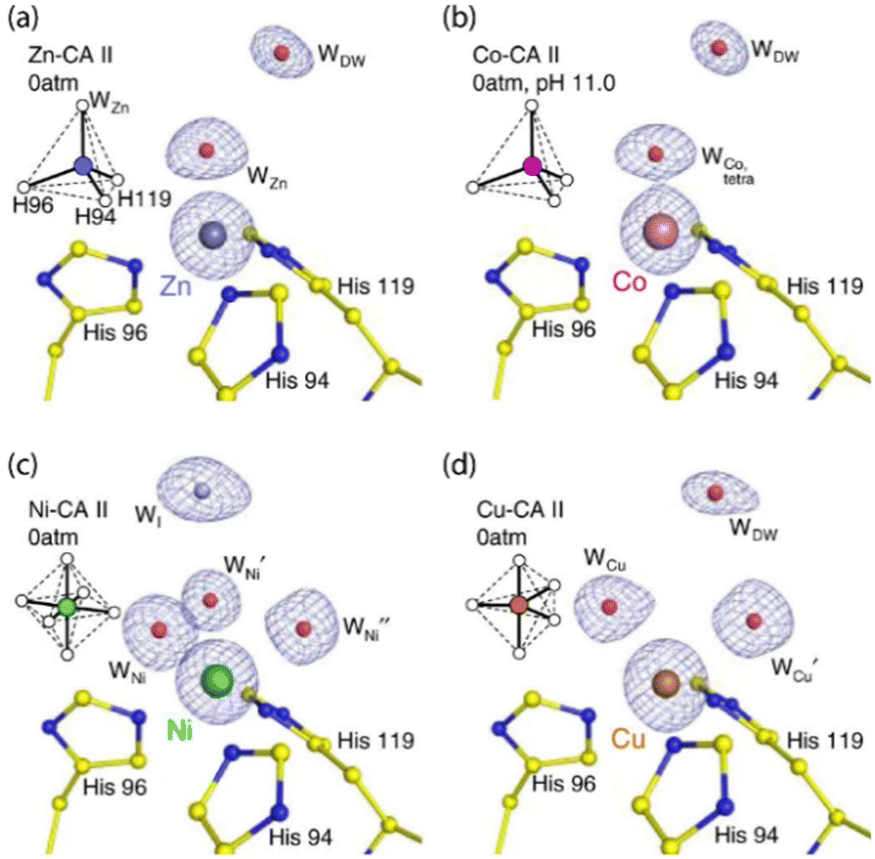 | ||
| Fig. 1 Crystal structure coordinates of the active sites of various metal-coordinated CA II proteins without CO2 pressurization: (a) tetrahedrally coordinated Zn–CA II (b) tetrahedrally coordinated Co–CA II (c) octahedrally coordinated Ni–CA II and (d) trigonal bipyramidal coordinated Cu–CA II. The structures in (a), (c) and (d) were obtained at pH 7.8 while that for (b) was obtained at pH 11.0. The intermediate water (WI) in (c) is coloured in steel blue for clarity. Adapted with permission from ref. 76. Copyright 2020. Nature Publishing House. | ||
Role of non-native metal substitution in engineered carbonic anhydrase mimics
Metal complexes as CA models
Modelling CA using metal complexes started several decades ago and was pioneered by Vahrenkamp and Kimura using macrocyclic ligand systems.77–81 Thus, Zn(II) complexes of the cyclen, 1,4,7,10-tetraazacyclododecane (Fig. 2(a)) and 1,5,9-triazacyclododecane (Fig. 2(b)) have been extensively studied to mimic the hydration of CO2. These complexes are still widely used in industry for CO2 hydration processes. The zinc(II)–cyclen complexes have lower kinetic rate than native CA and exhibit about 30% of its catalytic activity. Despite the fact that the zinc(II)–cyclen system has so widely been used, surprisingly its mechanism of action is still ambiguous. Moreover, the Zn–cyclen complexes become inactive in the presence of a high concentration of amines in the solution, which block the active site of the mimic in the solution state.82–87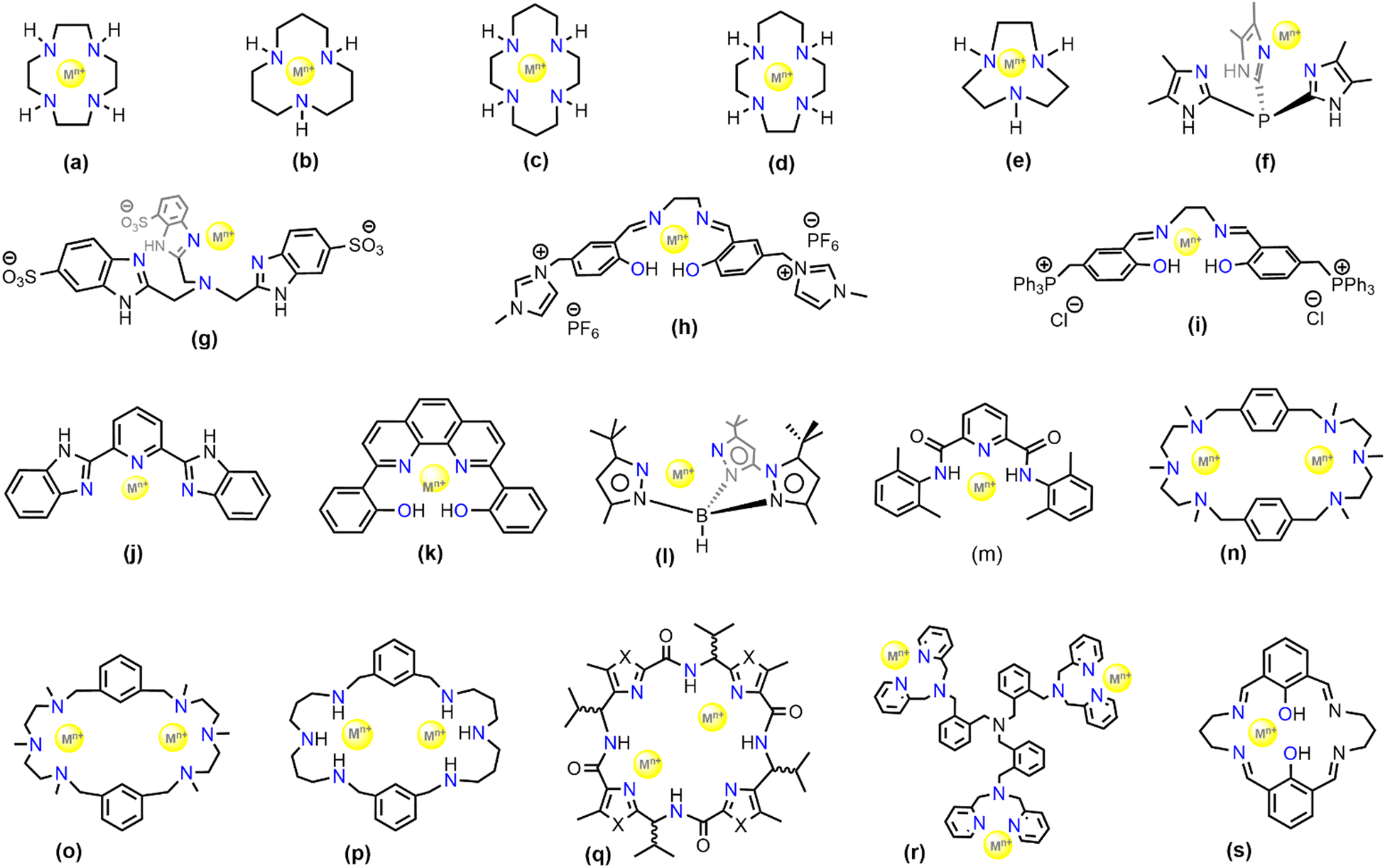 | ||
| Fig. 2 Selection of ligand frameworks (labelled as a–s) for biomimetic models used for CA activity studies. | ||
A series of five zinc complexes with different aza-macrocycles (Fig. 2(a)–(e)) were employed in the work of Koziol et al.58 The work established a correlation between the zinc–bicarbonate bond dissociation energy with the reaction rates, which suggests that the bicarbonate release is the rate-determining step in the catalytic cycle. Quantum mechanical studies have shed more light on the dependence of the coordination geometry and metal–bicarbonate binding on the reaction rates and catalytic efficiency with different systems (shown in Fig. 2(a), (b), (f) and (g)).88 As such, a comparison between zinc and cobalt in similar systems led to the conclusion that the Co-catalysed reaction is less efficient than the zinc-containing catalysts. The Co-containing mimics were observed to be aiding in CO2 complexation but their structural disadvantage results in slow release of the bicarbonate from the Co–cyclen complexes. The pKa value of the Co–cyclen complex was found to be higher than that of the corresponding Zn–cyclen complex resulting in greater intrinsic rate constant. Due to the high pKa values, these cyclen complexes perform exceptionally well in industrial applications for CO2 separation processes.59
The zinc–cyclen complexes, albeit being extensively studied and utilized for CO2 hydration, are highly susceptible to inhibitors. The highly electrophilic Zn(II) centre strongly coordinates with the anionic bicarbonate species in order to maintain charge balance thereby making the bicarbonate release very difficult. Under industrial carbon capture conditions, primary amine-based solvents are used. However, there are a few challenges of using homogeneous catalysts in primary amine-based solvents for CO2 capture such as water-solubility of the catalysts, stability under aerobic conditions, and requirement of redox-inert behaviour. To address such problems, Lippert et al. facilitated the use of water-soluble and stable salen-based complexes of Zn(II) and Co(II) (Fig. 2(h) and (i)). These ligands are anionic, electron donating and water soluble,89 which should result in a higher electron density around the metal ion. A similar Zn–salen based complex (Fig. 2(k)) employed by Kelsey et al. was also found to be highly suitable for CO2 adsorption in highly concentred primary amine solvents.90 Although η2-coordination of bicarbonate to the zinc ion is possible, it is energetically higher than the η1-ligation. Studies have shown that the η1-coordination motif is crucial to facilitate the bicarbonate release.67 This was again observed for a tris(pyrazolyl)hydroborato–Zn complex (TPHB) (Fig. 2(l)) synthesized by Parkin and co-workers as a CA mimic.70 The zinc–aqua complex of 2l was found unreactive towards CO2. However, its deprotonated zinc–hydroxide complex was found to be in rapid equilibrium with the bicarbonate derivative, resulting in a bridging carbonate condensation product which is highly water sensitive leading to regeneration of the hydroxide derivative. Analogues to the CA enzyme, where the deprotonation of the zinc–aqua complex is a key step to initiate the catalytic cycle, a similar study of the reversible protonation and deprotonation was conducted with model complexes.91 The same ligand system when impregnated with other transition metals like Fe(II), Co(II), Mn(II) and Cu(II) were found to have similar catalytic activity with the Zn–TPHB towards bicarbonate formation and release. Further computational studies such as DFT revealed that the pKa remains unchanged when Zn(II) was substituted with Co(II) thereby supporting the notion of cobalt being a successful substitute of zinc in CA enzyme. Further modifications when performed on the Co–TPHB complex revealed that the hydroxylated Co–TPHB, although having lower activity shows greater bidentate coordination of the HCO3−.
