
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
N-Heterocycle-coordinated λ5-iodanes as IBX alternatives for alcohol oxidations†
Nikita S.
Antonkin
a,
Yulia A.
Vlasenko
a,
Pim
Puylaert
 b,
Boris J.
Nachtsheim
*b and
Pavel S.
Postnikov
*a
b,
Boris J.
Nachtsheim
*b and
Pavel S.
Postnikov
*a
aResearch School of Chemistry and Applied Biomedical Sciences, Tomsk Polytechnic University, Tomsk 634050, Russian Federation. E-mail: postnikov@tpu.ru
bInstitut für Organische und Analytische Chemie, Universität Bremen, Leobener Straße NW2C, 28359 Bremen, Germany. E-mail: nachtsheim@uni-bremen.de
First published on 9th December 2024
Abstract
We report the synthesis of novel N-coordinated λ5-iodanes as a unique class of hypervalent iodine compounds. X-ray diffraction analysis revealed their intriguing (pseudo)cyclic structure, showcasing distinctive N⋯I-secondary bonding interactions. We demonstrate the in situ generation of reactive diacetoxy derivatives, which exhibits remarkable efficacy in alcohol oxidation reactions. Thermal stability assessments using TGA/DSC analysis provide crucial insights into the handling and potential applications of these compounds. This work expands the frontier of hypervalent iodine chemistry, offering new tools for selective oxidation reactions.
λ 5-Iodanes are widely applied in selective oxidations of alcohols,1,2 amines3 and phenols.4 The pentavalent iodine derivatives can be considered as the close-to-perfect metal-free oxidants possessing a high potential for selective dehydrogenations of carbonyl compounds5 or N-heterocycles,6 the oxidative cleavage of unsaturated bonds,7 and many more.8,9
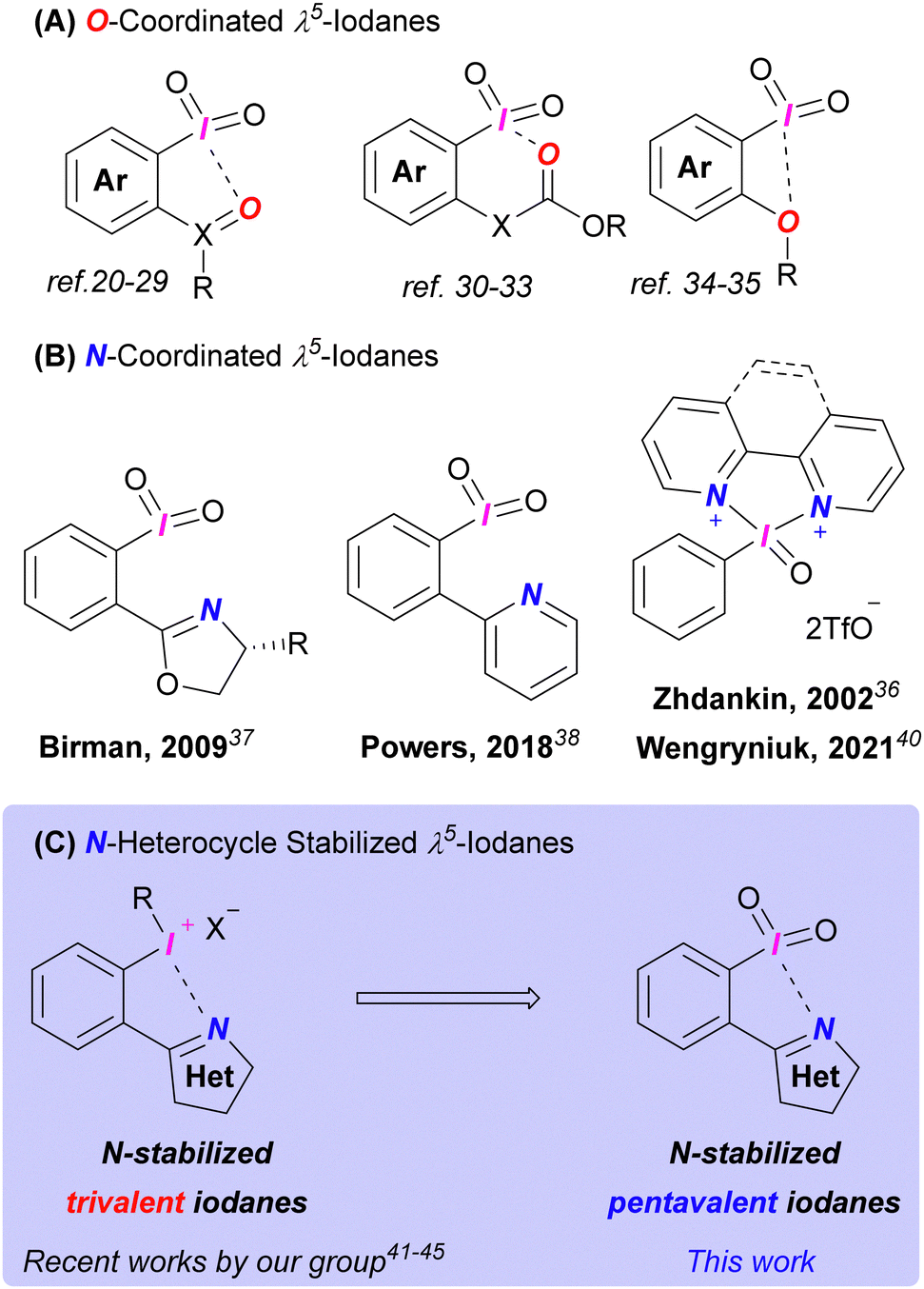
The majority of λ5-iodanes are O-tethered species.8 2-Iodoxybenzoic acid (IBX) and Dess–Martin periodinane (DMP) are the most widespread derivatives among them due to their established preparation methods and predictable selectivity. Despite its widespread use, IBX's limited solubility and low ambient reactivity remain challenging. The development of new λ5-iodanes remains crucial, as even DMP, a more soluble and active IBX analogue, suffers from moisture sensitivity.10 Different approaches were proposed to overcome these downsides. The functionalization of both C-ligands10–15 and O-ligands16–18 was used to improve reactivity and solubility of the iodanes. Another route to advanced hypervalent iodine reagents is the (pseudo)cyclic derivatives exhibiting secondary bonding interactions (e.g., halogen bonding) which partially break problematic polymeric structures and hence increase solubility.19 Many examples of (pseudo)cyclic λ5-iodanes have been introduced over the past years and their majority utilizes an oxygen atom as an electron donor for these secondary interactions (Fig. 1A).20–35