A Ni(II) based CA mimic was developed by Huang et al. wherein N,N′-2,6-dimethylphenyl-2,6-pyridinecarboamidate dianion (2m) was utilized as the ligand backbone.92 The mimic showed rapid conversion of CO2 to HCO3− in DMF solvent and exhibited rapid kinetics – about 100 times faster than any other metal mediated synthetic systems. The difference in absorbance of the reactant [Ni(pyN2Me2)(OH)]− and the product [Ni(pyN2Me2)(HCO3)]− in the UV-vis spectrum gives a clear indication of the progress of the reaction in DMF solvent (see Fig. 3). The metal complex [Ni(pyN2Me2)(OH)]− absorbs at 411 nm and a has a shoulder in the absorbance spectrum at 490 nm. Upon CO2 bubbling through the solution, the metal binds CO2 to form a monoanionic Ni–bicarbonate complex, that absorbs at 300 and 381 nm (shown by blue line in Fig. 3).92
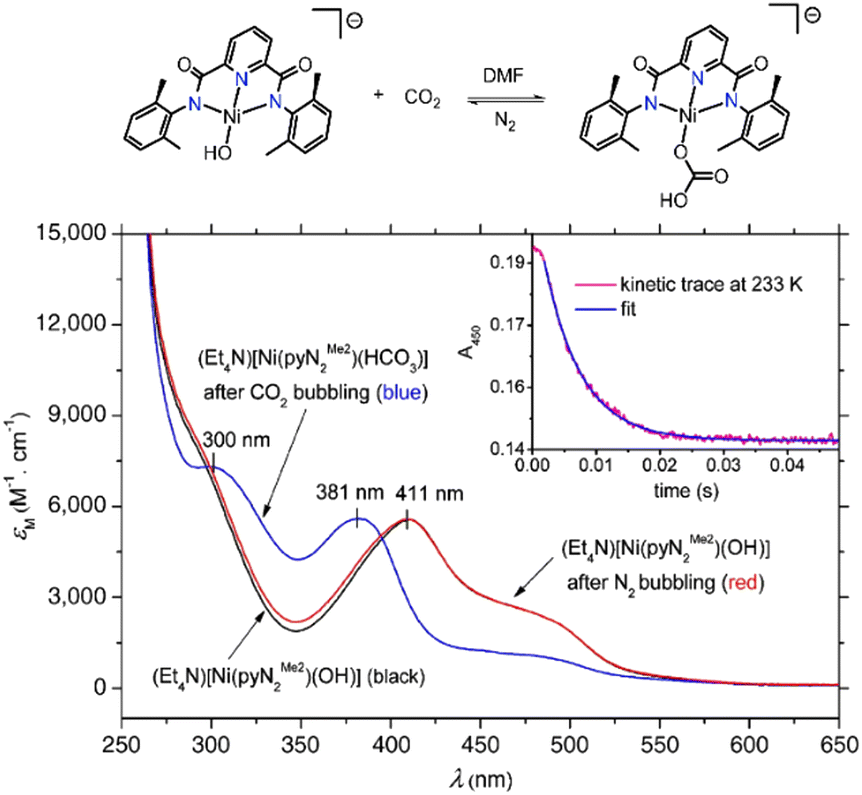 | ||
| Fig. 3 Absorption spectra in DMF: black line – absorption spectra of (Et4N)[Ni(pyN2Me2)(OH)]; blue line – after CO2 bubbling through the solution for 2 min formation of (Et4N)[Ni(pyN2Me2)(HCO3)]; red line – after vigorous N2 bubbling through (Et4N)[Ni(pyN2Me2)(HCO3)] for 20 min. Inset shows decay kinetics of the 450 nm band at 233 K. Adapted with permission from ref. 92. Copyright 2011. The National Academy of Sciences. | ||
Naturally occurring CA enzymes contain a single active site. However, recent optimisations have shown that organometallic complexes with a couple of active sites can also be used as CA mimics. Company et al. synthesized several Cu(II) containing binuclear complexes (Fig. 2: structures 2n, 2o, and 2p).93 These mimics have a mechanistic difference with CA wherein the CO2 directly gets attached to the hydroxyl ligand and generate HCO3− through ligand substitution reaction. The mechanism of CO2 hydration in these cases appears to be dependent on intermolecular or intramolecular interactions with the macrocyclic ligands.94
Comba et al. studied several Cu(II)-based binuclear biomimetic models of CA with a patellamide as the ligand framework (see Fig. 2, structure 2q).95 The characteristic feature of such models is that its catalytic activity is nearly fivefold greater than the ones with zinc–cyclen. However, the reaction mechanism (Scheme 3) is probably different from the one established for CA. In this case, the reaction mechanism is intramolecular and both the Cu(II) ions are involved in the complexation process. The catalyst showed a kcat value nearly equal to 7.3 × 10−3 s−1. The catalytic activity is further dependent upon the stereochemistry of the ligand system. It was observed that the ligand stereochemical configuration as R*, S*, R*, S* showed greater activity than the one with S*, S*, S*, S*.
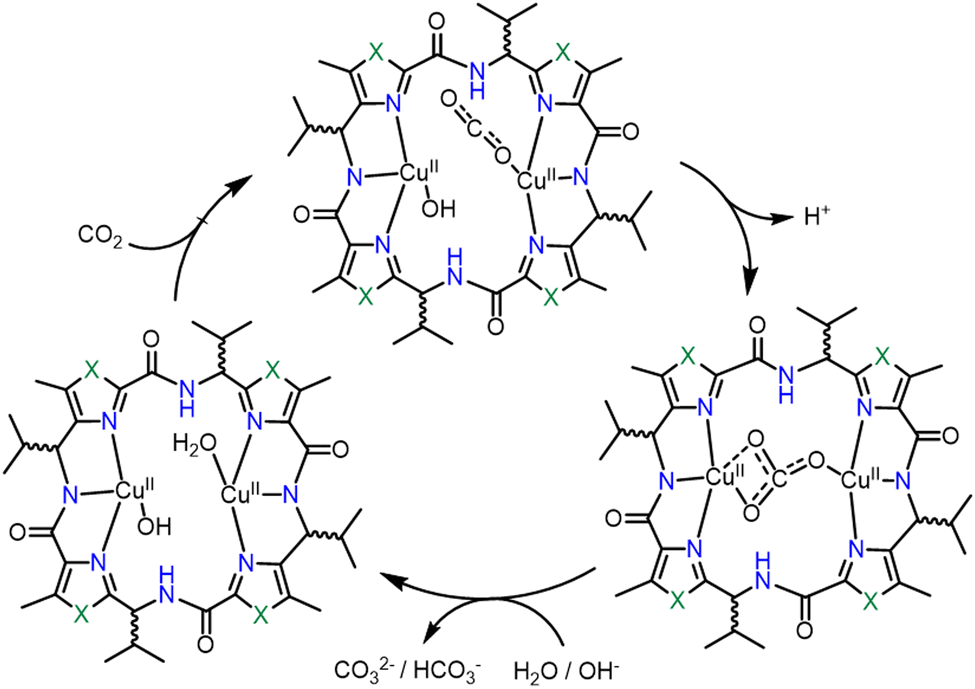 | ||
| Scheme 3 Catalytic cycle of a CA mimic designed by Comba and co-workers. Adapted from ref. 95. Copyright 2013. RSC publishing. | ||
Other than substrate approach to the active site, a crucial aspect of enzyme catalysis is the product release. In a recent report by Liu et al. it was observed that the simple change in the counter anion from triflate (coordinating as OTf) to perchlorate (coordinating as aqua ligands) invokes a feasible product release. Trinuclear zinc complexes (Fig. 2, structure 2r) synthesized by Liu et al. can capture μ3-oxoanions such as μ3-phosphate or μ3-carbonate and can be useful for their catalytic conversions.96 These oxo-anions impart structural conformity, wherein the structure could be sterically controlled. Although the stability of the Zn3–carbonate complex was considerable when the triflate counter anions were coordinated to the three Zn centres but the subsequent release of the carbonate was hindered by the steric fencing provided by those triflate ions (see Fig. 4). However, employment of the non-coordinating counterion, perchlorate, unfolded the cavity thereby leading to precipitation of CaCO3 in the presence of calcium perchlorate. This characteristic feature favoured the catalytic conversion of CO2.
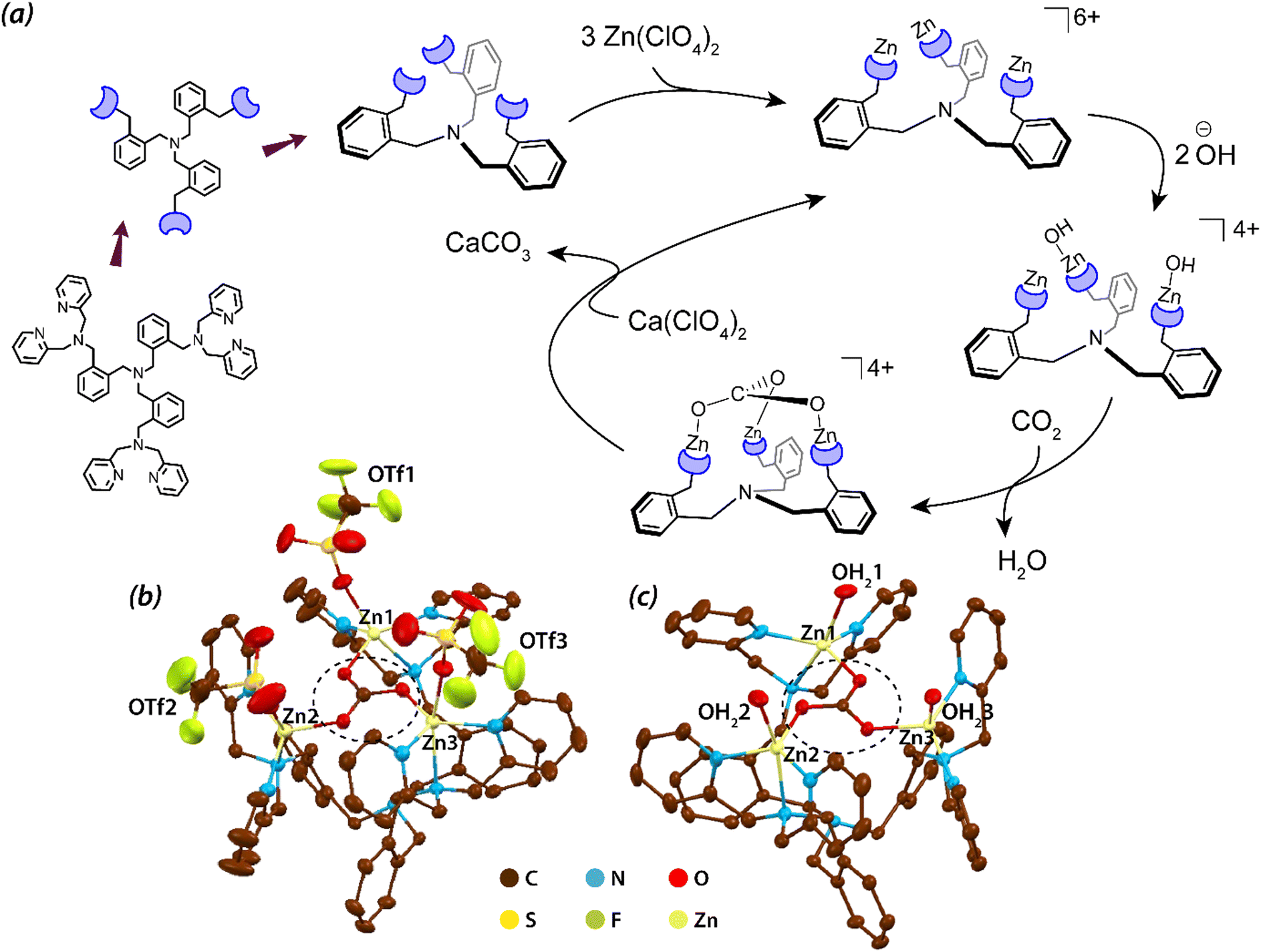 | ||
| Fig. 4 (a) Schematic representation of the tripodal ligand, formation of the trinuclear complex and the catalytic cycle of CO2 fixation; (b) and (c) X-ray structures of the trinuclear Zn complexes binding triflate and aqua ligands respectively. Also, the [Zn3(μ3-CO3)] can be seen in both the structures. The structures are reproduced from CCDC no. 931956 (b) and 931957 (c) respectively. Hydrogen atoms are omitted for clarity. Adapted with permission from ref. 96. Copyright 2013. Nature Publishing. | ||
An interesting CA mimic was recently reported by Lee and co-workers and contains self-assembled histidyl bolaamphiphiles as the ligand framework.60 Bolaamphiphiles are typical surfactant type of molecules having hydrophilic sites at either ends of a long hydrophobic (or, lipophilic) hydrocarbon chain. Keum et al. studied the effect of different transition metals, such as Zn(II), Co(II), Ni(II) and Cd(II) on the catalytic activity of CA mimics using such bolaamphiphiles.97 The catalytic activity was determined by para-nitrophenyl acetate (p-NPA) deacetylation experiments and comparing the kinetic parameters viz. turnover number (kcat) and Michaelis–Menten constant (KM) of the catalysts (Fig. 5). The calculated kcat/KM ratio gave a complete analysis of the performance of the catalysts on an industrial scale. The turnover number of these catalysts were found to be in the order of ZnII > CoII > CdII ≈ NiII (Fig. 5). According to the turnover number data, the Co(II)-containing complex had a catalytic activity comparable to that of Zn(II) due to the ability of the metal centre to mimic the tetrahedral coordination centre. On assessing the Michaelis–Menten constant of the complexes, that measure the binding affinity of the substrate towards the catalytic active site (and is largely dependent on their coordination sites), it was found that the KM values were in the order ZnII > CoII > CdII ≈ NiII. Another multinuclear CA mimic, [Zn9(Me2bta)12(OAc)6]·3DMF (Me2bta = 5,6-dimethyl-1,2,3-benzotriazole) developed by Huang et al. has similarity in the coordination sphere as CA.98 Not only this mimic was efficient in direct CO2 hydration to HCO3−, its CA assay activities along with the reusability parameters were found to be excellent. The complex was also synthesized as nanoparticle which was equally potent.