 | ||
| Fig. 1 Background and the concept of this work. | ||
To the best of our knowledge there are few examples of N-tethered (pseudo)cyclic λ5-iodanes in the literature (Fig. 1B).36–40 Derivatives introduced by Birman37 and Powers38 showed poor reactivity. A bipyridyl derivative established by Zhdankin in 2002 was not applied so far in synthetic applications,36 until a series of similar pentavalent iodanes were presented by Wengryniuk's lab.39,40 These derivatives possessed unique reactivity compared to O-ligated iodanes (e.g., oxidation of electron-deficient phenols)39 alongside with common oxidative properties toward alcohols and sulfides.40 However, these compounds are not “bench-stable” and there is no experimental evidence of N⋯I coordination in any of the abovementioned iodanes. Based on our previous work41–45 and Wengryniuk's recent findings,39,40 we herein developed novel N-heterocycle-coordinated (pseudo)cyclic λ5-iodanes and report their structure, reactivity in alcohol oxidations, and thermal stability.
Initially, we synthesized λ5-iodanes 2a and 2b using Oxone® in MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (Scheme 1). Iodane 2a was obtained in 5 h with full conversion, with a yield of 72–81% across multiple preparations. A gram-scale synthesis of 2a yielded 75% (1.02 g). For iodide 1b, incomplete conversion resulted in 50% yield of 2b. Extending the reaction to 10 h improved the yield to 64% but further extension to 15 h reduced it to 48% due to decomposition. X-Ray analysis confirmed the structures of iodanes 2a–b, revealing N⋯I secondary interactions with contact lengths of 2.649 Å and 2.662 Å, respectively (ESI,† Section S7). These distances are comparable to those in five-membered (pseudo)cyclic O-tethered iodanes (2.40–3.04 Å), confirming (pseudo)cyclic nature of 2a–b.19 This work presents the first confirmed (pseudo)cyclic λ5-iodanes with intramolecular N⋯I interaction. Unlike the previously reported intermolecular example that forms less soluble polymeric structures,35 our (pseudo)cyclic iodanes 2a–b show improved solubility in water, organic solvents, and weak acids, likely due to the basic tertiary nitrogen in the heterocycles.
:H2O (Scheme 1). Iodane 2a was obtained in 5 h with full conversion, with a yield of 72–81% across multiple preparations. A gram-scale synthesis of 2a yielded 75% (1.02 g). For iodide 1b, incomplete conversion resulted in 50% yield of 2b. Extending the reaction to 10 h improved the yield to 64% but further extension to 15 h reduced it to 48% due to decomposition. X-Ray analysis confirmed the structures of iodanes 2a–b, revealing N⋯I secondary interactions with contact lengths of 2.649 Å and 2.662 Å, respectively (ESI,† Section S7). These distances are comparable to those in five-membered (pseudo)cyclic O-tethered iodanes (2.40–3.04 Å), confirming (pseudo)cyclic nature of 2a–b.19 This work presents the first confirmed (pseudo)cyclic λ5-iodanes with intramolecular N⋯I interaction. Unlike the previously reported intermolecular example that forms less soluble polymeric structures,35 our (pseudo)cyclic iodanes 2a–b show improved solubility in water, organic solvents, and weak acids, likely due to the basic tertiary nitrogen in the heterocycles.
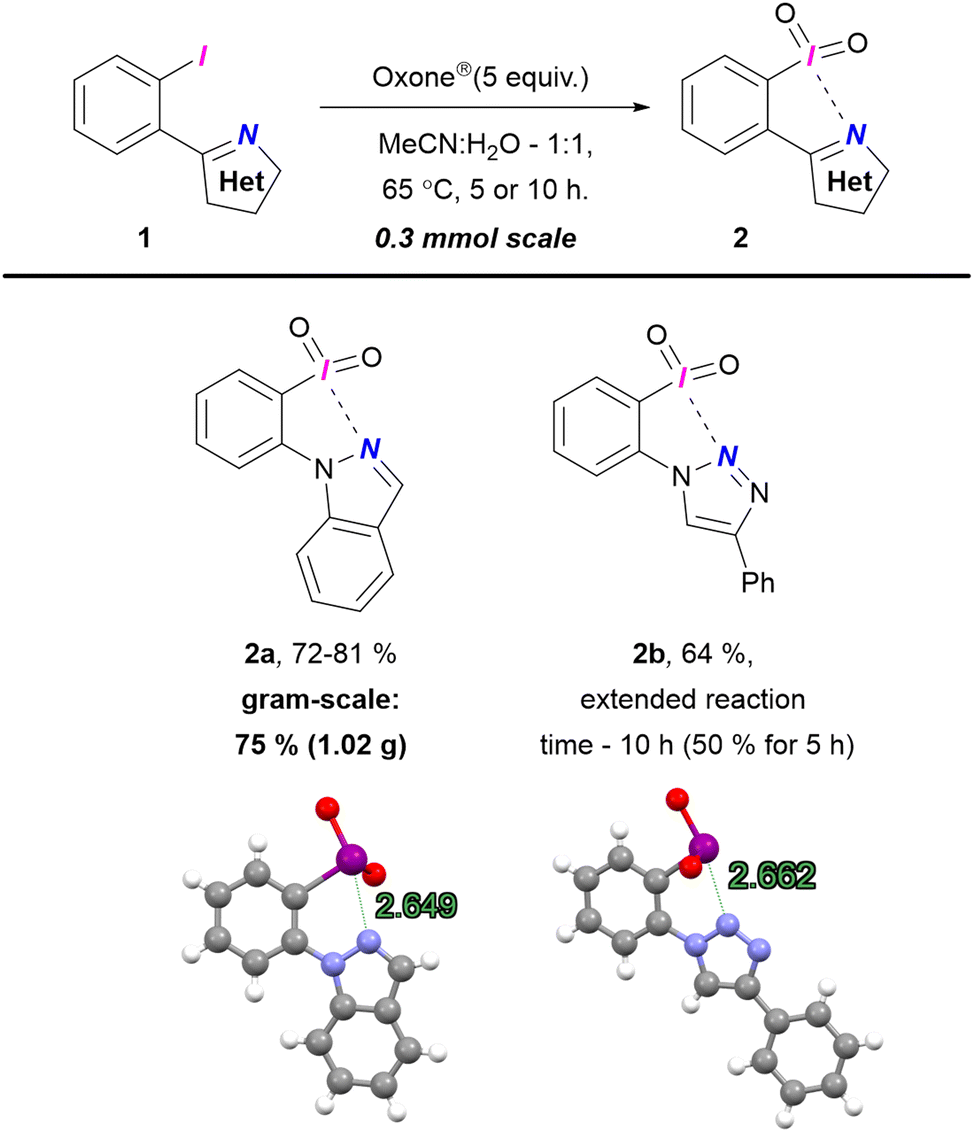 | ||
| Scheme 1 Preparation of λ5-iodanes 2a–b. | ||
TGA/DSC analysis revealed the thermal stability of iodanes 2a–b compared to IBX (ESI,† Section S6). Triazole derivative 2b showed a decomposition energy of 823.2 J g−1 at 218.5 °C, exploding violently. Derivative 2a decomposed at 202.6 °C with 396.3 J g−1, lower than IBX (506.8 J g−1 at 220.1 °C), suggesting 2a is safer regarding explosive properties. Both new derivatives meet the “100 K rule” for thermal stability, indicating safe use under ambient conditions.46 It is also worth mentioning, that no explosions of both 2a and 2b were experienced under severe impact with a metal hammer. However, for 2a–b as well as for IBX we observed explosion under rapid heating (e.g., with a heat gun). Therefore, these compounds should be operated with precautions.