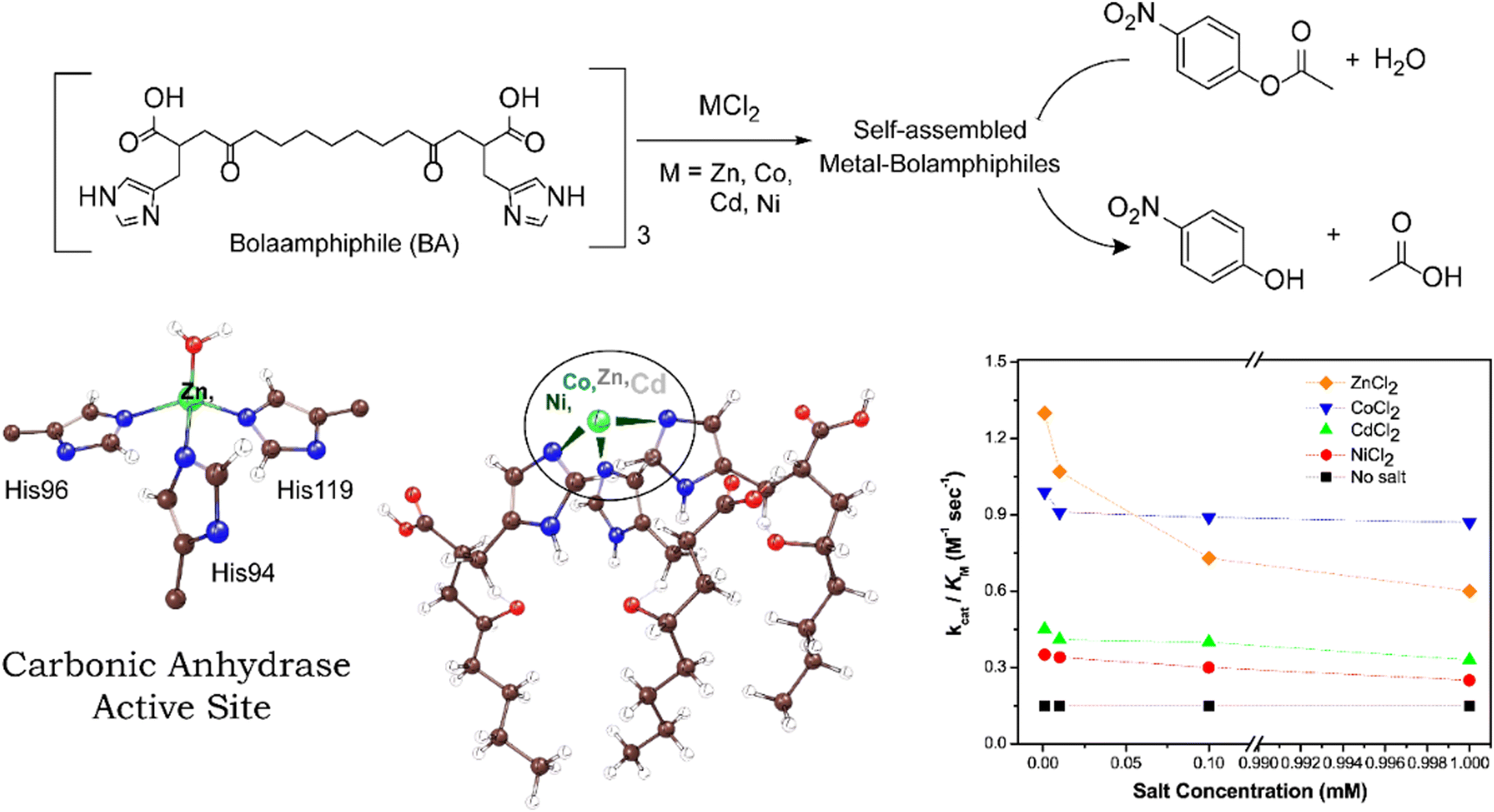 | ||
| Fig. 5 Schematic illustration of bolaamphiphiles and their self-assembled form that mimics the active site structure of CA. The self-assembled metal-bolaamphiphiles can hydrolyze p-nitrophenyl acetate into p-nitrophenol. Catalytic efficiency of different metal cofactors on self-assembled bolaamphiphiles is shown in the right. Adapted with permission from ref. 97. Copyright 2015. Elsevier Publishing. | ||
Apart from transition metal-substituted CA mimics, there have also been reports on lanthanide-incorporation complexes. Lanthanide(III) complexes with a mononuclear metal centre were developed by Bag et al.66 The complexes [Ln(LH2)(H2O)3Cl] (ClO4)2 [Ln = La, Sm, Gd, Tb, Nd, Eu] (Fig. 2, structure 2s) when suspended in a methanol–water mixture were found to regulate the CO2 fixation cycle by forming an exotic carbonato-bridged trinuclear complex (see Fig. 6). Further, the kinetic study of these complexes demonstrated enhanced rate of CO2 capture.
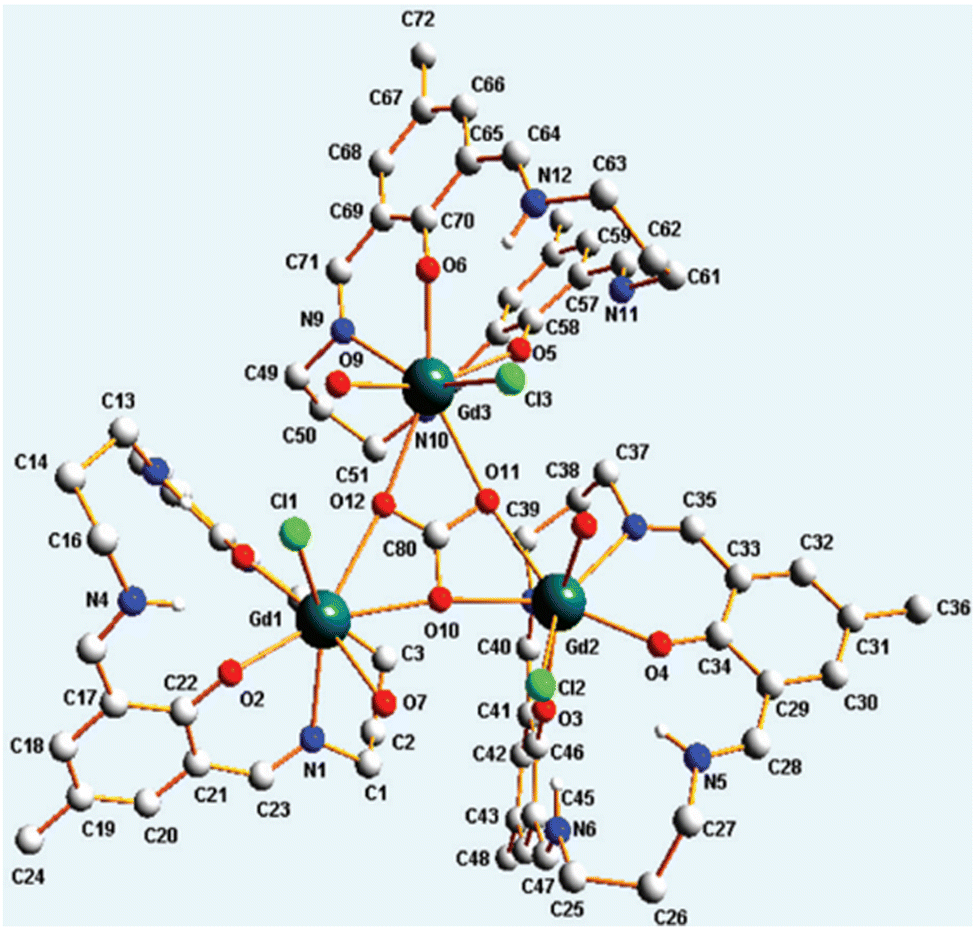 | ||
| Fig. 6 Trinuclear Gd complex cation capturing μ3-CO3. Adapted with permission from ref. 66. Copyright 2012, the Royal Society of Chemistry. | ||
Metal organic framework (MOFs) as CA models
MOFs are a relatively new hybrid class of compounds that bridge organic and inorganic chemistry.99–103 In catalysis, MOFs are generally synthesized for their significant potential for reusability and enhanced turnover number (TON).104–107 Synthesis of MOFs for biocatalytic purposes have mainly two strategies: enzyme immobilization and active site installation. Enzyme immobilization refers to the fixing or insertion of enzymes onto the surface or inside a carrier, such as MOFs, to create composite sites for catalysis while preserving (or, augmenting) their activity and stability.108,109 Over the years, carbonic anhydrases from various sources such as bovine erythrocytes, thermophilic organisms, and recombinant bacterial and human CAs have been immobilized on different materials like mesoporous molecular sieves, aluminium oxide, TiO2, Au, and Ag nanoparticles, iron fillings, hydrogels, etc. Traditional immobilization techniques are (a) adsorption, (b) surface covalent modification, (c) entrapment within a polymer, and (d) cross-linked enzyme aggregates.110 Enzyme immobilization stabilises through the formation of covalent bonding by using the side chains of various acidic or basic amino acid residues with a free –COOH or, –NH2 group (see Scheme 4).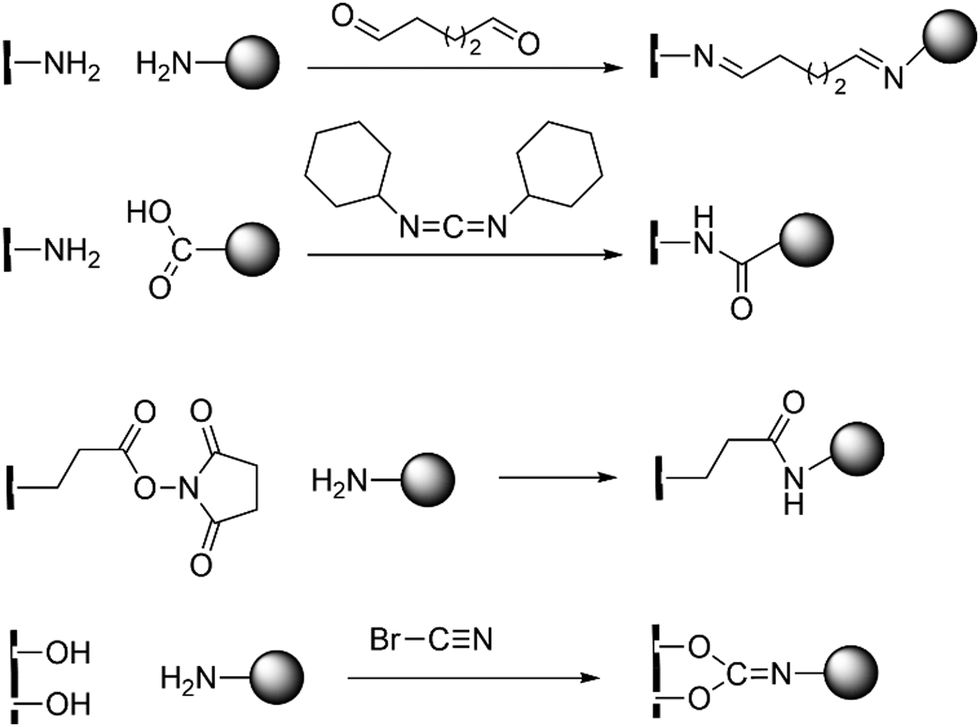 | ||
| Scheme 4 Different strategies of enzyme immobilization by covalent modification (a) amine–amine reaction through glutaraldehyde, (b) carbodiimide activation for amine–carboxylic acid reaction, (c) 4-N-hydroxysuccinimide–amine reaction and (d) cyanogen bromide activation for the hydroxylamine reaction. | ||
In many cases non-covalent interactions like van der Waals interactions and hydrophobic interactions are proposed to play a crucial role in catalysis. The advantage of this method is that the MOFs protects the integrity of the enzyme under harsh reaction conditions, rendering the enzymes easily accessible for the substrate and thereby enhancing the catalytic activity of the enzyme. A detailed compilation on the manifold strategies of CA immobilization has been reported in the form of mini-review by Shao et al.111
Active site installation on the other hand refers to the incorporation of an active site inside the MOFs. The active sites in this case have a range of different metal centres. The first chiral MOF, CFA-1, was synthesized and reported by Schmieder et al.112 The MOFs [Zn5(OAc)4(BIBTA)3] (CFA-1), and [Zn(CoZn3)(OAc)4(BIBTA)3] [BIBTA = 5,5′-bibenzotriazole] are structurally representative to that of the active centre of CA. In this case, Kuratowskii type secondary building unit (SBU) was utilized and an achiral ligand, namely BIBTA2−, was incorporated to produce the first generation of this type of CA mimics (see Scheme 5). Introduction of a redox-active metal like Co(II) on the periphery of the MOFs enhanced the microporous network of the MOF. This led to an increase in the potential to mimic the activity of CA.112 Bien et al. did some post-synthetic ligand modification to [Zn(ZnOAc)4(BIBTA)3] by exchanging acetate with bicarbonate and subsequent thermal activation to indirectly produce a nucleophilic Zn–OH moiety analogues to the Zn–OH moiety of α-CA (Fig. 7).113
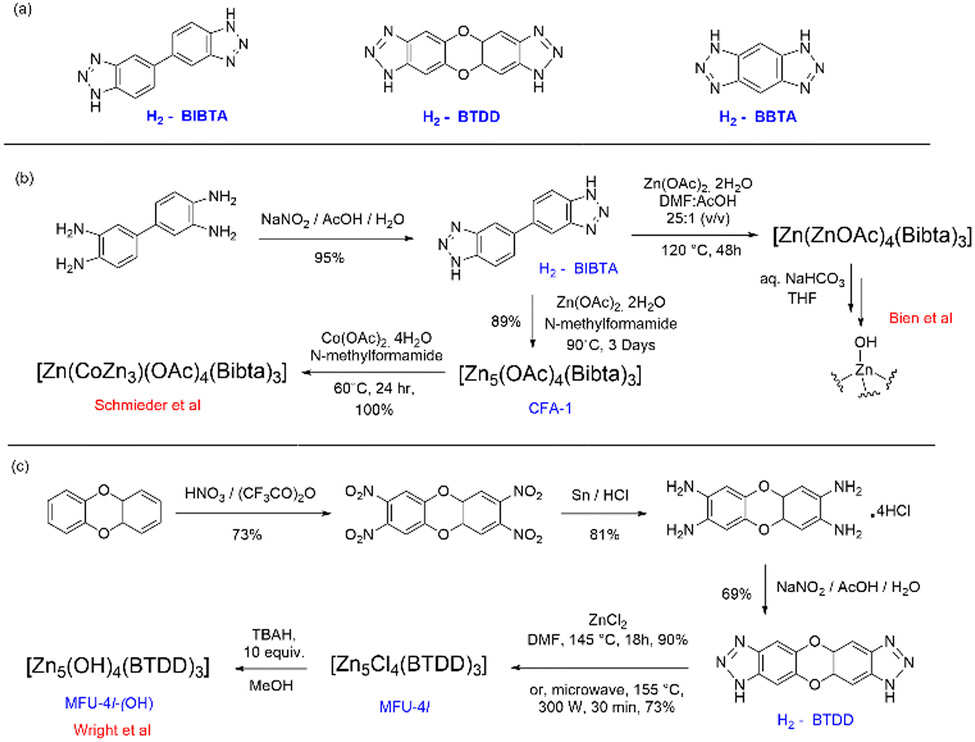 | ||
| Scheme 5 (a) Building block units of MOFs discussed herein; (b) synthesis of the MOFs [Zn(OAc)4(BIBTA)3] and [Zn(CoZn3)(OAc)4(BIBTA)3]; (c) synthesis of MOF MFU-4l and MFU-4l-(OH). | ||
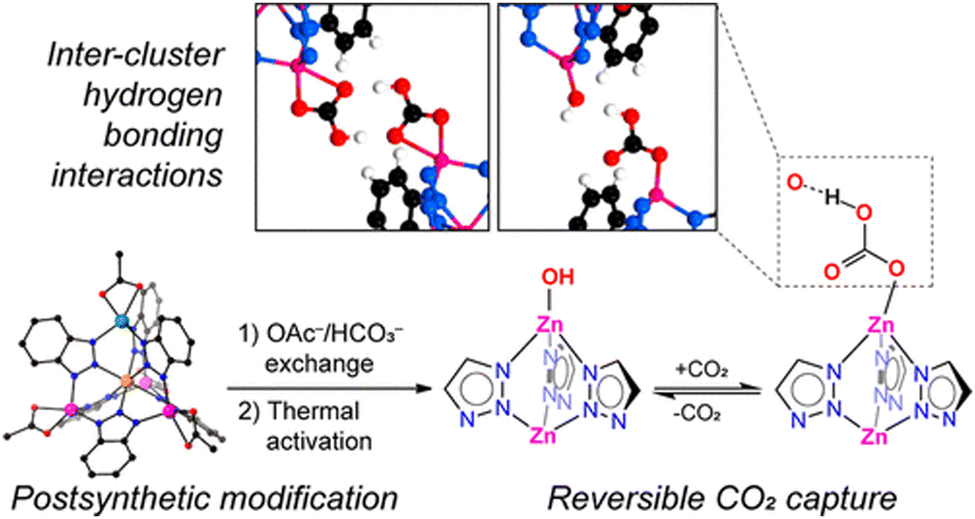 | ||
| Fig. 7 Synthetic methodology, structure and reversible CO2 fixation mechanism of [Zn(ZnOAc)4(BIBTA)3]. Image adapted with permission from ref. 113. Copyright 2018 American Chemical Society. | ||
These nucleophilic centres augmented the inter-cluster hydrogen bonding interactions and mimic the active site of the α-CA, and therefore, exhibit excellent catalytic activity for capturing CO2. [Zn5Cl4(BTDD)] (BTDD = bis(1H-1,2,3-triazolo[4,5-b]-[4′,5′i]) dibenzo-[1,4]-dioxin) (MFU-4l), is an isoreticular MOF designed to mimic the catalytic activity of CA through different pore sizes available in the structure of the framework. The various pore sizes as well as the cubic structure not only regulate the catalytic efficacy but also control the thermal stability of the mimic.114
Dinca and co-workers improved MFU-4l by anion metathesis of the terminal chloride with hydroxide using tetrabutylammonium hydroxide.115 The MOF was found to exhibit efficient catalytic CA mimetic activity. Reactions of MFU-4l-(OH) with H218O led to an equilibrium mixture of CO2 isotopologues (C16O2, C16,18O2 and C18O2) within 5 h, whereas it took 10 h for MFU-4l. As can be seen from Fig. 8, the CO2 adsorption rate was excellent within a pressure range of 0–17 torr with an adsorption of almost 1 mmol of CO2 per mmol of MOF, however the remaining CO2 can be seen to be adsorbed over an extended pressure range of 17–800 torr with an overall capacity of 3.41 mmol g−1 at 800 torr, i.e. 4.05 mmol of CO2 per mole of the MOF. The catalyst was also found to catalyse the hydrolysis reaction of p-nitrophenyl acetate analogous to the CO2 hydration activity of CA. Other than high CO2 uptake capacity, a porous CA mimicking MOF should exhibit high CO2/N2 selectivity, good recycling ability and functionality under wet flue gas conditions.
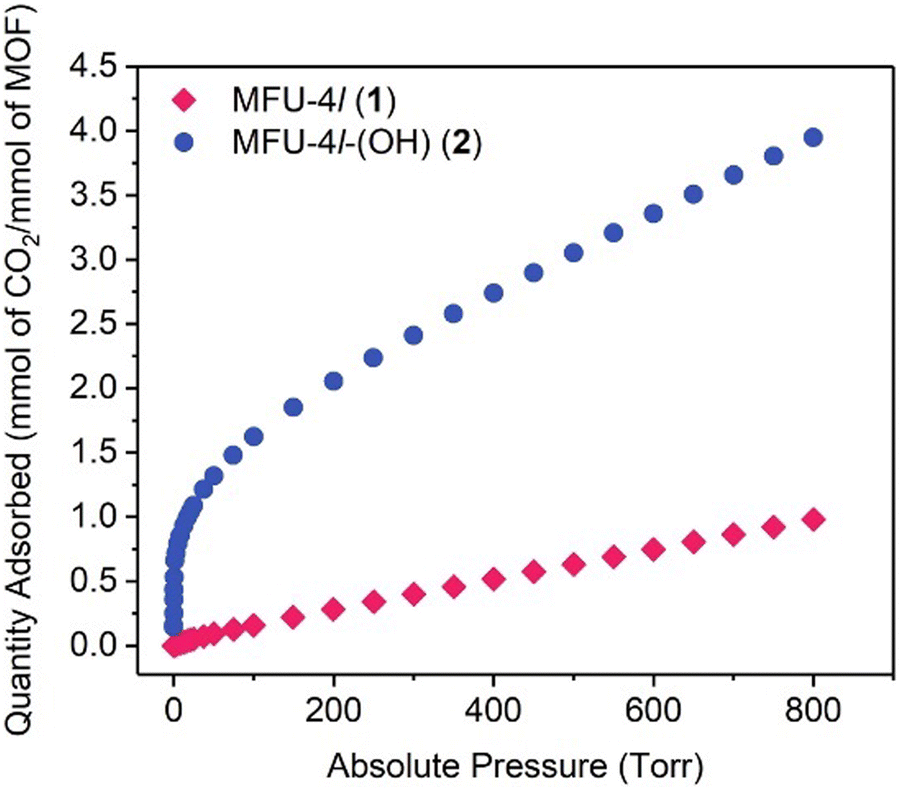 | ||
| Fig. 8 CO2 isotherm of MFU-4l and MFU-4l-(OH) at 298 K. Reprinted with permission from ref. 115. Copyright 2018 Elsevier Inc. | ||
A series of a porous coordination polymers were synthesized and investigated for CO2 adsorption, [M2Cl2 (BBTA)] (BBTA – benzo bis-triazole) having a monodentate hydroxide ligand and the active site substituted with redox metals like Mn(II) and Co(II).61 CO2 sorption abilities were found to be 5.36 and 4.24 mmol g−1 for [Mn2Cl2(BBTA)] and [Co2Cl2(BBTA)], respectively, at 298 K and 1 bar pressure. Infrared (IR) spectroscopy data revealed that the MOFs contained a similar intermediate as the bicarbonate-bound ion present in the catalytic cycle of CA. Furthermore, the adsorption of CO2 into the active site in the presence of moisture, i.e. under high-humidity flue gas conditions, is quite challenging due to a competition between CO2 and H2O binding. [Co2Cl2(BBTA)] was found to be inefficient toward CO2 capture from a wet–gas mixture. However, [CoIICoIII(OH)Cl2(BBTA)] retained its activity in both dry and wet conditions at a humidity level of 82%.
A pincer-based Co containing complex, 2,6-bis (2-benzimidazolyl)pyridine (Co-BBP) was designed and synthesized and encapsulated inside a mesoporous terbium MOF cage of pertinent dimension (Fig. 9), which produced a catalytic activity almost similar [≈100%] to that of native CA.116 The benzimidazole moieties present in the ligand mimic the histidine scaffolds of CA. The homogeneous complexes in solution are susceptible to dimerization, which in turn, leads to decrease in the catalytic efficacy. Therefore, a hydrophobic environment was hypothesized to be favourable to pocket the active centre. Hence to prevent dimerization and add stability to the MOF, Co-BPP was encapsulated in Tb(NO3)3·5H2O and the product Co-BPP@Tb-MOF when analysed for p-NPA hydrolysis activity, was found to give ∼14% increase in catalytic activity. This process of introducing functional linkers instead of entire enzyme into the MOFs was found to produce enhanced catalytic performances.