We investigated the oxidation of 1-phenylethanol as a model reaction to evaluate iodanes 2a–b (ESI,† Section S4 and Table S1 and Note S1 for details on optimization and preparation of carbonyl compounds 3). Iodane 2a demonstrated superior reactivity compared to 2b. Then various additives were explored, particularly anhydrides, which could potentially form more reactive carboxy-ligated iodane species.10 Using a three-fold excess of Ac2O relative to 2a in CDCl3 provided 3i in quantitative yield (Scheme 2), while lower anhydride ratios reduced yields. Although TFAA proved to be highly active for the oxidation, rapid alcohol acylation diminished the yield of ketone 3i. A control experiment using twice the amount of AcOH compared to Ac2O yielded only traces of 3i, indicating the in situ formation of (diacetoxy)iodylbenzene 2a′ from 2a and Ac2O rather than acid-catalyzed activation.
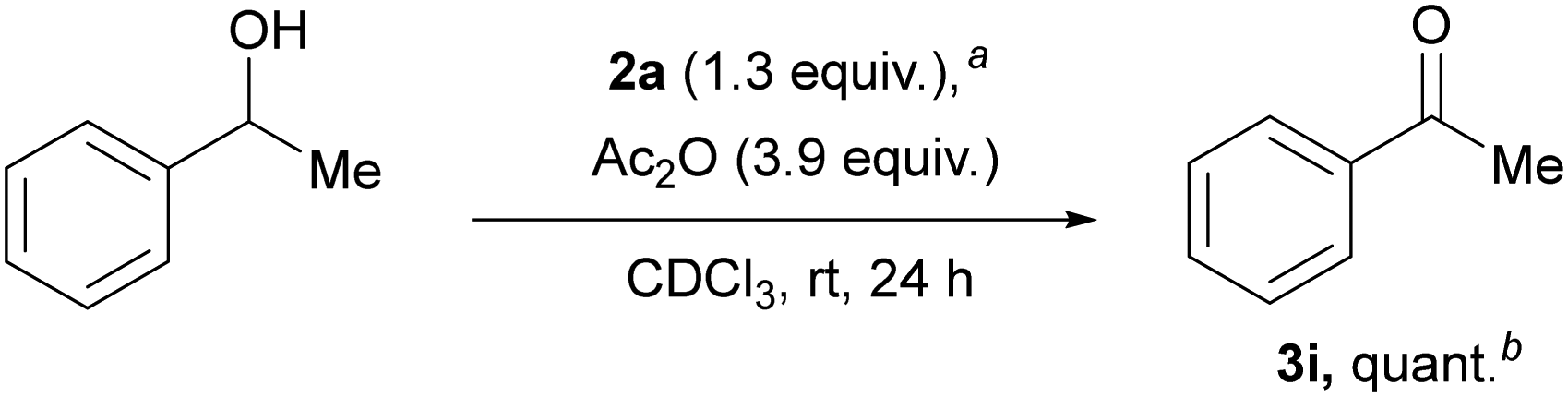 | ||
| Scheme 2 Optimized conditions of the alcohol oxidation with iodane 2a.aReaction conditions: 0.1 mmol of 1-phenylethanol (0.1 mmol, 12 mg), 2a (0.13 mmol, 46 mg), Ac2O (0.39 mmol, 36.6 μL), CDCl3 (2 mL). bYield was determined by NMR with 1,2-dibromoethane as an internal standard. | ||
We successfully generated diacetate 2a′ from treating iodane 2a with Ac2O (Scheme 3). 1H NMR confirmed full conversion of 2a. Despite high moisture sensitivity preventing isolation, we confirmed the structure of 2a′ by X-Ray diffraction, revealing a (pseudo)cyclic structure with an N⋯I contact distance of 2.655 Å (ESI,† Section S7).