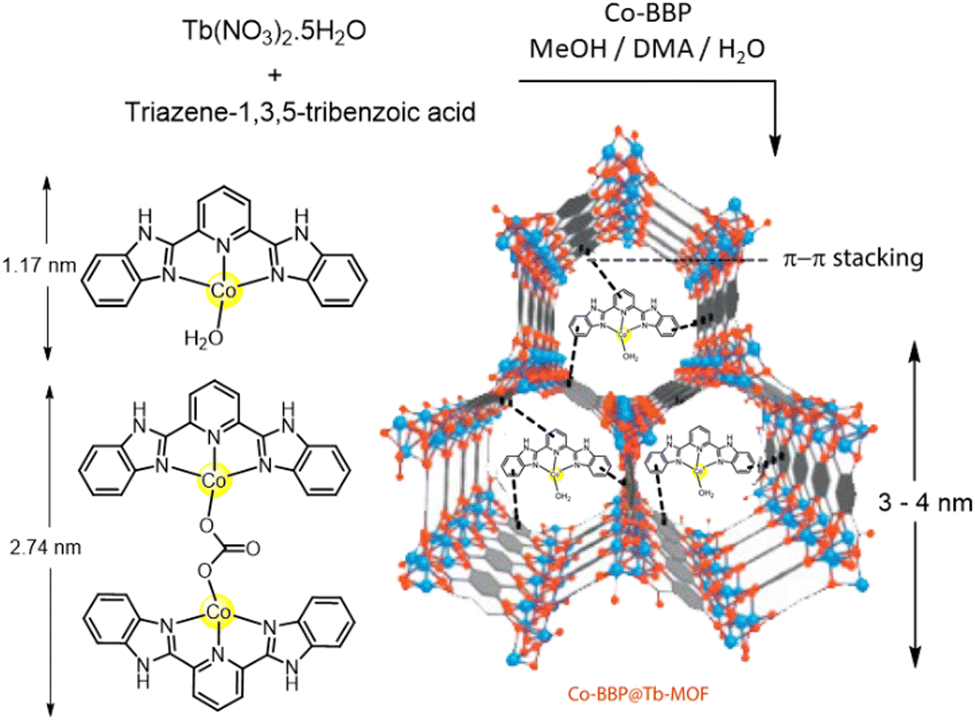 | ||
| Fig. 9 Pictorial illustration of the average sizes of the Tb-MOF cage, Co-BBP (monomer and dimer) and its encapsulation in Tb-MOF. Adapted with permission from ref. 116. Copyright 2013. Elsevier Publishing. | ||
Zeolitic imidazole framework (ZIFs) as CA models
In the endeavour of CO2 fixation, apart from MOFs, topological isomorphs of zeolites called zeolitic imidazole frameworks (ZIFs) were synthesized for sequestration and catalytic conversion of CO2. ZIFs are structural mimics of zeolites and contain a porous crystalline framework. However, unlike the aluminosilicate framework in zeolites, transition metals like Zn or Co occupy the tetrahedron in ZIFs, and imidazole groups replace the oxygen bridges. Attributing to their low cytotoxicity and good biocompatibility, biomacromolecules are often immobilized and encapsulated into ZIFs. In fact, the immobilization of several enzymes onto ZIFs exhibit superior enzyme activity and stability, e.g. cytochrome c onto ZIF-8, glucose oxidase onto ZIF-8, and catalase onto ZIF-90.117–119A number of ZIFs were synthesized and characterised and found to exhibit high thermal and chemical stability resulting in enhanced efficacy in catalytic conversion of CO2.120,121 Further, immobilization of CA onto ZIFs were done on CA/ZIF-L where polyvinylpyrrolidone was used to disperse CA into the solution and enhance its activity.122 The encapsulated system CA-ZIF-L was shown to enhance the thermal stability as well as the catalytic activity of the mimic in comparison to the pure ZIF-L (Fig. 10). The chemical adsorption and hydration of CO2 onto ZIF-L was amplified by the nucleophilic characteristic of the imidazole. ZIF-L also influenced the catalytic reusability by forming a protective layer over the nanocomposite.
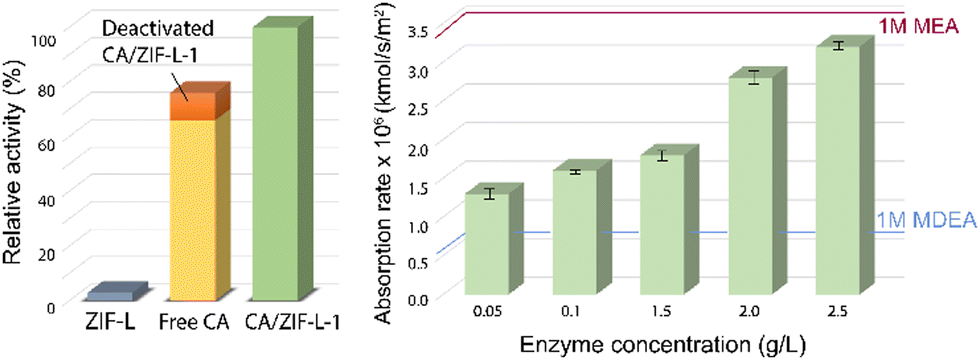 | ||
| Fig. 10 (left) Relative activity among ZIF-L, free enzyme, deactivated CA/ZIF-L-1 and activated CA/ZIF-L-1. It shows the sum of the CO2 hydration activities of the free CA enzyme and the inactive CA/ZIF-L-1 was lower than that of the active CA/ZIF-L-1, indicating the synergy between CA and ZIF-L support. The activities were plotted with respect to 100% activity of CA/ZIF-L-1. (right): CO2 absorption of various concentrations of CA/ZIF-L at 40 °C at a CO2 partial pressure of 15 kPa. The red and blue lines represent the CO2 adsorption rate into 1 M MEA and MDEA solutions at 40 °C, respectively. | ||
The structural features of the ZIF-8-L includes four 2-methylimidazole and a Zn(II) centre. Geometrically, the synthesized ZIF-8 was found to have missed coordination with 2-methylimidazole in certain parts of the framework matrix, resulting in the formation of an interstitial site which becomes a favourable centre for CO2 as well as H2O adsorption. These sites give rise to active Zn(2-mIM)nO units.123 Aiming towards improvising the structural attributes of ZIFs, Yu and co-workers devised an environmentally sustainable methodology to develop ZIF-8 nano-enzymes encapsulated with amino acids (Fig. 11).124 The role of amino acid was to influence the morphology and size of the ZIF-8 moiety in order to modulate the catalytic activity of the enzyme. The quantified presence of Zn2+ on the outer surface of ZIF-8 calculated through TEM results was responsible for the increase in catalytic activity of the nano-enzyme almost comparable to that of biologically available CA.
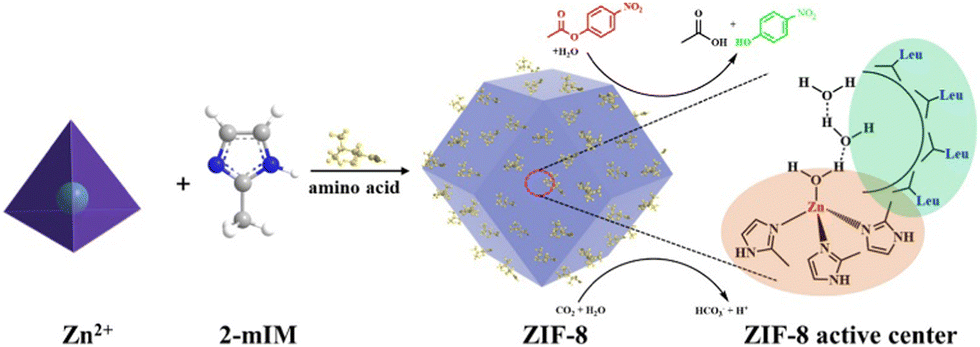 | ||
| Fig. 11 Pictorial illustration of the synthesis of ZIF-8 and its resemblance with the structure and function of CA (2-mIM = 2-methylimidazole). Adapted with permission from ref. 124. Copyright 2022. American Chemical Society. | ||
An iron-containing ZIF, i.e. ZIF-8@Fe3O4-carbonic anhydrase composite, was synthesized with the aim to aid CO2 adsorption into methyldiethanolamine (MDEA) due to facile recovery from the solution. The mimic exhibited 95.2% CO2 conversion rate and remarkable recyclability.125 Further investigation with the iron-containing ZIFs showed potential as CA mimics, wherein iron(II) was inserted into ZIFs to examine the catalytic activity of the mimics. FeX@CN-Mg viz. Fe3@CN-Mg, Fe10@CN-Mg and Fe20@CN-Mg were developed where X represents the mole ratio of zinc(nitrate)(hexahydrate) with respect to that of ferrous(sulphate)(heptahydrate). A comparative study based on parameters like pore size, availability of catalytic sites, percentage content of Fe and its effect in catalytic activity were conducted and the catalytic efficacy of Fe10@CN-Mg was found to be superior to other metal incorporated frameworks. These systems offer low cost and lesser metal content while having comparable kinetic constants like other models (KM = 6.37 mM and Vmax = 30.74 mM min−1).126
Among all the transition metals, the only metal that has a similar tendency to form tetrahedral co-ordinated structures similar to zinc is cobalt. As such, it is the optimal choice for the metal centre in the primary metal coordination sphere. Moreover, Co is also advantageous and a suitable substitute as it enhances the catalytic activity simultaneously improving the structural stability of the framework. A series of bimetallic ZIFs with different percentages of the metal centres were synthesized. Utilizing the properties of cobalt infused ZIFs, Huang and co-workers havesynthesized a series of Co/ZIF-8 framework by mixing Co(OAc)2·4H2O and Zn(OAc)2·2H2O in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, 1:3, 1:2, 1:1, 2:1, 4:1, and 1:0 ratio. With the increase in the percentage of the Co(II) ion, the kinetic parameters of p-NPA hydrolysis in terms of Vmax and KM, increase and decrease, respectively. The Km of Co/ZIF-8 was found to be higher than the natural enzyme while the Vmax values were found to be comparable to the natural CA with a steady increase in the Co-doping ratio.62 A similar study was again conducted by the same group using Cu(II) and Ni(II) doping experiments into bimetallic ZIF-8 nanostructures while maintaining the pore size uniformity.
:4, 1:3, 1:2, 1:1, 2:1, 4:1, and 1:0 ratio. With the increase in the percentage of the Co(II) ion, the kinetic parameters of p-NPA hydrolysis in terms of Vmax and KM, increase and decrease, respectively. The Km of Co/ZIF-8 was found to be higher than the natural enzyme while the Vmax values were found to be comparable to the natural CA with a steady increase in the Co-doping ratio.62 A similar study was again conducted by the same group using Cu(II) and Ni(II) doping experiments into bimetallic ZIF-8 nanostructures while maintaining the pore size uniformity.
As can be seen from Fig. 12, incorporation of either Ni or Cu have improved upon the esterase activity in comparison to the undoped ZIF-8. With an increase in the doping percentage, the catalytic activity was found to have enhanced. The Vmax values for ZIF-8, Co50%/ZIF-8, Ni50%/ZIF-8, and Cu50%/ZIF-8 were observed to be 321.87, 461.43, 460.27 and 506.13 nM s−1 respectively.62 At 80 °C, these mixed systems displayed excellent thermal stability and a substantially higher esterase activity profiles (see Fig. 12). Also, higher the doping ratio of specific metal ions, greater is the reverse hydration ratio. Further, unlike natural CA enzymes that are susceptible towards temperature, these bimetallic hybrid systems are quite temperature resistant and shows esterase activity even at 500 °C (Fig. 12(c)). Also, in terms of reusability, these bimetallic systems are comparable to the parent ZIF-8 up to 5 cycles of reusability.127
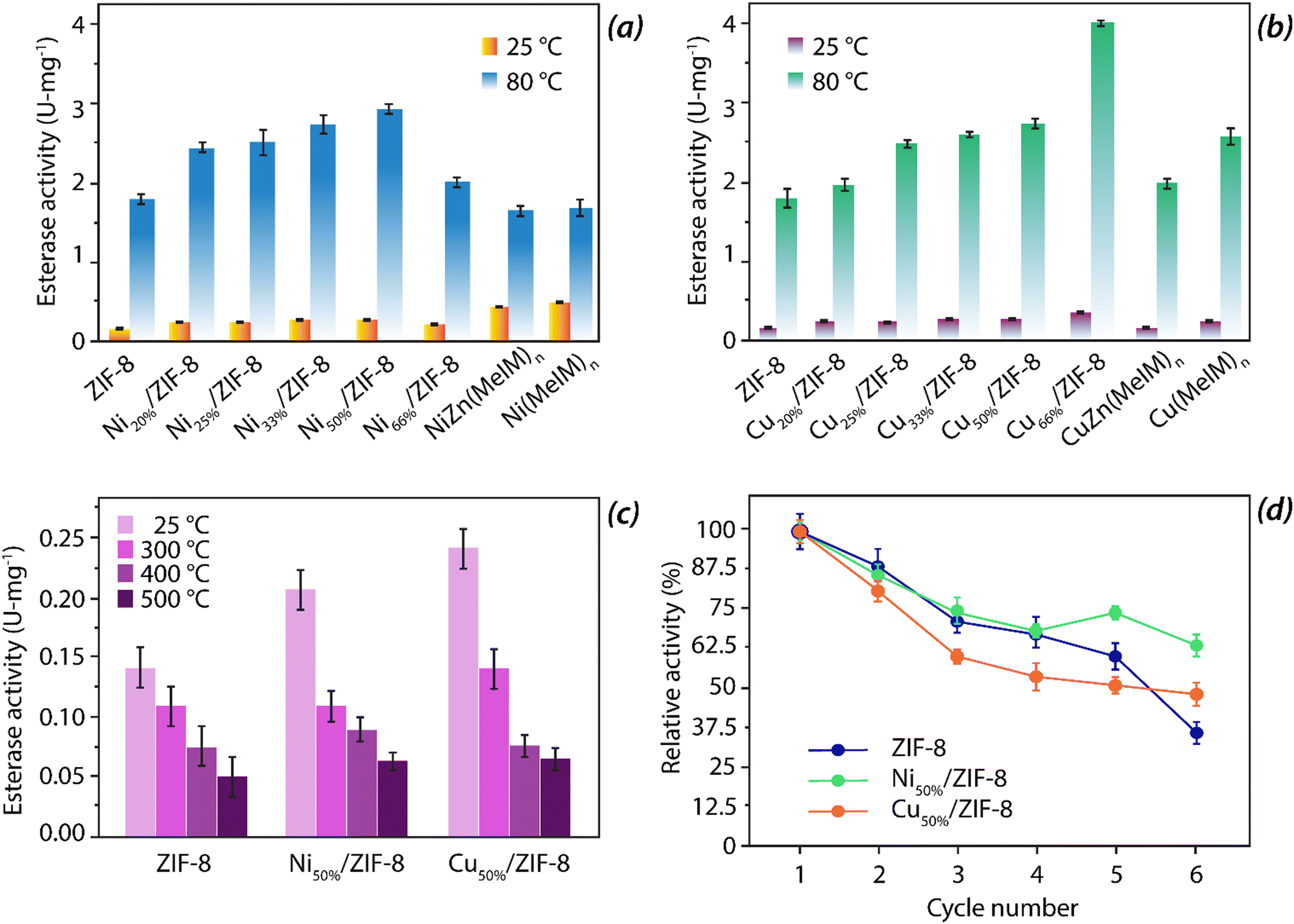 | ||
| Fig. 12 (a) Variation in esterase activity of various Ni-ZIF-8 mimics at 25 °C and 80 °C; (b) variation in esterase activity of various Cu-ZIF-8 mimics at 25 °C and 80 °C; (c) comparative study of esterase activity of ZIF-8, 50% Ni doped ZIF-8 and 50% Cu doped ZIF-8 mimics at 25 °C, 300 °C, 400 °C and 500 °C and (d) reusability cycle parameter of ZIF-8, 50% Ni doped ZIF-8 and 50% Cu doped ZIF-8. | ||
Amine based solvents have great credibility in post combustion carbon capture through the wet scrubbing method. In particular, monoethanolamine (MEA, 2-aminoethan-1-ol) has become a favourable industrial solvent for CO2 sequestration due to its high reactivity with CO2 and cost efficiency. Having considered that, Ji et al. developed ZIF-90 and ZIF-90@MEA to mimic CA to study the effect of amine-based solvents on the efficacy of CO2 hydration by ZIF-90.128 ZIF-90 has a Zn(2-ICA)3 structural unit (ICA = imidazole-2-carboxaldehyde, shown in Fig. 13) which is almost similar to that of CA. The free aldehyde groups react with monoethanolamine to form a condensed imine product, ZIF-90@MEA. The incorporation of MEA into ZIF-90 led to greater structural durability and increased catalytic activity and similar kinetic behaviour to the natural CA. The enhanced CO2 uptake capacity of alkanol–amine solvents along with lower percentage of environmental hazard have made these type of solvents preferentially more favoured by industries.129,130
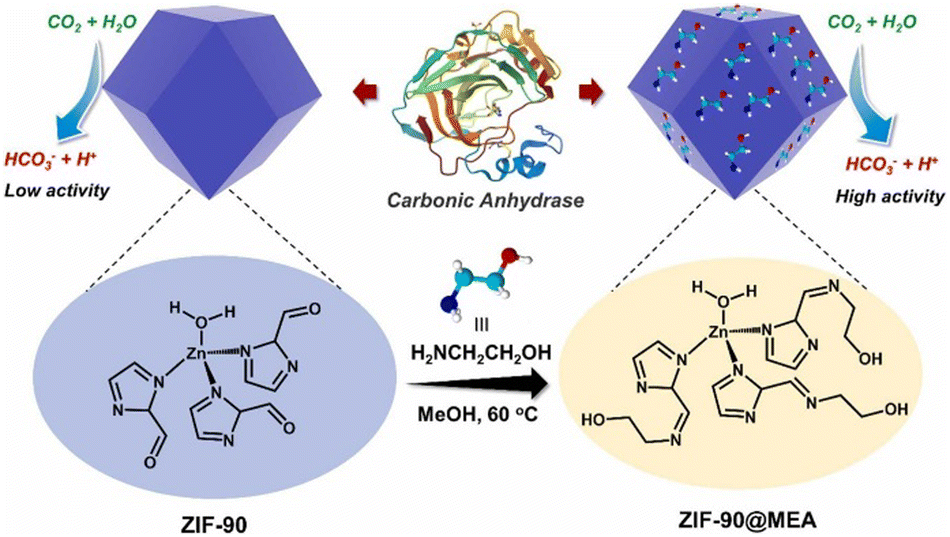 | ||
| Fig. 13 Schematic illustration of the synthesis of ZIF-90@MEA from ZIF-90. Reprinted with permission from ref. 128. Copyright 2023 Elsevier Publishing. | ||
Non-natural reactions by engineered carbonic anhydrase – artificial enzymes
Over the last few decades, the field of artificial metalloenzymes have attracted a lot of attention. Incorporation of an abiotic metal cofactor inside a protein scaffold gives an opportunity to tune the vast plethora of chemical catalysis with an added advantage of selectivity provided by the protein scaffold. Detailed review articles are in place describing the strategic synthesis, utilities and fundamentals of artificial metalloenzymes.131–135 Several new-to-nature reactions are being carried out with the help of these hybrid systems. Enantioselective styrene epoxidation was carried out by employing apoCA (1 equiv.) with Mn(II) (0.5 equiv.).136 The bovine CA and recombinant human CA has 81% sequence identity. Upon addition of bicarbonate (48 equiv.) and H2O2 (57 equiv.) in phosphate buffer, (R)-styrene oxide was formed with up to 50% enantiomeric excess. Control experiments without the apoCA yielded no epoxidation product inferring that the activity arises from the presence of Mn(II) in the active pocket of the enzyme. In the absence of Mn(II), the Zn-containing CA resulted into only 5% epoxidation with no enantioselectivity.In another endeavour, Ward and co-workers incorporated piano-stool complexes of iridium and ruthenium inside human CA, thereby generating artificial metalloenzymes for asymmetric transfer hydrogenation of imines.137,138 Capitalising on the high affinity of arylsulfonamides towards Zn(II), ligands were prepared with an appended arylsulfonamide anchor to bind to the Zn(II) of CA (Scheme 6). With the help of an Ir piano-stool complex meticulously placed at the entrance of the funnel-shaped sulphonamide binding site, asymmetric transfer hydrogenation was observed with an ee of 68%. Further, engineering the wild type enzyme by mutations (191A and K170A) enlarged the active binding pocket leading to less substrate inhibition and higher catalytic conversions.
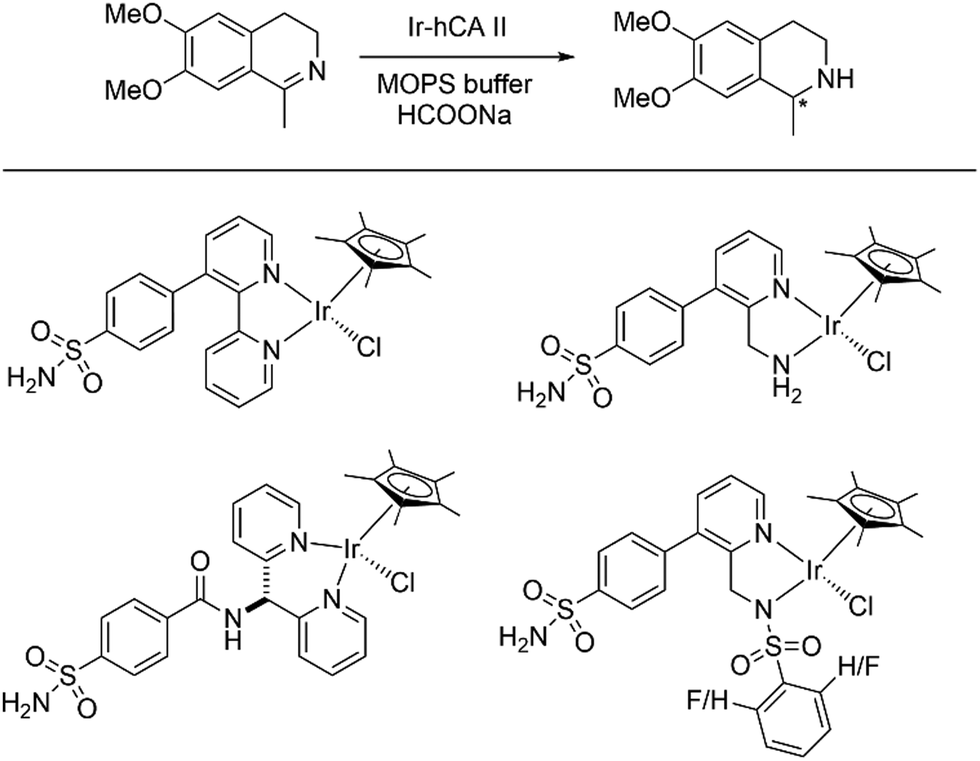 | ||
| Scheme 6 (top) Asymmetric transfer hydrogenation catalysed by Ir-hCA II artificial enzyme; (bottom) Piano-stool Ir complexes incorporated into the CA scaffold. | ||
The use of CA as peroxidases have been achieved by Kazlauskas and co-workers exchanging Zn with Mn into CA.139 Dialysis of the enzyme with 2,6-pyridinedicarboxylate in acetate buffer (pH 5.5) removes almost 95% of Zn(II) while further dialysis of the apoCA with MnCl2 at pH 6.95 yields the Mn-incorporated CA (80 mol% Mn and 5–10 mol% Zn).139 Peroxidase activity of the Mn–CA artificial metalloenzyme was tested using o-dianisidine and H2O2 as shown in Scheme 7. The peroxidase activity of this hybrid system (kcat = 140 s−1, kcat/KM = 1.4 × 106 M−1 s−1) was almost comparable to horseradish peroxidase (kcat = 630 s−1, kcat/KM = 57 × 106 M−1 s−1).
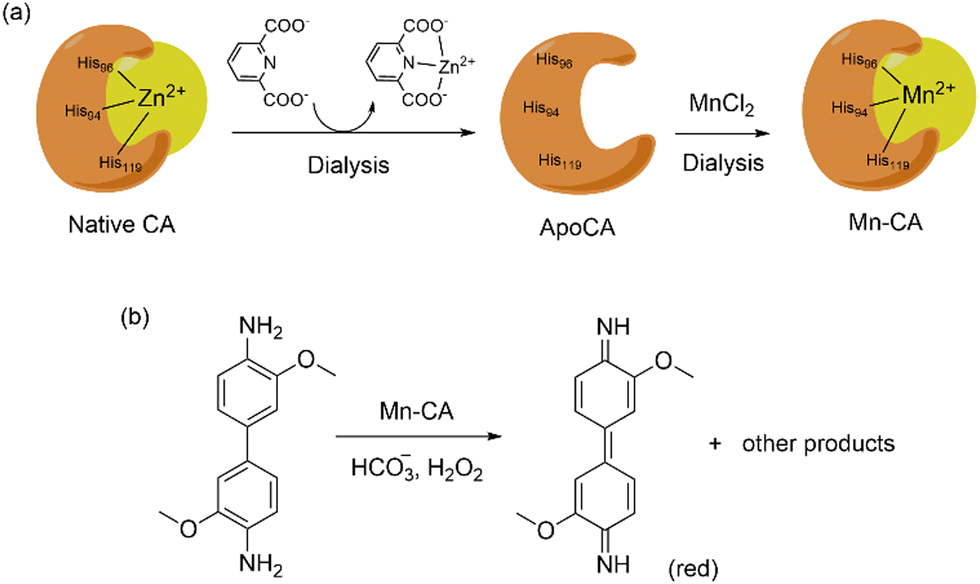 | ||
| Scheme 7 (top) Substitution of Zn(II) with Mn(II) in CA by dialysis. (bottom) Peroxidase activity of Mn–CA artificial enzyme using o-dianisidine as substrate. | ||
The same group has also reported the regioselective hydroformylation of styrene using the Rh-incorporated CA (Scheme 8).140,141 A similar dialysis procedure was again employed to substitute Zn with Rh using [Rh(CO)2(acac)]. A diethyl pyrocarbonate (DEPC)-modified human CA II mutant (DEPC-H4/10R + H17F-Rh) shows the best result with 66.2% of linear aldehyde and a regioselectivity (linear/branched product ratio) of 8.4.