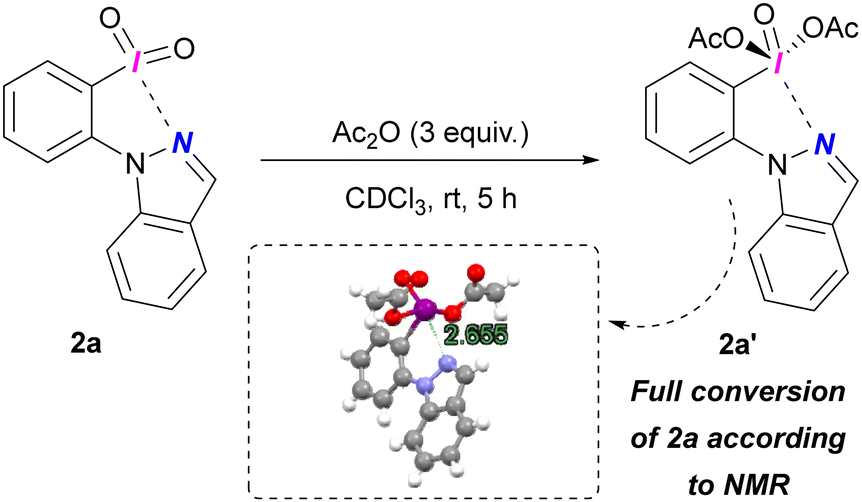 | ||
| Scheme 3 Preparation of diacetate 2a'. | ||
A reactivity comparison of iodanes 2a–b with IBX under optimal conditions (Fig. 2) (conditions from Scheme 2) revealed that 2a is superior to IBX in oxidizing 1-phenylethanol to acetophenone 3i. This enhanced performance is likely due to the easier formation of acetoxy derivative 2a′ compared to acetoxylated IBX derivatives, which require substantial heating, p-TsOH catalysis, and Ac2O as a solvent.47 When conducted open to atmosphere, the reaction yielded 96% of 3i, compared to quantitative yield in a closed vessel (ESI,† Section S5, Note S2). This suggests the discrepancy between quantitative yield during optimization and 86% yield in comparison experiments stems from unreacted iodane 2a present in samples during the initial hours. It is also noteworthy that the iodane 2a′ is more active than acyclic (diacetoxy)iodylbenzene since the latter one gave no oxidation product after 24 hours.40 We linked that fact with the increased solubility of (pseudo)cyclic derivative 2a′. Furthermore, the iodane 2a′ in contrast to diacetate of IBX ester introduced by Zhdankin do not require activation by TFA.26
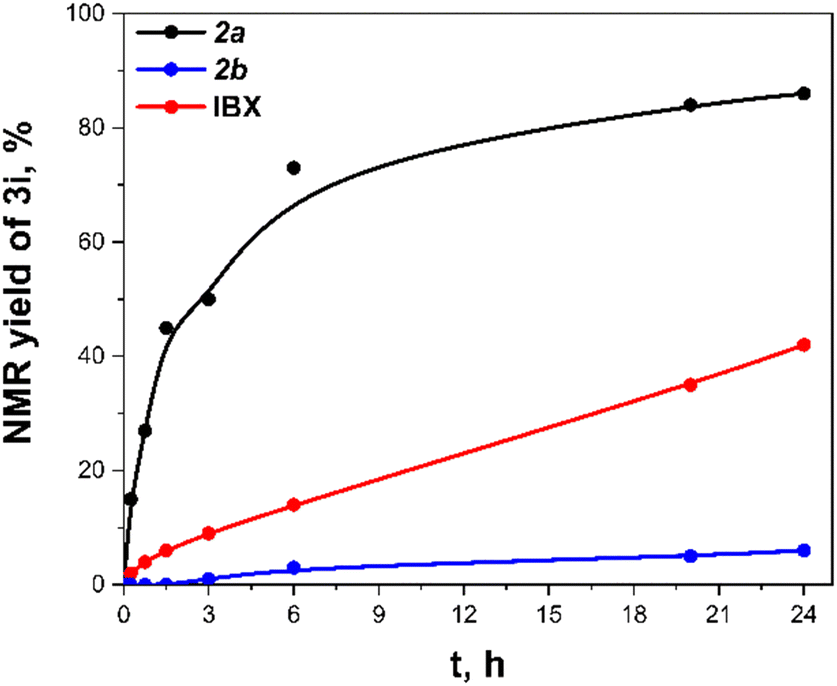 | ||
| Fig. 2 Reactivity comparison between 2a–b and IBX. | ||
Finally, under our optimal conditions, we investigated the oxidation of a variety of alcohols using 2a (Scheme 4). The oxidation generally yielded carbonyl compounds 3 in good to excellent yields. The observed moderate yields for 3k and 3n were due to isolation challenges, while lower yields of aldehydes 3d, 3e, and 3g were primarily attributed to product volatility rather than incomplete conversion or overoxidation. For alcohols containing basic nitrogen atoms (aldehydes 3o–p), we used TFA instead of Ac2O to avoid acylation side products. This approach gave 57% yield for 3o and 78% for 3p. Primary alcohols were oxidized to aldehydes 3q and 3r in 40% and 31% yields, respectively, though complete conversion required more than 24 h. Beyond alcohol oxidation, iodane 2a effectively converted thioanisole to sulfoxide 4 in 75% yield (Scheme 5). Attempts to synthesize α,β-unsaturated ketones from 4-methylcyclohexanone were unsuccessful under various conditions (ESI,† Section S5).
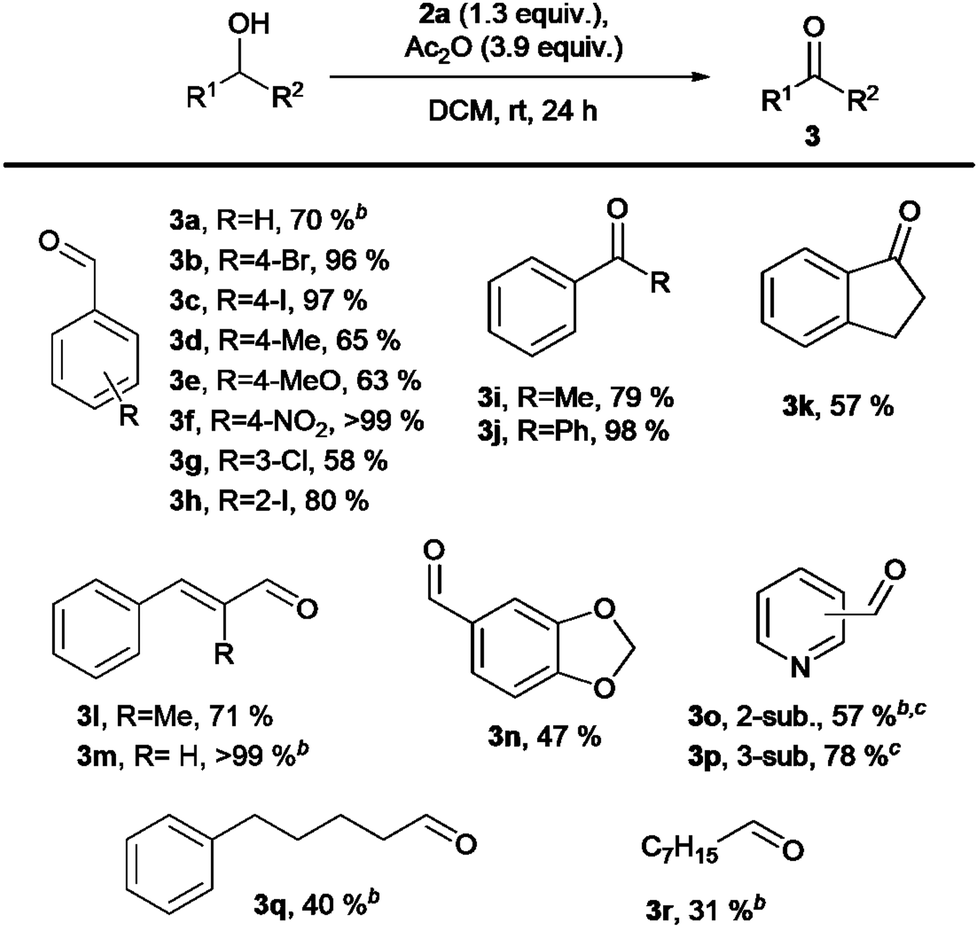 | ||
| Scheme 4 Reaction scope. aIsolated yields unless otherwise stated. Reaction conditions: 0,1 mmol of an alcohol, 0.13 mmol of λ5-iodane, λ5-iodane was premixed with Ac2O (0.39 mmol, 37 μL) in 1 mL of the solvent for 30 minutes and alcohol was added afterwards as the solution (1 mL, 2 mL for a reaction in total). bYields were determined by NMR with 1,2-dibromoethane as an internal standard. cTFA (0.39 mmol, 30 μL) instead of Ac2O. | ||
 | ||
| Scheme 5 Oxidation of thioanisole. aIsolated yield. | ||
In conclusion, we developed novel N-heterocycle coordinated (pseudo)cyclic λ5-iodanes, providing the first experimental evidence of intramolecular N-coordination in pentavalent iodine derivatives via X-Ray data. Iodanes 2a–b show unique solubility and form reactive species like diacetate 2a′. The in situ generated 2a′ oxidizes alcohols to carbonyl compounds 3 in 47% to quantitative yields, outperforming IBX under similar conditions. Thermal stability analysis suggests 2a as a potentially safer, less explosive alternative to IBX.
This work was supported by the Russian Science Foundation (No. 23-73-10091). NSA is personally thankful to the DAAD Research Grants – Short-Term Grants program.
Data availability
The data supporting this article have been included as part of the ESI† (NMR spectra). Crystallographic data for 2a, 2a′ and 2b compounds has been deposited at the CCDC under 2364396, 2364397, 2364398 correspondingly.Conflicts of interest
There are no conflicts to declare.Notes and references
- H. Tohma and Y. Kita, Adv. Synth. Catal., 2004, 346, 111–124 CrossRef CAS.
- M. Uyanik and K. Ishihara, Chem. Commun., 2009, 2086–2099 RSC.
- K. C. Nicolaou, C. J. Mathison and T. J. Montagnon, J. Am. Chem. Soc., 2004, 126, 5192–5201 CrossRef CAS.
- S. Quideau, L. Pouységu, P. A. Peixoto and D. Deffieux, in Hypervalent Iodine Chemistry. Topics in Current Chemistry, ed. T. Wirth, Springer, Cham, 1st edn, 2016, 373, 25–74 Search PubMed.
- K. C. Nicolaou, T. Montagnon and P. S. Baran, Angew. Chem., Int. Ed., 2002, 41, 993–996 CrossRef CAS PubMed.
- S. Hati, U. Holzgrabe and S. Sen, Beilstein J. Org. Chem., 2017, 13, 1670–1692 CrossRef CAS PubMed.
- J. N. Moorthy and K. N. Parida, J. Org. Chem., 2014, 79, 11431–11439 CrossRef CAS PubMed.
- V. V. Zhdankin, J. Org. Chem., 2011, 76, 1185–1197 CrossRef CAS.
- Y. Venkata Durga Nageswar, K. Ramesh and K. Rakhi, Front. Chem., 2022, 10, 841751 CrossRef.
- D. B. Dess and J. C. Martin, J. Am. Chem. Soc., 1991, 113, 7277–7287 Search PubMed.
- S. H. Stickley and J. C. Martin, Tetrahedron Lett., 1995, 36, 9117–9120 CrossRef CAS.
- A. P. Thottumkara and T. K. Vinod, Tetrahedron Lett., 2002, 43, 569–572 CrossRef CAS.
- A. Kommreddy, M. S. Bowsher, M. R. Gunna, K. Botha and T. K. Vinod, Tetrahedron Lett., 2008, 49, 4378–4382 CrossRef CAS.
- R. D. Richardson, J. M. Zayed, S. Altermann, D. Smith and T. Wirth, Angew. Chem., Int. Ed., 2007, 46, 6529–6532 CrossRef CAS.
- J. N. Moorthy, N. Singhal and K. Senapati, Tetrahedron Lett., 2008, 49, 80–84 CrossRef CAS.
- A. Y. Koposov, D. N. Litvinov, V. V. Zhdankin, M. J. Ferguson, R. McDonald and R. R. Tykwinski, Eur. J. Org. Chem., 2006, 4791–4795 CrossRef CAS.
- M. S. Yusubov, P. S. Postnikov, R. Y. Yusubova, A. Yoshimura, G. Jürjens, A. Kirschning and V. V. Zhdankin, Adv. Synth. Catal., 2017, 359, 3207–3216 CrossRef CAS.
- M. S. Yusubov, N. S. Soldatova, P. S. Postnikov, R. R. Valiev, A. Yoshimura, T. Wirth and V. V. Zhdankin, Chem. Commun., 2019, 55, 7760–7763 RSC.
- A. Yoshimura, M. S. Yusubov and V. V. Zhdankin, Org. Biomol. Chem., 2016, 14, 4771–4781 RSC.
- D. Macikenas, E. Skrzypczak-Jankun and J. D. Protasiewicz, Angew. Chem., Int. Ed., 2000, 39, 2007–2010 CrossRef CAS.
- V. V. Zhdankin, A. Y. Koposov, B. C. Netzel, N. V. Yashin, P. Brian, M. J. Rempel, R. R. Ferguson and Tykwinski, Angew. Chem., Int. Ed., 2003, 42, 2194–2196 CrossRef CAS.
- V. A. Nikiforov, V. S. Karavan, S. A. Miltsov, S. I. Selivanov, E. Kolehmainen, E. Wegelius and M. Nissinen, ARKIVOC, 2003, 6, 191–200 Search PubMed.
- V. V. Zhdankin, D. N. Litvinov, A. Y. Koposov, T. Luu, M. J. Ferguson, R. McDonald and R. R. Tykwinski, Chem. Commun., 2004, 106–107 RSC.
- A. Y. Koposov, V. N. Nemykin and V. V. Zhdankin, New J. Chem., 2005, 29, 998–1000 RSC.
- B. V. Meprathu, M. W. Justik and J. D. Protasiewicz, Tetrahedron Lett., 2005, 46, 5187–5190 CrossRef CAS.
- V. V. Zhdankin, A. Y. Koposov, D. N. Litvinov, M. J. Ferguson, R. McDonald, T. Luu and R. R. Tykwinski, J. Org. Chem., 2005, 70, 6484–6491 CrossRef CAS.
- B. V. Meprathu and J. D. Protasiewicz, Tetrahedron, 2010, 66, 5768–5774 CrossRef CAS.
- A. Bredenkamp, F. Mohr and S. F. Kirsch, Synthesis, 2015, 1937–1943 CAS.
- H.-J. Shen, Y.-N. Duan, K. Zheng and C. Zhang, J. Org. Chem., 2019, 84, 14381–14393 CrossRef CAS PubMed.
- U. Ladziata, A. Y. Koposov, K. Y. Lo, J. Willging, V. N. Nemykin and V. V. Zhdankin, Angew. Chem., Int. Ed., 2005, 44, 7127–7131 CrossRef CAS PubMed.
- A. K. Mailyan, I. M. Geraskin, V. N. Nemykin and V. V. Zhdankin, J. Org. Chem., 2009, 74, 8444–8447 CrossRef CAS PubMed.
- S. M. Altermann, S. Schäfer and T. Wirth, Tetrahedron, 2010, 66, 5902–5907 CrossRef CAS.
- J. N. Moorthy, K. Senapati and K. N. Parida, J. Org. Chem., 2010, 75, 8416–8421 CrossRef CAS.
- A. Y. Koposov, R. R. Karimov, I. M. Geraskin, V. N. Nemykin and V. V. Zhdankin, J. Org. Chem., 2006, 71, 8452–8458 CrossRef CAS.
- A. Yoshimura, C. T. Banek, M. S. Yusubov, V. N. Nemykin and V. V. Zhdankin, J. Org. Chem., 2011, 76, 3812–3819 CrossRef CAS.
- V. V. Zhdankin, A. Y. Koposov and N. V. Yashin, Tetrahedron Lett., 2002, 43, 5735–5737 CrossRef CAS.
- J. K. Boppisetti and V. B. Birman, Org. Lett., 2009, 11, 1221–1223 CrossRef CAS.
- A. Maity, S.-M. Hyun, A. K. Wortman and D. C. Powers, Angew. Chem., Int. Ed., 2018, 57, 7205–7209 CrossRef CAS.
- X. Xiao, N. S. Greenwood and S. E. Wengryniuk, Angew. Chem., Int. Ed., 2019, 131, 16327 CrossRef.
- X. Xiao, J. M. Roth, N. S. Greenwood, M. K. Velopolcek, J. Aguirre, M. Jalali, A. Ariafard and S. E. Wengryniuk, J. Org. Chem., 2021, 86, 6566–6576 CrossRef CAS PubMed.
- Y. A. Vlasenko, P. S. Postnikov, M. E. Trusova, A. Shafir, V. V. Zhdankin, A. Yoshimura and M. S. Yusubov, J. Org. Chem., 2018, 83, 12056–12070 CrossRef CAS PubMed.
- A. Boelke, E. Lork and B. J. Nachtsheim, Chem. – Eur. J., 2018, 24, 18653–18657 CrossRef CAS PubMed.
- A. Boelke, Y. A. Vlasenko, M. S. Yusubov, B. J. Nachtsheim and P. S. Postnikov, Beilstein J. Org. Chem., 2019, 15, 2311–2318 CrossRef CAS PubMed.
- Y. A. Vlasenko, T. J. Kuczmera, N. S. Antonkin, R. R. Valiev, P. S. Postnikov and B. J. Nachtsheim, Adv. Synth. Catal., 2023, 365, 535–543 CrossRef CAS.
- T. J. Kuczmera, P. Puylaert and B. J. Nachtsheim, Beilstein J. Org. Chem., 2024, 20, 1677–1683 CrossRef CAS.
- Recommendations from Committee of Experts on the Transport of Dangerous Goods and on the Globally Harmonized System of Classification and Labelling of Chemicals, https://unece.org/DAM/trans/doc/2018/dgac10c3/ST-SG-AC.10-C.3-2018-78e.pdf (accessed 22/08/2024).
- R. E. Ireland and L. Liu, J. Org. Chem., 1993, 58, 2899 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2364396–2364398. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc05058d |
| This journal is © The Royal Society of Chemistry 2025 |