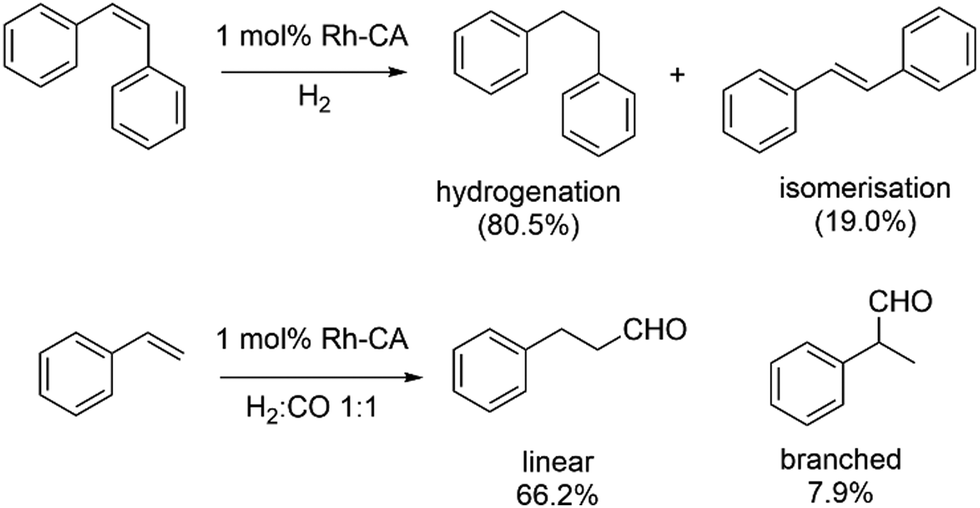 | ||
| Scheme 8 Hydrogenation of cis-stilbene (top) and hydroformylation of styrene (bottom) catalysed by Rh–CA artificial enzymes. | ||
Conclusions
Overall, a comprehensive compilation of the employment of different transition metal ions for the biomimetic activity of Carbonic Anhydrase has been presented here. This study includes the enzymatic modifications (in terms of metal ion substitution in natural enzymes), and incorporation of different bivalent metal ions in synthetic model systems such as metal complexes and porous or self-assembling materials. Also, the final section introduces the usage of CA in artificial metalloenzymes, where different non-native metal cofactors (Mn, Ru, Ir, Rh) are impregnated inside the enzyme scaffold to deliver new-to-nature reactivity. It is worth notifying that Zn(II) and Co(II) exhibit a higher binding affinity and carbonic anhydrase activity in comparison to other metals, such as Ni(II), Hg(II), Cd(II), Cu(II), and Mn(II). Under anaerobic conditions, it has also been observed in certain cases, that Fe(II) can exhibit enhanced catalytic activity in comparison to Zn(II). Structural analysis of non-native metal substitutions also revealed that the tetrahedral structure of Zn(II) plays a significant role in the activity of CA, and the activity decreases as we move from tetrahedral (ZnII) to octahedral (CoII, NiII) to trigonal bipyramidal (CuII) geometry (tetrahedral > octahedral > trigonal bipyramidal).Over time, various research groups have explored the potential of various metal complexes using small molecules and MOFs to diversify their applications and/or enhance their enzymatic potential. Different immobilization strategies and encapsulation by porous polymeric materials have indeed improved the thermal stability and catalytic efficacy of the hybrid systems in comparison to the direct usage of the enzymes towards industrial CO2 capture and hydration. Non-native metal substitutions in CA have led to better understanding of the enzyme activity. Further, the evolution of the field of artificial metalloenzymes have presented CA in a new outlook by improvising its reactivity by tuning its cofactor. In a nutshell, CA mimics have paved an avenue as a diverse class of biomimetic models, that have versatile applications. The choice of metal ions in imitating the activity of a natural enzyme is very crucial. However, based on this detailed discussion on the implications of non-native metal ions to mimic the activity of carbonic anhydrase, it is imperative that the choice of the metal ion (Zn) made by nature is immaculate. Although in several instances, Co can be used as a substitute, the pKa of the metal–aqua complex and its geometry is extremely important in the CO2 hydration activity of either an engineered enzyme or an engineered biomimetic model system. Furthermore, it has also been observed that the passage of CO2 to the vicinity of the metal ion requires a hydrophobic channel and the elimination of the bicarbonate/carbonate ion also needs to be facile and unhindered by steric fencing. A successful design of a CA mimic must be done taking all these parameters into consideration to achieve a reversible CO2 uptake and release and match CA's hydration rates – which is one of the fastest among all enzymes in nature.
Author contributions
DB, SdV and GM conceptualised the study and prepared the draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GM and DB acknowledges the DST-INSPIRE Faculty fund (DST/INSPIRE/04/2022/002659) for research grant and fellowship.References
- A. Das, C. Hessin, Y. Ren and M. Desage-El Murr, Chem. Soc. Rev., 2020, 49, 8840 RSC.
- A. C. Ghosh, C. Duboc and M. Gennari, Coord. Chem. Rev., 2021, 428, 213606 CrossRef CAS.
- M. D. White and E. Flashman, Curr. Opin. Chem. Biol., 2016, 31, 126 CrossRef CAS.
- C. R. Zwick III and H. Renata, Nat. Prod. Rep., 2020, 37, 1065 RSC.
- H. Tao and I. Abe, Eng. Microbiol., 2023, 3, 100062 CrossRef CAS.
- R. Ushimaru and I. Abe, ACS Catal., 2022, 13, 1045 CrossRef.
- R. Ushimaru, Y. Ding, T. Mori, K. Miyamoto, M. Uchiyama and I. Abe, J. Am. Chem. Soc., 2023, 145, 21966 CrossRef CAS PubMed.
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841 CrossRef CAS.
- Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature, ed. S. P. de Visser and D. Kumar, Royal Society of Chemistry, 2011 Search PubMed.
- H. M. Girvan and A. W. Munro, Curr. Opin. Chem. Biol., 2016, 31, 136 CrossRef CAS PubMed.
- N. P. Dunham and F. H. Arnold, ACS Catal., 2020, 10, 12239 CrossRef CAS PubMed.
- Handbook of Porphyrin Science: With Applications To Chemistry, Physics, Materials Science, Engineering, Biology And Medicine, ed. K. M. Kadish, R. Guilard and K. M. Smith, World Scientific, 2010, vol. 6–10 Search PubMed.
- M. Atanasov, P. Comba, S. Hausberg and B. Martin, Coord. Chem. Rev., 2009, 253, 2306 CrossRef CAS.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152 CrossRef CAS PubMed.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942 CrossRef CAS PubMed.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2018, 51, 2014 CrossRef CAS PubMed.
- L. Fu, Z. Ren, W. Si, Q. Ma, W. Huang, K. Liao, Z. Huang, Y. Wang, J. Li and P. Xu, J. CO2 Util., 2022, 66, 102260 CrossRef CAS.
- C. W. Jones, JACS Au., 2023, 3, 1536 CrossRef CAS PubMed.
- W. D. Jones, J. Am. Chem. Soc., 2020, 142, 4955 CrossRef CAS.
- F. Nocito and A. Dibenedetto, Curr. Opin. Green Sustainable Chem., 2020, 21, 34 CrossRef CAS.
- L. Lombardo, Y. Ko, K. Zhao, H. Yang and A. Züttel, Angew. Chem., Int. Ed., 2021, 60, 9580 CrossRef CAS PubMed.
- B. Yoon and G. A. Voth, J. Am. Chem. Soc., 2023, 145, 15663 CrossRef CAS.
- J. Han, X. Bai, X. Xu, A. Husile, S. Zhang, L. Qi and J. Guan, Chem. Sci., 2024, 15, 7870 RSC.
- A. Ogawa, K. Oohora, W. Gu and T. Hayashi, Chem. Commun., 2019, 55, 493 RSC.
- S. Fukuzumi, Y.-M. Lee, H. S. Ahn and W. Nam, Chem. Sci., 2018, 9, 6017 RSC.
- H. Rao, J. Bonin and M. Robert, J. Phys. Chem. B, 2018, 122, 13834 CAS.
- F. Franco, M. F. Pinto, B. Royo and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2018, 130, 4693 CrossRef.
- X. F. Liu, X. Y. Li and L. N. He, Eur. J. Org. Chem., 2019, 2437 CrossRef CAS.
- S. Fernández, F. Franco, M. Martínez Belmonte, S. Friães, B. Royo, J. M. Luis and J. Lloret-Fillol, ACS Catal., 2023, 13, 10375 CrossRef.
- P. A. Davethu and S. P. de Visser, J. Phys. Chem. A, 2019, 123, 6527 CrossRef CAS.
- C. Zhu, C. D’Agostino and S. P. de Visser, Chem. – Eur. J., 2023, 29, e202302832 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 1 Search PubMed.
- S. Bierbaumer, M. Nattermann, L. Schulz, R. Zschoche, T. J. Erb, C. K. Winkler, M. Tinzl and S. M. Glueck, Chem. Rev., 2023, 123, 5702 CrossRef CAS.
- A. Bottoni, C. Z. Lanza, G. P. Miscione and D. Spinelli, J. Am. Chem. Soc., 2004, 126, 1542 CrossRef CAS.
- Y. Demir, N. Demir, H. Nadaroglu and E. Bakan, Prep. Biochem. Biotechnol., 2000, 30, 49 CrossRef CAS PubMed.
- D. Neri and C. T. Supuran, Nat. Rev. Drug Discovery, 2011, 10, 767 CrossRef CAS.
- S. Akocak and C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2019, 34, 1652 CrossRef CAS PubMed.
- Y. Xu, L. Feng, P. D. Jeffrey, Y. Shi and F. M. Morel, Nature, 2008, 452, 56 CrossRef CAS PubMed.
- Cadmium: from Toxicity to Essentiality, ed. A. Sigel, H. Sigel and R. K. Sigel, Springer, 2013 Search PubMed.
- S. Del Prete, D. Vullo, G. M. Fisher, K. T. Andrews, S.-A. Poulsen, C. Capasso and C. T. Supuran, Bioorg. Med. Chem. Lett., 2014, 24, 4389 CrossRef CAS.
- M. S. Kimber and E. F. Pai, EMBO J., 2000, 19, 1407 CrossRef CAS PubMed.
- S. B. Roberts, T. W. Lane and F. M. Morel, J. Phycol., 1997, 33, 845 CrossRef CAS.
- Handbook of Metalloproteins, ed. Y. Xu, C. T. Supuran and F. M. Morel, 2006 Search PubMed.
- C. T. Supuran, J. Enzyme Inhib. Med. Chem., 2012, 27, 759 CrossRef CAS PubMed.
- S. R. MacAuley, S. A. Zimmerman, E. E. Apolinario, C. Evilia, Y.-M. Hou, J. G. Ferry and K. R. Sowers, Biochemistry, 2009, 48, 817 CrossRef CAS.
- G. Mukherjee, C. W. Lee, S. S. Nag, A. Alili, F. G. C. Reinhard, D. Kumar, C. V. Sastri and S. P. de Visser, Dalton Trans., 2018, 47, 14945 RSC.
- G. Mukherjee, A. Alili, P. Barman, D. Kumar, C. V. Sastri and S. P. de Visser, Chem. – Eur. J., 2019, 25, 5086 CrossRef CAS PubMed.
- W. N. Oloo and L. Que Jr., Acc. Chem. Res., 2015, 48, 2612 CrossRef CAS.
- G. Mukherjee and C. V. Sastri, Isr. J. Chem., 2020, 60, 1032 CrossRef CAS.
- G. Mukherjee, J. K. Satpathy, U. K. Bagha, M. Q. E. Mubarak, C. V. Sastri and S. P. de Visser, ACS Catal., 2021, 11, 9761 CrossRef CAS.
- G. Mukherjee, G. Velmurugan, M. Kerscher, J. Kumar Satpathy, C. V. Sastri and P. Comba, Chem. – Eur. J., 2024, 30, e202303127 CrossRef CAS.
- U. K. Bagha, J. K. Satpathy, G. Mukherjee, C. V. Sastri and S. P. de Visser, Org. Biomol. Chem., 2021, 19, 1879 RSC.
- S. P. de Visser, G. Mukherjee, H. S. Ali and C. V. Sastri, Acc. Chem. Res., 2021, 55, 65 CrossRef PubMed.
- N. Proos Vedin and M. Lundberg, J. Biol. Inorg. Chem., 2016, 21, 645 CrossRef CAS.
- L. Vicens, G. Olivo and M. Costas, ACS Catal., 2020, 10, 8611 CrossRef CAS.
- A. R. McDonald and L. Que Jr., Coord. Chem. Rev., 2013, 257, 414 CrossRef CAS.
- S. P. de Visser, Chem. – Eur. J., 2020, 26, 5308 CrossRef CAS.
- L. Koziol, C. A. Valdez, S. E. Baker, E. Y. Lau, W. C. Floyd III, S. E. Wong, J. H. Satcher Jr., F. C. Lightstone and R. D. Aines, Inorg. Chem., 2012, 51, 6803 CrossRef CAS PubMed.
- R. Ma, G. F. Schuette and L. J. Broadbelt, Appl. Catal., A, 2015, 492, 151 CrossRef CAS.
- M. C. Kim and S. Y. Lee, Chem. – Eur. J., 2014, 20, 17019 CrossRef CAS.
- P.-Q. Liao, H. Chen, D.-D. Zhou, S.-Y. Liu, C.-T. He, Z. Rui, H. Ji, J.-P. Zhang and X.-M. Chen, Energy Environ. Sci., 2015, 8, 1011 RSC.
- Y. Xiang, D. Yu, C. Qin, J. Deng, X. Wang, B. Ge and F. Huang, Colloids Surf., A, 2024, 685, 133227 CrossRef CAS.
- S. Lindskog, Pharmacol. Ther., 1997, 74, 1 CrossRef CAS.
- X. Niu, X. Li, Z. Lyu, J. Pan, S. Ding, X. Ruan, W. Zhu, D. Du and Y. Lin, Chem. Commun., 2020, 56, 11338 RSC.
- L. Li, W. Xu, Z. Wu, W. Geng, S. Li, S. Sun, M. Wang, C. Cheng and C. Zhao, Small, 2024, 20, 2307537 CrossRef CAS.
- P. Bag, S. Dutta, P. Biswas, S. K. Maji, U. Flörke and K. Nag, Dalton Trans., 2012, 41, 3414 RSC.
- J. Coleman, Nature, 1967, 214, 193 CrossRef CAS.
- J. Vidgren, A. Liljas and N. P. Walker, Int. J. Biol. Macromol., 1990, 12, 342 CrossRef CAS PubMed.
- B. C. Tripp, C. B. Bell, F. Cruz, C. Krebs and J. G. Ferry, J. Biol. Chem., 2004, 279, 6683 CrossRef CAS PubMed.
- J. G. Ferry, Biochim. Biophys. Acta, 2010, 1804, 374 CrossRef CAS.
- T. Marino, N. Russo and M. Toscano, J. Am. Chem. Soc., 2005, 127, 4242 CrossRef CAS.
- J. Led and E. Neesgaard, Biochemistry, 1987, 26, 183 CrossRef CAS PubMed.
- K. A. Kogut and R. Rowlett, J. Biol. Chem., 1987, 262, 16417 CrossRef CAS.
- H. Irving and R. Williams, J. Chem. Soc., 1953, 3192 RSC.
- G. P. Lisi, R. P. Hughes and D. E. Wilcox, J. Biol. Inorg. Chem., 2016, 21, 659 CrossRef CAS PubMed.
- J. K. Kim, C. Lee, S. W. Lim, A. Adhikari, J. T. Andring, R. McKenna, C.-M. Ghim and C. U. Kim, Nat. Commun., 2020, 11, 4557 CrossRef CAS PubMed.
- H. Vahrenkamp, Acc. Chem. Res., 1999, 32, 589 CrossRef CAS.
- R. Alsfasser, M. Ruf and H. Vahrenkamp, Chem. Ber., 1993, 126, 703 CrossRef CAS.
- E. Kimura, T. Shiota, T. Koike, M. Shiro and M. Kodama, J. Am. Chem. Soc., 1990, 112, 5805 CrossRef CAS.
- E. Kimura, T. Koike and K. Toriumi, Inorg. Chem., 1988, 27, 3687 CrossRef CAS.
- J. Suh, S. J. Son and M. P. Suh, Inorg. Chem., 1998, 37, 4872 CrossRef CAS.
- X. Zhang, R. van Eldik, T. Koike and E. Kimura, Inorg. Chem., 1993, 32, 5749 CrossRef CAS.
- M. Bräuer, J. L. Pérez-Lustres, J. Weston and E. Anders, Inorg. Chem., 2002, 41, 1454 CrossRef.
- R. Davy, Energy Procedia, 2009, 1, 885 CrossRef CAS.
- W. C. Floyd III, S. E. Baker, C. A. Valdez, J. K. Stolaroff, J. P. Bearinger, J. H. Satcher Jr. and R. D. Aines, Environ. Sci. Technol., 2013, 47, 10049 CrossRef.
- R. Ma, G. F. Schuette and L. J. Broadbelt, Int. J. Chem. Kinet., 2014, 46, 683 CrossRef CAS.
- X. Zhang and R. van Eldik, Inorg. Chem., 1995, 34, 5606 CrossRef CAS.
- E. Y. Lau, S. E. Wong, S. E. Baker, J. P. Bearinger, L. Koziol, C. A. Valdez, J. H. Satcher Jr., R. D. Aines and F. C. Lightstone, PLoS One, 2013, 8, e66187 CrossRef CAS.
- C. A. Lippert, K. Liu, M. Sarma, S. R. Parkin, J. E. Remias, C. M. Brandewie, S. A. Odom and K. Liu, Catal. Sci. Technol., 2014, 4, 3620 RSC.
- R. A. Kelsey, D. A. Miller, S. R. Parkin, K. Liu, J. E. Remias, Y. Yang, F. C. Lightstone, K. Liu, C. A. Lippert and S. A. Odom, Dalton Trans., 2016, 45, 324 RSC.
- C. Bergquist, T. Fillebeen, M. M. Morlok and G. Parkin, J. Am. Chem. Soc., 2003, 125, 6189 CrossRef CAS PubMed.
- D. Huang, O. V. Makhlynets, L. L. Tan, S. C. Lee, E. V. Rybak-Akimova and R. H. Holm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1222 CrossRef CAS PubMed.
- A. Company, J.-E. Jee, X. Ribas, J. M. Lopez-Valbuena, L. Gomez, M. Corbella, A. Llobet, J. Mahia, J. Benet-Buchholz and M. Costas, Inorg. Chem., 2007, 46, 9098 CrossRef CAS.
- R. Menif, A. E. Martell, P. J. Squattrito and A. Clearfield, Inorg. Chem., 1990, 29, 4723 CrossRef CAS.
- P. Comba, L. R. Gahan, G. R. Hanson, M. Maeder and M. Westphal, Dalton Trans., 2014, 43, 3144 RSC.
- X. Liu, P. Du and R. Cao, Nat. Commun., 2013, 4, 2375 CrossRef.
- C. Keum, M.-C. Kim and S.-Y. Lee, J. Mol. Catal. A: Chem., 2015, 408, 69 CrossRef CAS.
- Y. Huang, S. Zhang, H. Chen, L. Zhao, Z. Zhang, P. Cheng and Y. Chen, Inorg. Chem., 2019, 58, 9916 CrossRef CAS.
- Y. Cheng, S. J. Datta, S. Zhou, J. Jia, O. Shekhah and M. Eddaoudi, Chem. Soc. Rev., 2022, 51, 8300 RSC.
- J. Fonseca, L. Meng, I. Imaz and D. Maspoch, Chem. Soc. Rev., 2023, 52, 2528 RSC.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed.
- A. E. Baumann, D. A. Burns, B. Liu and V. S. Thoi, Commun. Chem., 2019, 2, 86 CrossRef.
- Y. Li, R. Wang, X. Liu, K. Li and Q. Xu, Nanotechnology, 2023, 34, 202002 CrossRef CAS.
- M. Safaei, M. M. Foroughi, N. Ebrahimpoor, S. Jahani, A. Omidi and M. Khatami, Trends Anal. Chem., 2019, 118, 401 CrossRef CAS.
- D. Li, A. Yadav, H. Zhou, K. Roy, P. Thanasekaran and C. Lee, Global Chall., 2024, 8, 2300244 CrossRef.
- L. Chen and Q. Xu, Matter, 2019, 1, 57 CrossRef.
- S. Mubarak, D. Dhamodharan, P. N. Ghoderao and H.-S. Byun, Coord. Chem. Rev., 2022, 471, 214741 CrossRef CAS.
- Y. Yuan, F. Wang, H. Li, S. Su, H. Gao, X. Han and S. Ren, Process Biochem., 2022, 122, 214 CrossRef CAS.
- H. Rasouli, I. Iliuta, F. Bougie, A. Garnier and M. C. Iliuta, Sep. Purif. Technol., 2022, 287, 120505 CrossRef CAS.
- C. Molina-Fernández and P. Luis, J. CO2 Util., 2021, 47, 101475 CrossRef.
- P. Shao, J. Ye, Y. Shen, S. Zhang and J. Zhao, Gas Sci. Eng., 2024, 205237 CrossRef CAS.
- P. Schmieder, D. Denysenko, M. Grzywa, B. Baumgärtner, I. Senkovska, S. Kaskel, G. Sastre, L. van Wüllen and D. Volkmer, Dalton Trans., 2013, 42, 10786 RSC.
- C. E. Bien, K. K. Chen, S.-C. Chien, B. R. Reiner, L.-C. Lin, C. R. Wade and W. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662 CrossRef CAS PubMed.
- D. Denysenko, M. Grzywa, M. Tonigold, B. Streppel, I. Krkljus, M. Hirscher, E. Mugnaioli, U. Kolb, J. Hanss and D. Volkmer, Chem. – Eur. J., 2011, 17, 1837 CrossRef CAS PubMed.
- A. M. Wright, Z. Wu, G. Zhang, J. L. Mancuso, R. J. Comito, R. W. Day, C. H. Hendon, J. T. Miller and M. Dincă, Chem, 2018, 4, 2894 CAS.
- P. C. Sahoo, Y. N. Jang and S. W. Lee, J. Cryst. Growth, 2013, 373, 96 CrossRef CAS.
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761 CrossRef CAS.
- F.-K. Shieh, S.-C. Wang, C.-I. Yen, C.-C. Wu, S. Dutta, L.-Y. Chou, J. V. Morabito, P. Hu, M.-H. Hsu and K. C.-W. Wu, J. Am. Chem. Soc., 2015, 137, 4276 CrossRef CAS PubMed.
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408 RSC.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O’Keeffe and O. M. Yaghi, Science, 2008, 319, 939 CrossRef CAS.
- B. Wang, A. P. Côté, H. Furukawa, M. O’Keeffe and O. M. Yaghi, Nature, 2008, 453, 207 CrossRef CAS.
- S. Zhang, M. Du, P. Shao, L. Wang, J. Ye, J. Chen and J. Chen, Environ. Sci. Technol., 2018, 52, 12708 CrossRef CAS.
- J. Chen, L. Huang, Q. Wang, W. Wu, H. Zhang, Y. Fang and S. Dong, Nanoscale, 2019, 11, 5960 RSC.
- S. Sun, Z. Zhang, Y. Xiang, M. Cao and D. Yu, Langmuir, 2022, 38, 1621 CrossRef CAS.
- Q. Ying, H. Chen, P. Shao, X. Zhou, X. He, J. Ye, S. Zhang, J. Chen and L. Wang, J. CO2 Util., 2021, 49, 101565 CrossRef CAS.
- F. Zhu, H. Qiu, F. Wang, X. Zhang, G.-P. Lu, Y. Lin and H. Huang, ACS Sustainable Chem. Eng., 2023, 11, 7388 CrossRef CAS.
- Y. Xiang, D. Yu, H. Zhang, X. Wang, B. Ge and F. Huang, Colloids Surf., A, 2024, 689, 133711 CrossRef CAS.
- Z. Ji, Z. Zhang, W. Lv, Y. Shi, K. Wang, S. Ni, H. Ma and M. Cao, Colloids Surf., A, 2023, 677, 132338 CrossRef CAS.
- P. Luis, Desalination, 2016, 380, 93 CrossRef CAS.
- C. Ma, F. Pietrucci and W. Andreoni, J. Chem. Theory Comput., 2015, 11, 3189 CrossRef CAS PubMed.
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Kohler, J. C. Lewis and T. R. Ward, Chem. Rev., 2018, 118, 142 CrossRef CAS.
- H. J. Davis and T. R. Ward, ACS Cent. Sci., 2019, 5, 1120 CrossRef CAS PubMed.
- M. V. Doble, L. Obrecht, H.-J. Joosten, M. Lee, H. J. Rozeboom, E. Branigan, J. H. Naismith, D. B. Janssen, A. G. Jarvis and P. C. Kamer, ACS Catal., 2021, 11, 3620 CrossRef CAS.
- Z. Birch-Price, F. J. Hardy, T. M. Lister, A. R. Kohn and A. P. Green, Chem. Rev., 2024, 124, 8740 CrossRef CAS PubMed.
- J. C. Lewis, P. S. Coelho and F. H. Arnold, Chem. Soc. Rev., 2011, 40, 2003 RSC.
- A. Fernández-Gacio, A. Codina, J. Fastrez, O. Riant and P. Soumillion, ChemBioChem, 2006, 7, 1013 CrossRef.
- F. W. Monnard, T. Heinisch, E. S. Nogueira, T. Schirmer and T. R. Ward, Chem. Commun., 2011, 47, 8238 RSC.
- F. W. Monnard, E. S. Nogueira, T. Heinisch, T. Schirmer and T. R. Ward, Chem. Sci., 2013, 4, 3269 RSC.
- K. Okrasa and R. J. Kazlauskas, Chem. – Eur. J., 2006, 12, 1587 CrossRef CAS PubMed.
- Q. Jing and R. J. Kazlauskas, ChemCatChem, 2010, 2, 953 CrossRef CAS.
- Q. Jing, K. Okrasa and R. J. Kazlauskas, Chem. – Eur. J., 2009, 15, 1370 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |