
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Vitrimeric electrolytes – overview and perspectives
Zviadi
Katcharava†
 ab,
Anja
Marinow†
*b and
Wolfgang H.
Binder
*ab
ab,
Anja
Marinow†
*b and
Wolfgang H.
Binder
*ab
aDesign of 3D-Printable Polymers Based on Regional Resources, Just Transition Center, Martin Luther University Halle-Wittenberg, 06099 Halle, Germany
bMacromolecular Chemistry, Division of Technical and Macromolecular Chemistry, Faculty of Natural Sciences II (Chemistry, Physics, Mathematics), Institute of Chemistry, Martin Luther University Halle-Wittenberg, von-Danckelmann-Platz 4, D-06120 Halle, Germany. E-mail: wolfgang.binder@chemie.uni-halle.de; anja.marinow@chemie.uni-halle.de
First published on 17th January 2025
Abstract
Lithium batteries, essential for consumer electronics, transportation and the energy sector, still require further improvement in performance, safety, and sustainability. Traditonal organic solvent-based electrolytes, widely used in current systems, pose significant safety risks and restrict the development of next generation devices. Vitrimers are materials with unique physical and chemical properties, which offer a promising alternative to overcome these limitations, finally reaching processability and recyclability of solid electrolytes. Despite their potential a comprehensive overview of vitrimeric electrolytes’ design and application in lithium batteries is lacking. This review article summarizes the key concepts, design principles, and notable advancements in vitrimeric electrolytes. We will also discuss the challenges still restricting the widespread adoption of vitrimeric electrolytes and explore future perspectives for leveraging vitrimeric materials in high-performance, safer, and more sustainable lithium battery technologies.
Introduction
Modern society heavily relies on batteries due to their wide range of applications, from consumer electronics to electric vehicles and stationary energy storage devices.1,2 The urgent need to reduce fossil fuel consumption and shift towards renewable energy sources is driving a growing demand for improved battery technologies, which are essential for storing captured green energy before it is delivered to consumers. The most widely used type of rechargeable battery is the Li-ion battery which is schematically depicted in Fig. 1.3,4 Two electrodes are electrically isolated by a porous polyolefin separator, while the liquid electrolyte serves as the medium for transporting lithium ions during charging and discharging cycles. The rising demand for batteries requires several key shortcomings to be addressed in current technologies.5,6 The most pressing concern is safety,7 which poses significant danger to consumers and is associated with the use of flammable organic solvents in electrolytes. Thermal runaway, the primary cause of battery malfunction, can result in fire hazards due to irreversible exothermic chain of reactions.8 This can be triggered not only by electrical, mechanical, or thermal abuse of the device, but also by the formation/growth of dendrites.9 These needle-like structures of metallic lithium can randomly grow on the anode, pierce the separator over multiple cycles and lead to short circuit. Another significant challenge facing next-generation batteries, especially containing silicon-based anodes, is the drastic volumetric changes of the particles during charging/discharging cycles.10,11 This mechanical stress can lead to cracks, pulverization of the active material, and a shortened cycle life. The solid electrolyte interface (SEI), a protective layer between the electrolyte and anode, plays a crucial role in battery performance.12,13 Its degradation can significantly reduce battery functionality. To achieve long-term operation, stabilizing the SEI is essential. Ideally, batteries should have a stable interface that can withstand repeated cycling without compromising performance. As another important aspect we need to consider the responsible management of resources and the transition to a circular economy.6,14 Materials used in batteries should preferably be reprocessable and easier to recover.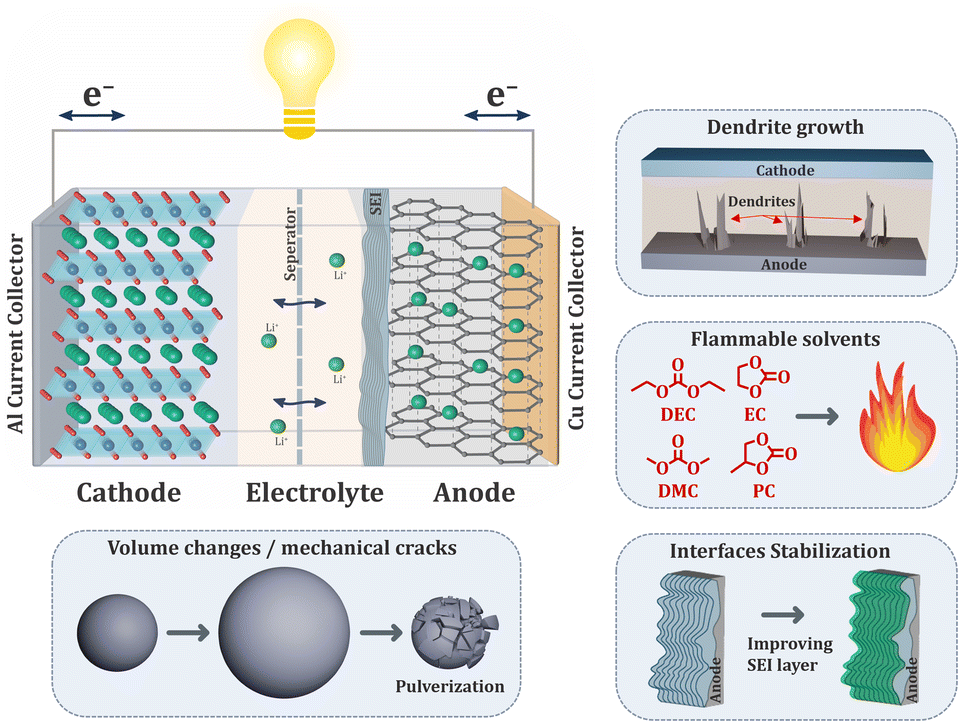 | ||
| Fig. 1 Schematic illustration of Li-ion battery working principle. Highlighted dendrite growth, flammable solvents used in electrolytes, volumetric changes leading to the pulverization and interface stabilization. | ||
One key strategy for enhancing Li-ion batteries is the replacement of flammable electrolytes with safer, more stable alternatives, while simultaneously addressing sustainability concerns.15,16 Polymer-based electrolytes (PEs), known for their inherent non-flammability, non-volatility, and enhanced mechanical properties, offer a promising solution. Not only can they overcome safety concerns, but they also enable the development of more flexible and stress-resistant devices. Polymeric materials enable the possibility to embed smart functionalities such as self-healing and reprocessability, which can be achieved by introducing dynamic interactions in the materials.17–19 Such PEs can extend battery lifespan and improve recyclability. Various comprehensive reviews described self-healable PEs in batteries and their potential benefits.17,20,21 In this highlight we focus on dynamic covalent interactions in PEs, which can undergo bond exchange reaction via an associative mechanism. These materials, known as vitrimers, were introduced following the seminal work of Leibler et al.22 The basic principle of vitrimeric exchange is depicted in Fig. 2: upon thermal activation a bond forms an associative state, an intermediate step before the bond exchange occurs.23,24 Below a certain temperature, known as the topology freezing temperature or vitrimeric temperature (Tv), the polymeric material's structure is considered frozen. Due to these unique properties, vitrimers are standing between thermosetting and thermoplastic materials. At the service temperatures, they exhibit enhanced mechanical properties and three-dimensional stability like thermosets. However, they are malleable and reprocessable at higher temperatures similar to thermoplastics. The application of the vitrimeric principle as reprocessable/recyclable thermosets thus holds great promise and opens the door to a range of other applications.25–27 Their incorporation into battery technologies therefore has emerged as a potential solution for safer, mechanically enhanced, recyclable, and thermally stable polymeric electrolytes for Li-ion batteries. Critical in this endeavour however is the compatibility of the known, often well-established vitrimeric exchange chemistry inside ion-conductive materials (with the inherent changes in e.g. vitrimeric temperatures in comparison to well established materials), requiring electrochemical stability, the quest to meet the necessary ion-transporting qualities, and finally allow the recyclability of the electrolytes via the dynamic bond exchange. It is herein that research is still in its infancy, although quite a significant number of materials are already known. This review will sort, classify and finally also report on the performance of such vitrimeric electrolytes in the context of their use as alternative electrolytes to existing, well established, but largely nonrecyclable alternatives. We will highlight the progress and advancement made in this area, which often relies on dynamic bonds illustrated in Fig. 2, either in combination or separately. Additionally, we will cover the use of vitrimeric binders for silicon-based anodes, compensating the volumetric changes and potentially enhancing battery lifecycles.
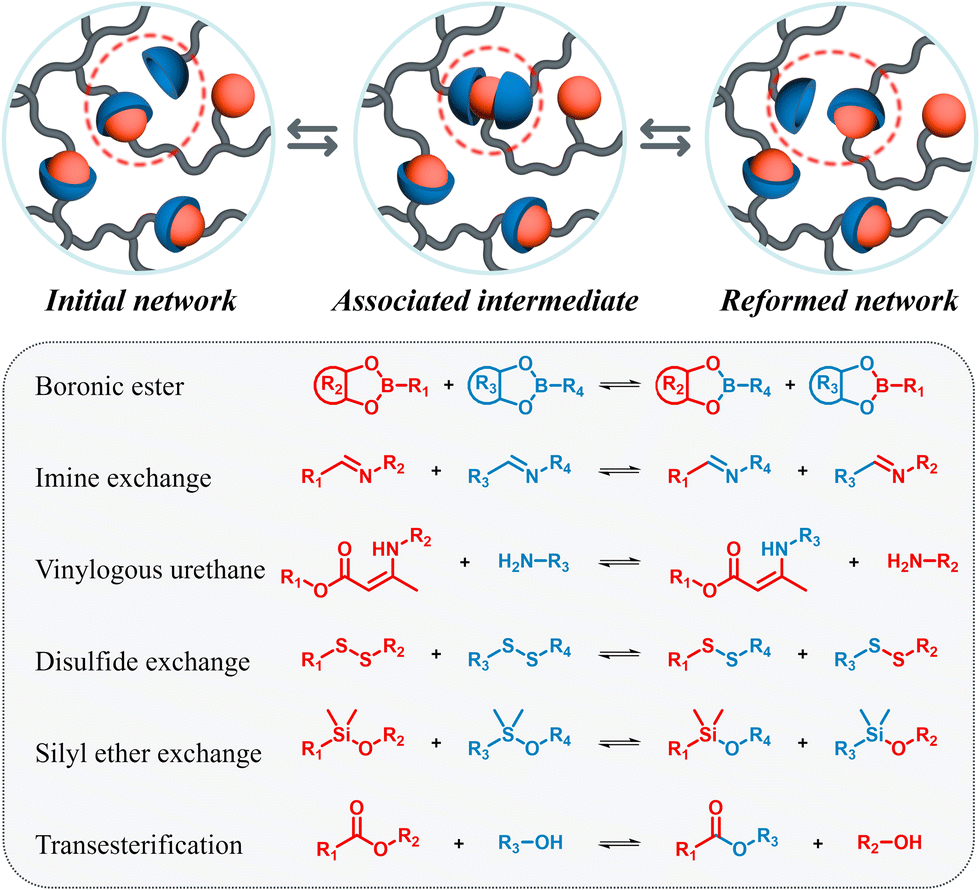 | ||
| Fig. 2 Schematic illustration of associative bond exchange in vitrimeric materials and common types of dynamic covalent bonds. | ||
Vitrimeric electrolytes
Vitrimers with boronic ester bonds
Knowing the potential of the poly(ethylene oxide) for application as PE, Evens and co-workers28 pioneered the use of terminal hydroxyl-groups of linear PEO as the dynamic binding sites for boric acid, thus generating catalyst-free dynamic boronic ester networks within the PEO/LITFSI polymer electrolyte (Fig. 3A). Rheological studies revealed a strong correlation between salt content and the dynamic of the covalent network. Higher salt concentrations accelerated dynamic exchange, indicated by a significant increase of stress relaxation speed (up to 2 orders of magnitude). Additionally, the formation of the B-TFSI intermediates improved the flow behaviour of the networks. By optimizing the LiTFSI content, they developed polymer electrolytes with moderate ionic conductivity (up to 3.5 × 10−4 S cm−1 at 90 °C), while exhibiting shear modulus in the range of 1 to 10 MPa, depending on salt content. The dynamic nature of the boronic ester network enabled self-healing at 60 °C, with 98% recovery achieved within 34 h without external pressure, or within just 10 min under a force of 19.6 N. Moreover, the network was fully reversible, dissolving in water to regenerate the original monomers, highlighting its potential as a sustainable electrolyte (Fig. 3A). Robust, self-standing solid polymer electrolyte (SPE) could be obtained via the photopolymerization of a macromolecular crosslinker (diGMPA) with boronic ester bonds and poly(ethylene glycol)diacrylate (PEGDA) (Fig. 3B).29 The incorporation of cyclic borates aimed to enhance the network's salt tolerance compared to boric acid, while the transesterification of boronic esters within the polymer electrolyte facilitated topology rearrangement. This process increased the mobility of diGMPA chains, allowing for more efficient lithium-ion conduction, in turn enhancing the overall ion transport within the system (Fig. 3B). These SPEs exhibited self-heling within 30 min at 60 °C without external pressure, in turn re-establishing B–O covalent bonds between the interfaces of the damaged surfaces and restoring the mechanical integrity of the SPE film. Although only moderate ion conductivities could be reached (around 10−5 S cm−1 at 30 °C), it was demonstrated that the transesterification of boronic ester groups could promote the ion conduction through accelerating the mobility of Li ions in the polymer matrix. Healed samples demonstrated minimal alteration in ionic conductivity compared to the original electrolyte, indicating that the ion channels could fully recover to their initial state. To improve the ionic conductivity of vitrimeric SPEs, we combined a thermally and electrochemically stable pyrrolidinium-based ionic liquid (IL) monomer with a styrene-based monomer featuring a terminal hydroxyl (OH) group, designed to serve as a dynamic crosslinking site (Fig. 3C).30 The resulting polymeric ionic liquids (PILs) were crosslinked with boric acid, producing catalyst-free boric ester vitrimeric SPEs. By adjusting the monomer ratio and crosslinking density, we were able to balance the trade-off between mechanical strength and ion conductivity. As anticipated, added salt increased ionic conductivity, while the temperature dependency of conductivity follows Vogel–Fulcher–Tammann behaviour, indicating a combination of long-range segmental motions and ion hopping mechanisms for charge transport. Under optimized conditions, we obtained stable films with conductivity up to 10−4 S cm−1 at 80 °C and E-modulus of 0.24 MPa. Furthermore, the addition of salt led to an increase in both the topology freezing temperature (Tv) and the corresponding activation energy required for dynamic bond exchange (from Tv = −71 °C, Ea = 61.7 kJ mol−1 for a pure vitrimer, to Tv = −64 °C, Ea = 70.9 kJ mol−1 for vitrimer with LiTFSI/EO = 0.1, respectively). This suggests that salt plays an important role in modulating the vitrimeric network's dynamics, enhancing thermal stability and influencing the overall reprocessing capabilities of the material. 3D printing via fused deposition modelling (FDM) was explored as a potential method for reprocessing vitrimeric PILs. The increased bond exchange rate at elevated temperatures reduced viscosity, improving flow and printability. Fig. 3C illustrates a successfully printed star-shaped structure with good adhesion between 7 printed layers. The material maintained its stable shape in a water-free environment, highlighting the potential of FDM for creating complex battery designs using vitrimeric PILs. The vitrimeric nature of the material allows for self-healing of mechanical damage via the dynamic boric ester bonds, which undergo bond exchange and reformation, effectively repairing cracks or imperfections. We observed a remarkable 70% recovery in tensile strength within 30 minutes after self-healing. Additionally, these materials can be fully reprocessed at moderate temperatures (4 h at 70 °C) through hot pressing (Fig. 3C), further enhancing their sustainability and recyclability.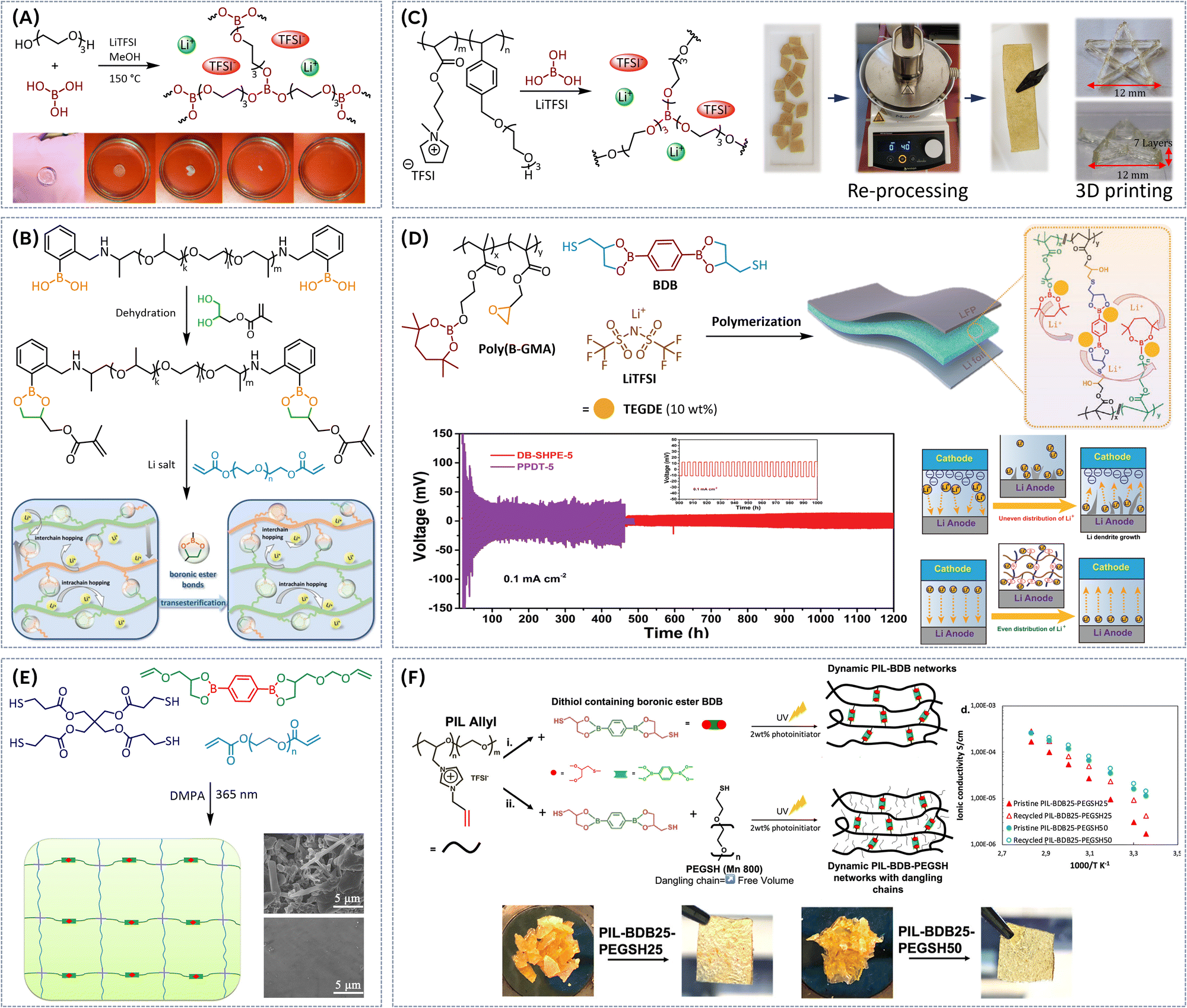 | ||
| Fig. 3 (A) Synthesis of PEO-based dynamic boronic ester networks and dissolution of vitrimeric sample in water in 30 min. Reproduced with the permission from ref. 28. Copyright © 2019 American Chemical Society. (B) Cyclic borate PEGDA-network based on poly(ethylene glycol). Reproduced from ref. 29 with permission from the Royal Society of Chemistry. (C) Pyrrolidinium-based catalyst free vitrimeric networks and corresponding reprocessing/3D printing. Reproduced from ref. 30 with permission from the Royal Society of Chemistry. (D) Synthesis of cyclic boric ester/epoxide-based electrolytes by RAFT polymerization, corresponding galvanostatic cycling performance of symmetric Li|Li cells with DB-SHPE-5 and PPDT-5 and schematics presenting Li deposition on Li metal with general polymer electrolytes and DB-SHPE-5. Reproduced with the permission from ref. 31. © 2023 Wiley-VCH GmbH (E) synthesis of self-healing gel polymer electrolyte using a thiol–ene click reaction and SEM images of Li anodes of Li/LE (liquid electrolyte)/Li cell and Li/PBAD6-PEG4/Li cell after cycling. Reproduced with the permission from ref. 32. © 2023 Published by Elsevier Ltd. (F) Synthesis of vitrimeric polymeric ionic liquid containing a copolymer rubber of poly(epichlorohydrin) and PEO, reprocessing process and corresponding conductivities before and after recycling. Reproduced with the permission from ref. 33. Copyright © 2022 The Authors. Published by American Chemical Society. | ||
Another promising approach involves copolymers combining cyclic borate monomers and epoxide-containing ones (Fig. 3D).31 A thermally initiated ring-opening reaction between thiol and epoxy groups, using 2,2′-(1,4-phenylene)-bis[4-mercaptan-1,3,2-dioxaborolane] (BDB) as crosslinker, introduces additional dynamic boronic ester groups. This results in dynamic boronic ester based self-healing polymer electrolytes with excellent mechanical properties, exhibiting increasing tensile strengths (up to 0.35 MPa) with higher BDB content. The SPEs containing boronic ester bonds can not only allow for topology rearrangements via boronic ester transesterification and demonstrate excellent self-healing capabilities (efficiency of 87% after 3 h at 60 °C), but also the presence of abundant boronic ester bonds with unoccupied p-orbital enables homogeneous deposition of Li ions on the Li metal through the Lewis acid–base interactions between boron atoms and salt anions, resulting in enhanced interfacial stability (Fig. 3D). Furthermore, the boronic ester bonds can endow the SPE with reprocessability and recyclability taking advantage of associative transesterification reaction (successful remolding after compression at 80 °C for 1 h in hot press). Most notably, the Li/SPE/Li symmetric cells exhibit a stable voltage plateau after cycling for 1200 h and the LiFePO4/SPE/Li batteries present enhanced cycling performance, attributed to the cross-linked structure with abundant boron moieties and the excellent interfacial stability between SPE film and Li metal (Fig. 3D). Different type of boronic ester based self-healing gel polymer electrolyte was prepared using a thiol–ene click reaction between a functional monomer, PBAD, containing two dynamic boronic ester bonds and two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, pentaerythritol tetra(3-mercaptopropionate) (PETMP), and polyethylene glycol diacrylate (PEGDA). This network was further plasticized with 1 M lithium bis(fluorosulfonyl)imide (LiFSI) in a dimethyl carbonate (DMC) and ethylene carbonate (EC) mixture (Fig. 3E).32 The boronic ester bonds as highly reversible covalent bonds, endow vitrimeric electrolyte with good flexibility and remarkable self-healing ability at RT. The mechanical damage was rapidly healed within 1 h at RT, while ionic conductivities and mechanical properties were effectively restored. Vitrimeric electrolyte possesses high ionic conductivity of 8.67 × 10−4 S cm−1 at RT, while Lewis acid–base interactions between anions and boron atoms of the vitrimer contributed to high lithium-ion transference number of 0.71. Additionally, significant resistance to the growth of Li dendrites was observed, improving the safety profile of this electrolyte for use in batteries (Fig. 3E). When incorporated into LiFePO4 (LFP)/Li cells, the boronic ester-based vitrimeric electrolyte enabled high cycling stability, maintaining a discharge specific capacity of 124.7 mA h g−1 after 150 cycles, with a capacity retention rate of 92.6%. Another notable example of using thiol–ene click chemistry to prepare vitrimeric electrolytes involves polymeric ionic liquid containing a copolymer rubber of poly(epichlorohydrin) and poly(ethylene oxide) (PEO) as a backbone and bearing allyl functional groups (Fig. 3F).33 The PIL was then cross-linked with dynamic boronic ester cross-linkers BDB through thiol–ene “click” photoaddition. Additionally, PEO dangling chains were introduced to act as free volume enhancers, improving the material's ionic conductivity and mechanical flexibility. By adjusting the cross-linker and dangling chain content, the properties of the all-solid PIL networks could be tailored. The resulting ionoelastomers were soft (0.2 MPa), stretchable (300%), and conducting (1.6 × 10−5 S cm−1 at 30 °C). The associative exchange reaction of BDB imparted vitrimer properties to these materials, including self-healing (within 1 h at RT) and reprocessability (hot pressing at 120 °C under pressure). Remarkably, the recycled materials retained their original modulus and ionic conductivity (Fig. 3F), demonstrating the potential for sustainable and circular material usage.
C double bonds, pentaerythritol tetra(3-mercaptopropionate) (PETMP), and polyethylene glycol diacrylate (PEGDA). This network was further plasticized with 1 M lithium bis(fluorosulfonyl)imide (LiFSI) in a dimethyl carbonate (DMC) and ethylene carbonate (EC) mixture (Fig. 3E).32 The boronic ester bonds as highly reversible covalent bonds, endow vitrimeric electrolyte with good flexibility and remarkable self-healing ability at RT. The mechanical damage was rapidly healed within 1 h at RT, while ionic conductivities and mechanical properties were effectively restored. Vitrimeric electrolyte possesses high ionic conductivity of 8.67 × 10−4 S cm−1 at RT, while Lewis acid–base interactions between anions and boron atoms of the vitrimer contributed to high lithium-ion transference number of 0.71. Additionally, significant resistance to the growth of Li dendrites was observed, improving the safety profile of this electrolyte for use in batteries (Fig. 3E). When incorporated into LiFePO4 (LFP)/Li cells, the boronic ester-based vitrimeric electrolyte enabled high cycling stability, maintaining a discharge specific capacity of 124.7 mA h g−1 after 150 cycles, with a capacity retention rate of 92.6%. Another notable example of using thiol–ene click chemistry to prepare vitrimeric electrolytes involves polymeric ionic liquid containing a copolymer rubber of poly(epichlorohydrin) and poly(ethylene oxide) (PEO) as a backbone and bearing allyl functional groups (Fig. 3F).33 The PIL was then cross-linked with dynamic boronic ester cross-linkers BDB through thiol–ene “click” photoaddition. Additionally, PEO dangling chains were introduced to act as free volume enhancers, improving the material's ionic conductivity and mechanical flexibility. By adjusting the cross-linker and dangling chain content, the properties of the all-solid PIL networks could be tailored. The resulting ionoelastomers were soft (0.2 MPa), stretchable (300%), and conducting (1.6 × 10−5 S cm−1 at 30 °C). The associative exchange reaction of BDB imparted vitrimer properties to these materials, including self-healing (within 1 h at RT) and reprocessability (hot pressing at 120 °C under pressure). Remarkably, the recycled materials retained their original modulus and ionic conductivity (Fig. 3F), demonstrating the potential for sustainable and circular material usage.
While boronic ester-based vitrimeric electrolytes offer several advantages, their moderate ionic conductivity compared to commercial liquid electrolytes remains a significant drawback.
However, their reprocessability and recyclability at conventional LIB operating temperatures (around 60 °C) make them promising candidates for sustainable energy storage solutions. The ability to reprocess these polymer electrolytes through dynamic boronic ester bond exchange offers a means to avoid environmental impacts and extend their lifespan. Additionally, the integration of additive manufacturing (AM) techniques can further enhance the sustainability and performance of batteries, while enabling the design of more complex battery architectures. By incorporating multiple functionalities, such as electrode interface stabilization, boronic ester-based vitrimers can emerge as promising alternatives for next-generation lithium metal batteries.
Vitrimers with amine/imine bonds
To address the limitations of PEO-based electrolytes, such as inefficient Li-ion conduction at room temperature, narrow electrochemical stability windows, and poor contact with electrode interfaces, which negatively impact the performance of rechargeable lithium metal batteries, various strategies have been explored to incorporate dynamic imine bonds into PEO-based matrix. By introducing imine bonds, the electrolyte's polymer network is expected to gain enhanced flexibility and self-healing properties, allowing for better ion transport and improved interface stability with lithium metal electrodes. These dynamic bonds enable reversible crosslinking, promoting continuous restructuring of the polymer matrix under operating conditions, which in turn facilitates the mobility of lithium ions and ensures more stable contact with the electrodes.One of the earliest reported materials in this field was prepared via condensation polymerization of poly(ethylene glycol) diamine with 1,3,5-triformylbenzene in dimethylformamide (DMF) solvent (Fig. 4A).34 The dynamic imine bonds effectively reduced the crystallinity, significantly improving the ionic conductivity of the SPE and endowed the electrolyte with strong adhesion properties, which are beneficial for the effective contact between the electrolyte and electrodes. The resulting flexible electrolyte films show good mechanical properties (elongation above 500%, strain stress > 130 kPa) and self-heling ability (24 h at room temperature). More importantly, this SPE achieved high ionic conductivity (7.48 × 10−4 S cm−1 at 25 °C) and wide electrochemical stability window (5.0 V vs. Li/Li+). As a result, Li‖Li symmetrical cells with the SPE showed reliable stability in a >1200 h cycling test under room temperature (Fig. 4A), whereas the assembled Li|SPE|LiFePO4 cell maintained a discharge capacity of 126.4 mA h g−1 after 300 cycles (0.1C, 27 °C). Xie and coworkers35 employed a similar synthetic approach to systematically investigate the influence of the molecular weight of H2N-PEG-NH2 on the thermal, electrochemical and self-healing properties of the resulting dynamically crosslinked SPEs. SPE with PEG molecular weight of 3400 g mol−1 exhibited high ionic conductivity of 4.8 × 10−4 S cm−1, a wide electrochemical stability window (5.1 V) and good lithium ion transfer number (tLi+ = 0.31) with an ethylene oxide/Li+ ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at 25 °C. Excellent self-healing ability (10 min at room temperature, Fig. 4B), high mechanical strength and good adhesion were achieved. Furthermore, a Li|SPE-3400|LiFePO4 battery showed good reversible specific capacity of 125.1 mA h g−1 (at 0.1C) with a Coulombic efficiency of 97.9% after 100 cycles. A Schiff base reaction of lower molecular weight H2N-PEG-NH2 (Mn = 1000 g mol−1) with aldehyde groups of benzene-1,3,5-tricarbaldehyde (BTA) (1:1 ratio) resulted in rapidly self-healing electrolyte films (Fig. 4C).36 In BTA, aromatic ring, acting as an electron acceptor, facilitates stabilization of zwitterionic intermediate that has a negative charge on the imine nitrogen, generating strong imine bonds with bond energy of 615 kJ mol−1 enhancing the structural integrity of the SPE. At RT, the electrolyte achieved a self-healing efficiency of 87.9% after 60 min of healing, while at 60 °C full self-healing efficiency (100%) was reached within 30 min. To improve the wettability of electrode surface, the membrane was soaked in a mixture of organic solvent and salt, resulting in ionic conductivity of 4.79 × 10−3 S cm−1 at 30 °C. X-Ray photoelectron spectroscopy (XPS) measurements indicated that SPE promotes the formation of LiF-rich SEI, which plays an important role in suppression of Li-dendrite growth. Both before and after healing, the electrolyte film enables good cycle performance. The initial discharge capacity of LFP/healed SPE/Li cell was 142.2 mA h g−1 (at 1C, Fig. 4C), which is almost identical to that of LFP/original SPE/Li cell. After 80 cycles, the LFP/healed SPE/Li cell showed a discharge capacity of 139.0 mA h g−1, with a retention rate of 97.7%. To enhance the thermal stability of SPE while promoting the formation of LiF-rich SEI to suppress Li-dendrite growth, perfluoropolyether (PFPE)-based vitrimers were synthesized via thermally initiated polyaddition and in situ N-alkylation in the presence of a fluorinated cross-linker (Fig. 4D).37 These vitrimers exhibited a viscous flow activation energy of 161 kJ mol−1, with relaxation times varying from 2.5 h at 170 °C to 4 min at 210 °C. Thermal creep experiment and relaxometry identified the topology freezing transition temperatures to be around 110 °C. The vitrimer network is stable under acidic and basic environments and could be remolded at 170 °C and 200 bar for 18 h, recovering the mechanical properties (E = 0.36 MPa). Ionic conductivities for non-doped materials range from 0.5 × 10−6 to 1.1 × 10−6 S cm−1 at 27 °C. A novel approach toward dynamic imine bond-rich vitrimer polymer electrolytes, formed via Schiff base reaction between aldehyde and ammonia has been reported (Fig. 5A).38 Thereby, the amine group in the nucleophilic reagent Jeffamine ED-600 and branched polyethyleneimine (bPEI) undergoes nucleophilic addition with the carbonyl group of terephthalaldehyde (TA), and the intermediate product is further dehydrated to form the desired vitrimer. Hot pressing at 60 °C and 10 MPa for 10 minutes triggers an imine exchange reaction, enabling the material to be remoulded without compromising its integrity. Moreover, the material's self-healing properties are evident when cracked films are heated at 110 °C; the cracks gradually disappear due to the continuous formation and breaking of imine bonds. The dynamic polymer network exhibits solid-state plasticity because reversible imine bonding reactions enable the integrity of the polymer network to be maintained while altering the cross-linked points under external stimuli. Thus, this 3D dynamic cross-linked network not only effectively facilitates the polymer chain dynamics but also improves ionic conductivity. Additionally, dynamic bond exchange fosters electrolyte flowability and self-healing during lithium deposition, promoting a tightly integrated electrode/electrolyte interface while inhibiting the growth of lithium dendrites. Consequently, lithium symmetric cells utilizing the SPE demonstrate exceptional long-term cycling stability, surpassing 1100 h and exhibit good ionic conductivity of 2.7 × 10−4 S cm−1 even at room temperature (30 °C). The LiFePO4‖Li battery maintains stable cycling performance at both 40 °C and 60 °C. Another noteworthy application of Schiff base chemistry involves the polycondensation reaction between tetrapthalaldehyde, Jeffamine D2000 (polyether amine with propylene oxide soft segment), and poly ether amine T403 (Fig. 5B) at 120 °C.39 To enhance the ion conductivity and self-heling properties succinonitrile (SN) plasticizer and lithium salt were added. The ion conductivity shows an obviously increasing trend as the degree of cross-linking degreases and SN content rises, reaching a maximum value of 6.43 × 10−4 S cm−1 at 30 wt% LiTFSI and 50 wt% of SN at 60 °C. Notably, the addition of SN plasticizer lowered the activation energy for the dynamic imine bond exchange, enabling the self-healing of the SPE already at room temperature with 74% recovery of its original tensile strength (Fig. 5B). Furthermore, the electrochemical window was widened to 5.1 V vs. Li/Li+ with SN addition. The Li‖SPE‖LiFePO4 cell exhibited a stable discharge capacity of 100 mA h g−1 at 0.05C after 50th cycles, demonstrating the potential of this imine-based SPE for battery applications. Importantly, the SPEs membranes with imine bonds can heal damages outside or inside a battery without compromising the discharging capacity of lithium-ion battery. Acceleration of associative imine bond exchange kinetics can also be achieved by bottom-up synthesis of pseudo-cyclic vinylogous urethane (VU) vitrimers, where the cation-templated pseudo-crown ether building blocks are formed by the complexation of alkaline metal cations with pseudo-cyclic acetoacetate-terminated PEG-AA (Fig. 5C).40 The unique network strand topology of pseudocyclic vitrimers results in higher crosslinking density, enhanced mechanical properties and accelerated topological rearrangement compared to vitrimers based on linear building blocks. Due to high crosslinking density and intramolecular plasticization, the strength, stretchability, and toughness of the pseudo-cyclic vitrimer PEG-KPF6 are significantly increased compared to the traditional vitrimer (E = 1.24 MPa, elongation at break = 269% and toughness = 2.21 MJ m−3, respectively). Stress relaxation and creep recovery tests demonstrate that the topological rearrangement of pseudo-cyclic vitrimers is significantly accelerated, and the characteristic relaxation time is reduced by about an order of magnitude compared to the traditional vitrimer at the same temperature (τ80°C = 191.8 s vs. τ80°C = 743.0 s, respectively). This acceleration is attributed to a combination of factors: low viscosity facilitating segment movement, an orderly arranged network topology and high crosslinking density promoting exchange reactions, and the catalytic effect of alkaline metal salts on VU transamination. The resulting PEG-KPF6 vitrimer
exhibits a much better self-healing ability than traditional vitrimer with linear building blocks. A scratched PEG-KPF6 vitrimer can be completely self-healed at room temperature within 60 s, while its counterpart takes 6 h to recover at 120 °C. Additionally, successful reprocessing via compression molding (at 80 °C, 15 MPa for 15 min) has been reported (Fig. 5C). The binding mode and influence of LiTFSI salt on physical and chemical properties on vinylogous urethane vitrimers have been extensively studied.41 Viscoelastic measurements revealed a significant decrease in characteristic relaxation time (τ*) of VU vitrimers containing salt by a factor of ∼70 compared to neutral vitrimers. This reduction is attributed to Li-ion coordination and catalysis of VU bond exchange. 7Li NMR further elucidated that VU vitrimers with longer linker lengths prefer lithium-ethylene oxide (Li-EO) solvation, whereas shorter linkers are unable to adequately solvate the cation, leading to a preference of Li-VU coordination. This positive effect of LiTFSI on properties of VU is further confirmed by incorporation of associative dynamic vinylogous urethane motif into a poly(ethylene oxide) network electrolyte containing LiTFSI salt (Fig. 5D).42 The resulting dynamic covalent network electrolyte possesses an activation energy of 111 kJ mol−1, exhibiting modest ion conductivity (∼10−5 S cm−1 at room temperature) and good mechanical integrity (E ∼ 1 MPa). The vitrimers can be successfully reprocessed in hot press at 90 °C for 30 min. However, the stress–strain curves of recycled SPE indicated considerable deterioration in mechanical properties (Young's modulus of 0.23 MPa) (Fig. 5D). Nevertheless, the recycled vitrimer is still mechanically robust enough to be used as a separator. Recently, an efficient vitrimeric SPE was prepared trough the reaction between the hydroxyl groups of Salen and the epoxy groups of PEGDGE under sequential temperature variations from 50 to 160 °C in the presence of LiTFSI (Fig. 5E).43 The reversible imine reaction, involving hydrolysis, transamination, and imine metathesis, enables dynamic properties such as self-healing and recycling without catalyst. The dynamic imine bond of vitrimeric SPEs contributed significantly to the formation of flexible films with high mechanical properties due to the crosslinked structure, enhancing interfacial contact with electrodes, and inhibiting lithium dendrite growth through their self-healing properties. An optimized composition V-SPE_2000 (where MnPEG = 2000 g mol−1) demonstrated a required activation energy of 52 kJ mol−1 for exchange reactions. Additionally, this material can autonomously recover damage without additional stimuli at room temperature. These excellent mechanical and self-healing properties significantly contributed to the exceptional interfacial adhesion of the solid electrolyte, even during charge–discharge processes with lithium metal electrodes. V-SPE_2000 exhibited ionic conductivity of 3.25 × 10−4 S cm−1 at 60 °C. It also provided a wide electrochemical potential range and inhibited the growth of lithium dendrites, which was confirmed through LSV and galvanostatic cycling test (Fig. 5E). The LMB full cell employing V-SPE_2000 demonstrated a high initial discharge capacity of 154.7 mA h g−1 at 0.1C.
:1 at 25 °C. Excellent self-healing ability (10 min at room temperature, Fig. 4B), high mechanical strength and good adhesion were achieved. Furthermore, a Li|SPE-3400|LiFePO4 battery showed good reversible specific capacity of 125.1 mA h g−1 (at 0.1C) with a Coulombic efficiency of 97.9% after 100 cycles. A Schiff base reaction of lower molecular weight H2N-PEG-NH2 (Mn = 1000 g mol−1) with aldehyde groups of benzene-1,3,5-tricarbaldehyde (BTA) (1:1 ratio) resulted in rapidly self-healing electrolyte films (Fig. 4C).36 In BTA, aromatic ring, acting as an electron acceptor, facilitates stabilization of zwitterionic intermediate that has a negative charge on the imine nitrogen, generating strong imine bonds with bond energy of 615 kJ mol−1 enhancing the structural integrity of the SPE. At RT, the electrolyte achieved a self-healing efficiency of 87.9% after 60 min of healing, while at 60 °C full self-healing efficiency (100%) was reached within 30 min. To improve the wettability of electrode surface, the membrane was soaked in a mixture of organic solvent and salt, resulting in ionic conductivity of 4.79 × 10−3 S cm−1 at 30 °C. X-Ray photoelectron spectroscopy (XPS) measurements indicated that SPE promotes the formation of LiF-rich SEI, which plays an important role in suppression of Li-dendrite growth. Both before and after healing, the electrolyte film enables good cycle performance. The initial discharge capacity of LFP/healed SPE/Li cell was 142.2 mA h g−1 (at 1C, Fig. 4C), which is almost identical to that of LFP/original SPE/Li cell. After 80 cycles, the LFP/healed SPE/Li cell showed a discharge capacity of 139.0 mA h g−1, with a retention rate of 97.7%. To enhance the thermal stability of SPE while promoting the formation of LiF-rich SEI to suppress Li-dendrite growth, perfluoropolyether (PFPE)-based vitrimers were synthesized via thermally initiated polyaddition and in situ N-alkylation in the presence of a fluorinated cross-linker (Fig. 4D).37 These vitrimers exhibited a viscous flow activation energy of 161 kJ mol−1, with relaxation times varying from 2.5 h at 170 °C to 4 min at 210 °C. Thermal creep experiment and relaxometry identified the topology freezing transition temperatures to be around 110 °C. The vitrimer network is stable under acidic and basic environments and could be remolded at 170 °C and 200 bar for 18 h, recovering the mechanical properties (E = 0.36 MPa). Ionic conductivities for non-doped materials range from 0.5 × 10−6 to 1.1 × 10−6 S cm−1 at 27 °C. A novel approach toward dynamic imine bond-rich vitrimer polymer electrolytes, formed via Schiff base reaction between aldehyde and ammonia has been reported (Fig. 5A).38 Thereby, the amine group in the nucleophilic reagent Jeffamine ED-600 and branched polyethyleneimine (bPEI) undergoes nucleophilic addition with the carbonyl group of terephthalaldehyde (TA), and the intermediate product is further dehydrated to form the desired vitrimer. Hot pressing at 60 °C and 10 MPa for 10 minutes triggers an imine exchange reaction, enabling the material to be remoulded without compromising its integrity. Moreover, the material's self-healing properties are evident when cracked films are heated at 110 °C; the cracks gradually disappear due to the continuous formation and breaking of imine bonds. The dynamic polymer network exhibits solid-state plasticity because reversible imine bonding reactions enable the integrity of the polymer network to be maintained while altering the cross-linked points under external stimuli. Thus, this 3D dynamic cross-linked network not only effectively facilitates the polymer chain dynamics but also improves ionic conductivity. Additionally, dynamic bond exchange fosters electrolyte flowability and self-healing during lithium deposition, promoting a tightly integrated electrode/electrolyte interface while inhibiting the growth of lithium dendrites. Consequently, lithium symmetric cells utilizing the SPE demonstrate exceptional long-term cycling stability, surpassing 1100 h and exhibit good ionic conductivity of 2.7 × 10−4 S cm−1 even at room temperature (30 °C). The LiFePO4‖Li battery maintains stable cycling performance at both 40 °C and 60 °C. Another noteworthy application of Schiff base chemistry involves the polycondensation reaction between tetrapthalaldehyde, Jeffamine D2000 (polyether amine with propylene oxide soft segment), and poly ether amine T403 (Fig. 5B) at 120 °C.39 To enhance the ion conductivity and self-heling properties succinonitrile (SN) plasticizer and lithium salt were added. The ion conductivity shows an obviously increasing trend as the degree of cross-linking degreases and SN content rises, reaching a maximum value of 6.43 × 10−4 S cm−1 at 30 wt% LiTFSI and 50 wt% of SN at 60 °C. Notably, the addition of SN plasticizer lowered the activation energy for the dynamic imine bond exchange, enabling the self-healing of the SPE already at room temperature with 74% recovery of its original tensile strength (Fig. 5B). Furthermore, the electrochemical window was widened to 5.1 V vs. Li/Li+ with SN addition. The Li‖SPE‖LiFePO4 cell exhibited a stable discharge capacity of 100 mA h g−1 at 0.05C after 50th cycles, demonstrating the potential of this imine-based SPE for battery applications. Importantly, the SPEs membranes with imine bonds can heal damages outside or inside a battery without compromising the discharging capacity of lithium-ion battery. Acceleration of associative imine bond exchange kinetics can also be achieved by bottom-up synthesis of pseudo-cyclic vinylogous urethane (VU) vitrimers, where the cation-templated pseudo-crown ether building blocks are formed by the complexation of alkaline metal cations with pseudo-cyclic acetoacetate-terminated PEG-AA (Fig. 5C).40 The unique network strand topology of pseudocyclic vitrimers results in higher crosslinking density, enhanced mechanical properties and accelerated topological rearrangement compared to vitrimers based on linear building blocks. Due to high crosslinking density and intramolecular plasticization, the strength, stretchability, and toughness of the pseudo-cyclic vitrimer PEG-KPF6 are significantly increased compared to the traditional vitrimer (E = 1.24 MPa, elongation at break = 269% and toughness = 2.21 MJ m−3, respectively). Stress relaxation and creep recovery tests demonstrate that the topological rearrangement of pseudo-cyclic vitrimers is significantly accelerated, and the characteristic relaxation time is reduced by about an order of magnitude compared to the traditional vitrimer at the same temperature (τ80°C = 191.8 s vs. τ80°C = 743.0 s, respectively). This acceleration is attributed to a combination of factors: low viscosity facilitating segment movement, an orderly arranged network topology and high crosslinking density promoting exchange reactions, and the catalytic effect of alkaline metal salts on VU transamination. The resulting PEG-KPF6 vitrimer
exhibits a much better self-healing ability than traditional vitrimer with linear building blocks. A scratched PEG-KPF6 vitrimer can be completely self-healed at room temperature within 60 s, while its counterpart takes 6 h to recover at 120 °C. Additionally, successful reprocessing via compression molding (at 80 °C, 15 MPa for 15 min) has been reported (Fig. 5C). The binding mode and influence of LiTFSI salt on physical and chemical properties on vinylogous urethane vitrimers have been extensively studied.41 Viscoelastic measurements revealed a significant decrease in characteristic relaxation time (τ*) of VU vitrimers containing salt by a factor of ∼70 compared to neutral vitrimers. This reduction is attributed to Li-ion coordination and catalysis of VU bond exchange. 7Li NMR further elucidated that VU vitrimers with longer linker lengths prefer lithium-ethylene oxide (Li-EO) solvation, whereas shorter linkers are unable to adequately solvate the cation, leading to a preference of Li-VU coordination. This positive effect of LiTFSI on properties of VU is further confirmed by incorporation of associative dynamic vinylogous urethane motif into a poly(ethylene oxide) network electrolyte containing LiTFSI salt (Fig. 5D).42 The resulting dynamic covalent network electrolyte possesses an activation energy of 111 kJ mol−1, exhibiting modest ion conductivity (∼10−5 S cm−1 at room temperature) and good mechanical integrity (E ∼ 1 MPa). The vitrimers can be successfully reprocessed in hot press at 90 °C for 30 min. However, the stress–strain curves of recycled SPE indicated considerable deterioration in mechanical properties (Young's modulus of 0.23 MPa) (Fig. 5D). Nevertheless, the recycled vitrimer is still mechanically robust enough to be used as a separator. Recently, an efficient vitrimeric SPE was prepared trough the reaction between the hydroxyl groups of Salen and the epoxy groups of PEGDGE under sequential temperature variations from 50 to 160 °C in the presence of LiTFSI (Fig. 5E).43 The reversible imine reaction, involving hydrolysis, transamination, and imine metathesis, enables dynamic properties such as self-healing and recycling without catalyst. The dynamic imine bond of vitrimeric SPEs contributed significantly to the formation of flexible films with high mechanical properties due to the crosslinked structure, enhancing interfacial contact with electrodes, and inhibiting lithium dendrite growth through their self-healing properties. An optimized composition V-SPE_2000 (where MnPEG = 2000 g mol−1) demonstrated a required activation energy of 52 kJ mol−1 for exchange reactions. Additionally, this material can autonomously recover damage without additional stimuli at room temperature. These excellent mechanical and self-healing properties significantly contributed to the exceptional interfacial adhesion of the solid electrolyte, even during charge–discharge processes with lithium metal electrodes. V-SPE_2000 exhibited ionic conductivity of 3.25 × 10−4 S cm−1 at 60 °C. It also provided a wide electrochemical potential range and inhibited the growth of lithium dendrites, which was confirmed through LSV and galvanostatic cycling test (Fig. 5E). The LMB full cell employing V-SPE_2000 demonstrated a high initial discharge capacity of 154.7 mA h g−1 at 0.1C.
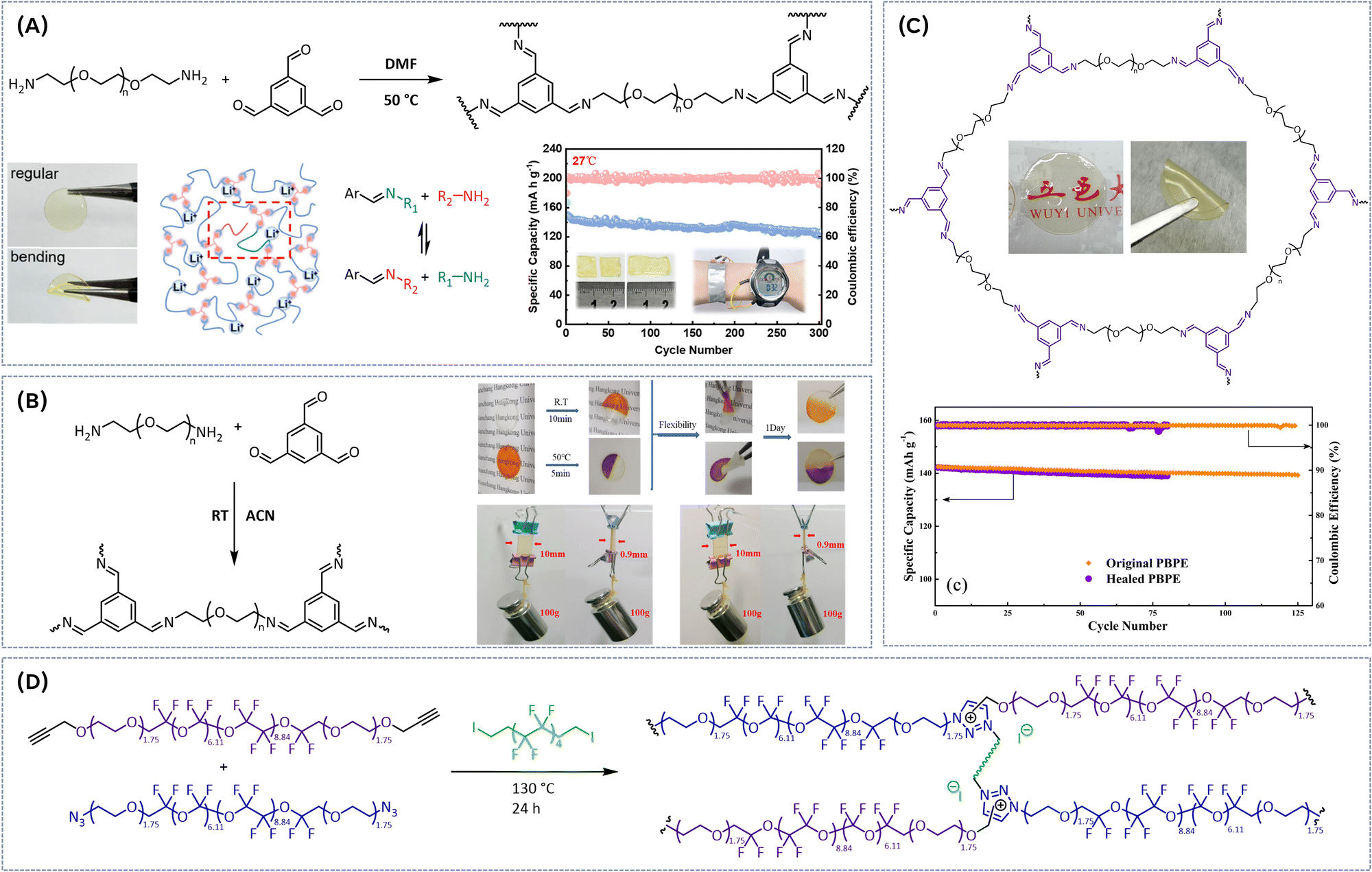 | ||
| Fig. 4 (A) Condensation polymerization of poly(ethylene glycol) diamine with 1,3,5-triformylbenzene and corresponding long-term cycling performance of the Li|SHSPE|LiFePO4 cell at 0.1C. Reproduced with the permission from ref. 34. Copyright © 2021, American Chemical Society. (B) Preparation of imine-exchange-based vitrimeric electrolyte via polycondensation, self-healing performance of SPE at room temperature and 50 °C, mechanical strength of original and healed electrolyte at room temperature. Reproduced with the permission from ref. 35. © 2022 Society of Industrial Chemistry. (C) A Schiff base reaction of lower molecular weight H2N-PEG-NH2 with aldehyde groups of benzene-1,3,5-tricarbaldehyde (BTA) and cycle performances of LFP/original PBPE/Li cell and LFP/healed BPE/Li cell at 1C rate. Reproduced with the permission from ref. 36. © 2021 Elsevier B.V. All rights reserved. (D) Synthesis of perfluoropolyether-based vitrimers via thermally initiated polyaddition and in situ N-alkylation in the presence of a fluorinated cross-linker. Reproduced with the permission from ref. 37. Copyright © 2019 American Chemical Society. | ||
 | ||
| Fig. 5 (A) Synthesis of dynamic imine bond-rich vitrimer polymer electrolytes, corresponding Li plating/stripping profiles of lithium symmetric cells with the V-SPE-PEG and SEM images of Li anode surface. Reproduced with the permission from ref. 38. © 2024 Elsevier B.V. All rights are reserved. (B) Preparation of Jeffamine D2000-based vitrimer and optical image of its self-healing process. Reproduced with the permission from ref. 39. © 2023 Elsevier Ltd. All rights reserved. (C) Synthesis of pseudo-cyclic vinylogous urethane vitrimer electrolyte and reprocessing demonstration. Reproduced with the permission from ref. 40. © 2022 Elsevier B.V. All rights reserved. (D) Synthesis of dynamic vinylogous urethane poly(ethylene oxide)-based network electrolyte, hot-press reprocessing and ionic conductivity and Young modulus of SPE after different times of reprocessing. The dashed line indicates the conductivity of 10−5 S cm−1. Reproduced with the permission from ref. 42. Copyright © 2022, American Chemical Society. (E) Synthesis of vitrimeric SPE trough the reaction between the hydroxyl groups of Salen and the epoxy groups of PEGDGE and corresponding imine dynamic bonds. Schematic of change in the electrolyte/electrode interface during cycling for V-SPE_2000 and conventional SPEs. And galvanostatic cycling curves of Li‖V-SPE_2000‖Li cell at a current density of 0.1 mA cm−2 at 60 °C. Reproduced with the permission from ref. 43. © 2024 Elsevier Ltd. All rights are reserved. | ||
Imine-based vitrimers represent the most extensively studied class of vitrimers for applications in lithium-ion batteries. Additionally, to their ion conductivities which are approximately an order of magnitude higher compared to those of boronic ester-based vitrimer electrolytes at room temperature, imine-based vitrimers offer a distinct advantage: their exceptional self-healing capability at room temperature. Additionally, this type of vitrimer effectively stabilizes the solid electrolyte interphase, which plays a crucial role in suppressing the formation and growth of lithium dendrites. However, the reprocessing of imine-based vitrimers requires more harsh conditions (elevated temperatures, external pressure) compared to boronic ester based vitrimers. These properties make imine-based vitrimers particularly promising for enhancing the performance and safety of next-generation LIBs.
Vitrimers with disulfide bonds
Disulfide dynamic bonds present a promising strategy towards self-healable, long-lasting and recyclable/reprocessable electrolytes for LIBs. Fig. 6A illustrates a vitrimeric electrolyte prepared with a rigid and flexible epoxy resin as a backbone and disulfide bonds as reversible crosslinking points.44 The epoxy resin imparts superior mechanical abilities (tensile strength >20 MPa) to the electrolytes, while the disulfide bonds provide excellent self-healing capabilities (healing efficiency >95% after 2 h at RT). The ionic conductivity of the SPEs with higher content of flexible PEGDGE chain segments reaches values higher as 10−3 S cm−1 at room temperature, and importantly, the multiple healing cycles do not impact the ionic conductivity (Fig. 6A). Furthermore, Li plating/stripping experiments demonstrate good cycling stability up to 1800 h at 1 mA h cm−2, indicating an inhibitory effect on the growth of lithium dendrites. The Li/LiFePO4 cell also exhibits good cycling stability and superior rate performance (capacity of 133 mA h g−1 at 0.1C). Rowan et al. presented dynamic network polymers of varying cross-link densities using a thiol oxidation reaction between a bisthiol monomer and tetrathiol cross-linker (Fig. 6B).45 At optimal LiTFSI salt loading, these vitrimers achieved ionic conductivities of 1 × 10−4 and 1 × 10−5 S cm−1 at 90 °C for the lowest and highest cross-linking density, respectively. Compared to nondynamic networks, these dynamic networks exhibited a notable increase in ion conductivity above 90 °C, attributed to enhanced ion transport facilitated by the dynamic nature of their disulfide bonds. Lap shear adhesion and conductivity tests on ITO-coated glass substrates demonstrated the superiority of the dynamic network. It exhibits a higher adhesive shear strength of 0.2 MPa (vs. 0.03 MPa for the nondynamic network) and higher ion conductivity after the exposure to external stimulus (UV light or heat). Moreover, both adhesive strength and conductivity remained stable through multiple debonding/rebonding cycles, highlighting the potential of these structurally dynamic networks as versatile solid polymer electrolyte adhesives.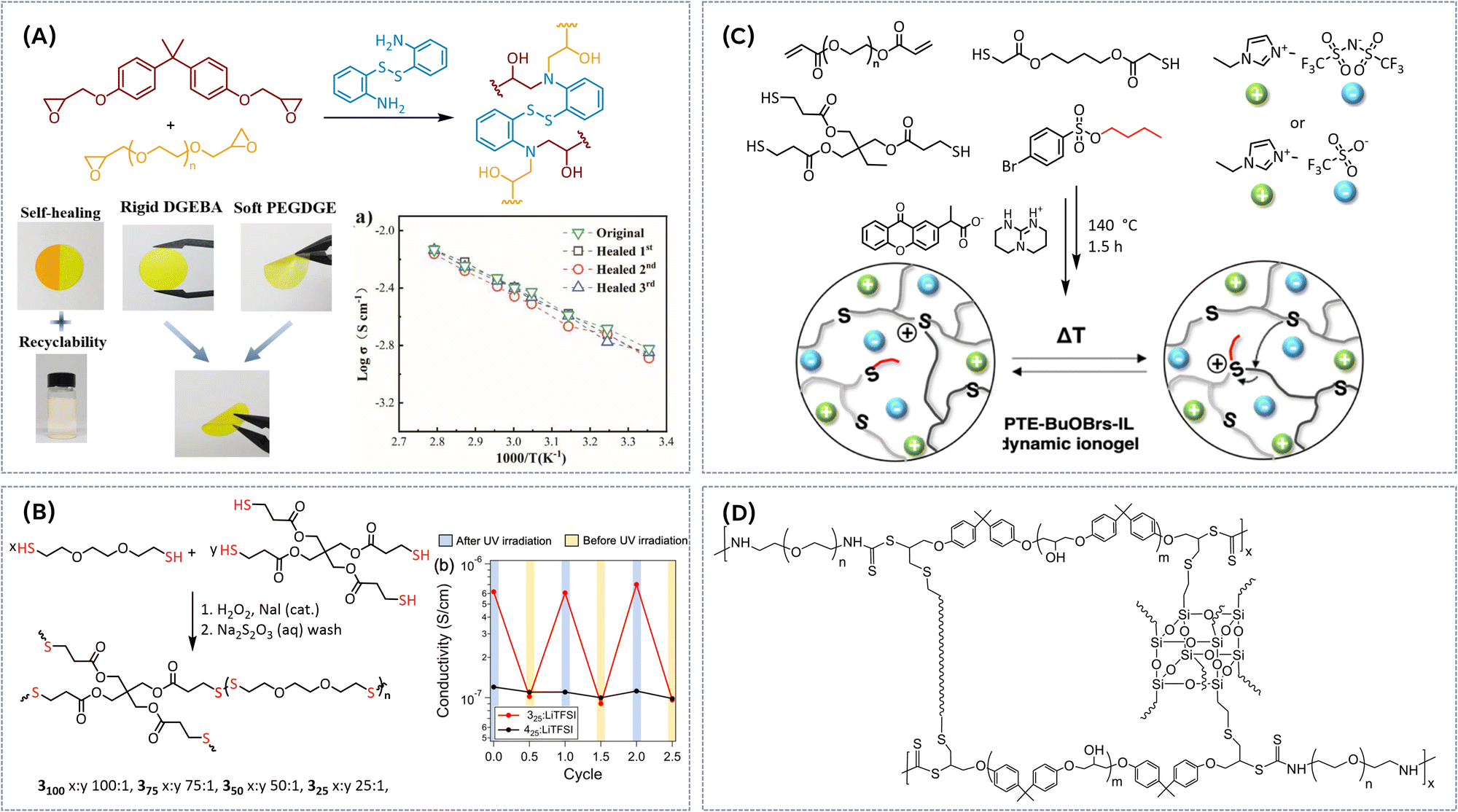 | ||
| Fig. 6 (A) Synthesis of a vitrimeric electrolyte with a rigid and flexible epoxy resin as a backbone and disulfide bonds as reversible crosslinking points, self-healing behaviour and temperature dependence of ionic conductivity of healed RFSPE-3. Reproduced with the permission from ref. 44. © 2021 Elsevier Ltd. All rights reserved. (B) Thiol oxidation reaction between a bisthiol monomer and tetrathiol cross-linker and room temperature ionic conductivity of dynamic 325:LiTFSI and nondynamic 425:LiTFSI polymers with Li salt (r = 0.1) over two and half cycles of debonding and rebonding by exposure to UV irradiation (ca. 350 nm) while pressed together (500 g weight, 0.136 MPa). To test the conductivity strength after debonding the adhesive-coated ITO slides are pressed together (500 g weight, 0.136 MPa) without light (or heat) before the measurements were done. Reproduced with the permission from ref. 45. Copyright © 2020, American Chemical Society. (C) Preparation of polythioether vitrimer networks. Reproduced from ref. 46 with permission from the Royal Society of Chemistry. (D) Preparation of POSS-vitrimeric composite electrolyte. Reproduced with the permission from ref. 47. © 2023 Elsevier Ltd. All rights reserved. | ||
Another promising approach involves utilizing vitrimeric polymers as a robust network backbone to embed additional liquid components, such as ionic liquids, to enhance the ion transport capabilities of the electrolyte.48,49 Ionic liquids have been successfully incorporated into various covalent polymer networks, forming iongels that not only improve conductivity but can also exhibit self-healing properties. Extending the iongel concept to dynamic vitrimeric networks could significantly boost material performance while enhancing reprocessability. Recently, polythioether vitrimer networks were synthesized by using the thiol–ene Michael addition between multifunctional thiol and diacrylate, initiated by a photobase generator (Fig. 6C).46 After thermal activation treatment, these networks exhibited vitrimeric properties, allowing the formation of alkylsulfonium salts. At elevated temperatures (140 °C), the networks demonstrated healing and stress relaxation due to the S-transalkylation exchange reaction between sulfonium salts and thioether nucleophiles. The activation energy for this dynamic bond exchange ranged between 60.6 kJ mol−1 and 73.6 kJ mol−1. Subsequently, vitrimeric ionogels were created by forming the dynamic network directly in the presence of IL (50 wt%). These ionogels demonstrated Young's modulus of 0.9 MPa and ionic conductivities in the order of 10−4 S cm−1 at room temperature. However, it was found that the addition of ILs modified the dynamic properties of the vitrimer networks.
This change is likely due to the dilution effect of the dynamic functions caused by the IL, as well as the interaction between alkyl sulfonium ions and the ions of the IL. As a result, the dynamic exchange kinetics slowed down, leading to an increase in both activation energy and the vitrification temperature. While this may reduce the efficiency of self-healing at a given temperature, it also provides more dimensionally stable networks at higher application temperatures, making them suitable for various performance-critical applications.
Disulfide bond-based vitrimers have not only been employed in the development of gel polymer electrolytes but also in the design of composite polymer electrolytes.47 For the preparation of the composite electrolyte linear multi-segmented poly(mercaptothiourethane-co-ethylene oxide) copolymer was synthesized via the step-growth polyaddition and in next step crosslinked with disulfide bonds via radical coupling reaction (Fig. 6D). The crosslinking density was further adjusted by incorporating octavinyl POSS via a thiol–ene radical addition reaction. The LiTFSI salt can be well dispersed into the networks and the crystallization of PEO was effectively suppressed. The ionic conductivity of the SPEs can be fine-tuned depending on the ethylene oxide to LiTFSI molar ratio and the percentage of POSS, achieving values as high as 2.1 × 10−5 S cm−1 at 27 °C. Although the introduction of POSS significantly increased the activation energy of dynamic bond exchange up to 155 °C, the polymer networks remained reprocessable at high temperatures ranging from 130 °C to 160 °C.
The incorporation of disulfide dynamic bonds into polymer networks provides enhanced mechanical integrity and stability, creating robust structures. However, this comes with certain drawbacks, including limited ion transport, resulting in lower ionic conductivities, and elevated reprocessing temperatures. A more promising approach for improving their application as electrolytes is to combine disulfide dynamic bonds with other reversible bonding systems, creating a dual network system. This strategy offers the potential to balance mechanical strength with improved ion transport, which will be explored in the following chapter.
Other types of dynamic bonds
Although less commonly explored, also other types of dynamic bonds have been used in the design of vitrimeric solid polymer electrolytes. One recent example involves a thiourea-based dynamic covalent ion conducting elastomer (Fig. 7A).50 The network was designed by crosslinking diamine-functionalized poly(ether-thiourea) containing triethylene glycol (TUEG) with poly(hexamethylene diisocyanate) (PHDI) as the cross-linker. The inclusion of thiourea moieties is particularly advantageous as they promote the formation of non-crystalline structures due to the zigzag arrangement of hydrogen bonding and dynamic covalent bond exchange. Under optimized LiTFSI salt content, vitrimeric films exhibit proper dimensional stability and ionic conductivity, reaching approximately 4.6 × 10−4 S cm−1 at 25 °C. Owing to the dissociative mechanism of thiourea moiety, vitrimer network can be reprocessed both through the hot-press (3 MPa, 140 °C, 10 min) and solution process (DMF, 140 °C, 2 h), without significantly compromising the ionic conductivity (Fig. 7A). Our working group developed a catalyst-free dynamic silyl ether covalent bond as a linkage to prepare self-healable, mechanically robust and fully reprocessable gel polymer electrolytes (Fig. 7B).51 By incorporating polymeric ionic liquids into the dynamically cross-linked networks, both ion conductivity and mechanical properties can be fine-tuned. Rheological analysis demonstrated the dynamic nature of the network, showing rubbery-like behavior at high frequencies and liquid-like behavior at low frequencies. The gel polymer electrolyte containing 80 wt% of a bis(trifluoromethansulfonamide) lithium/ionic liquid (LiTFSI/IL) mixture displayed good ion conductivities of 0.13 × 10−3 S cm−1 at 20 °C and 1.88 × 10−3 S cm−1 at 80 °C. The elastic modulus of the gel electrolyte could reach a value of 0.24 MPa. Moreover, the GPEs exhibited successful self-healing after mechanical damage, with 89% healing efficiency achieved after 12 hours at 180 °C under 1 MPa of pressure. Reprocessing was also effective, with efficiencies between 85–90% at 180 °C over a 6-hour period (Fig. 7B). The combination of tunable dynamic properties, high ionic conductivity, and strong mechanical performance highlights the potential of these dynamic silyl ether bonds for next-generation sustainable solid electrolytes.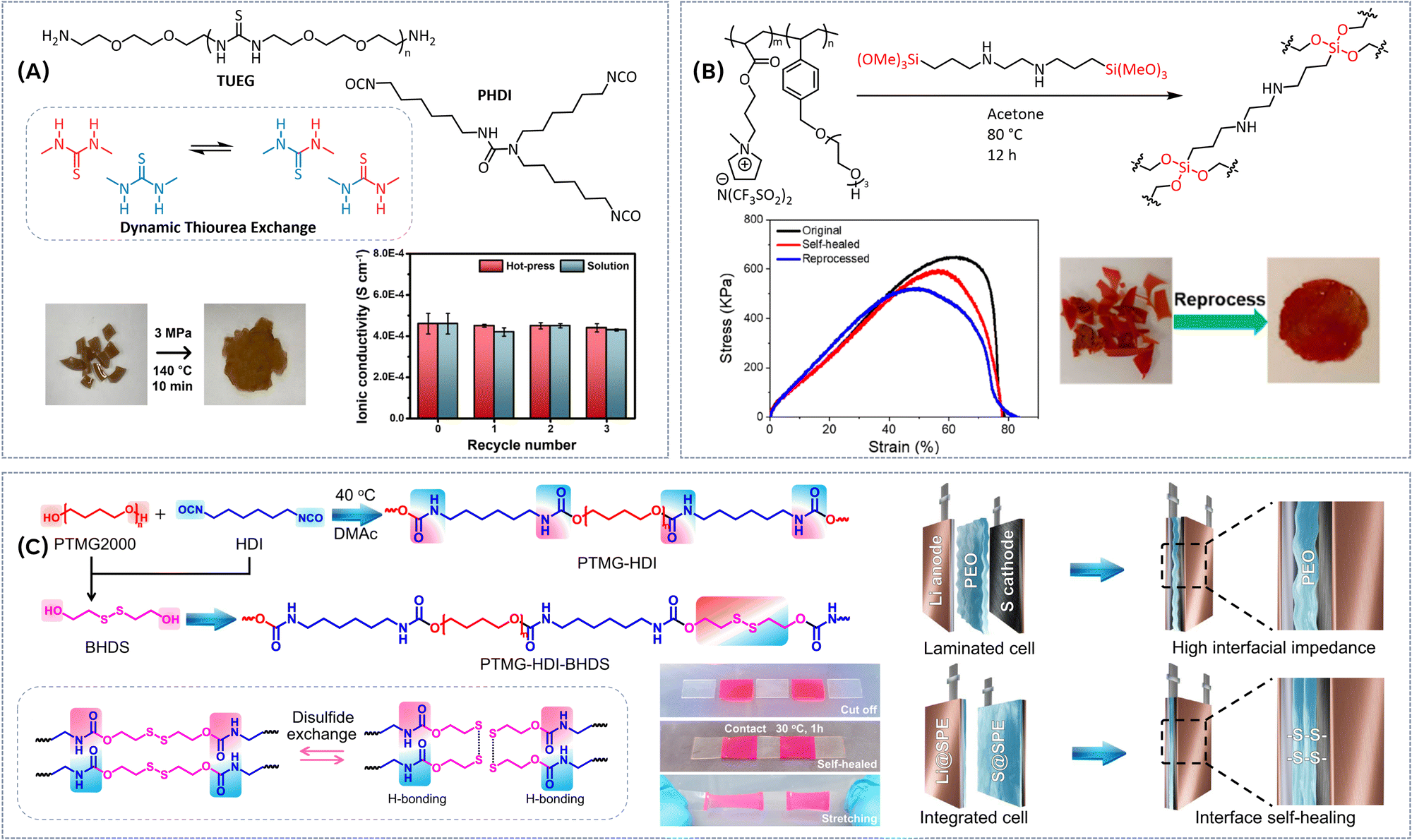 | ||
| Fig. 7 (A) Synthesis of a thiourea-based dynamic covalent ion conducting elastomer, hot-press reprocessing and ionic conductivity of SPE after different times of reprocessing (hot-press and solution processes). Reproduced with the permission from ref. 50. © 2023 Elsevier Ltd. All rights reserved. (B) Synthesis of catalyst-free dynamic silyl ether polymeric ionic liquid vitrimer electrolyte, reprocessing and tensile stress–strain curves of the original, self-healed, and reprocessed vitrimer samples. Reproduced with the permission from ref. 51. Copyright © 2023, American Chemical Society. (C) Preparation of poly(ether-urethane)-based SPEs incorporating both disulfide and hydrogen bonds, the interfacial self-healing process, and structure of the integrated electrode/electrolyte and solid-state Li–S battery. Reproduced with the permission from ref. 52. Copyright © 2024, The Author(s). | ||
Dual network systems
Extensive research has been devoted to addressing the limitations of vitrimeric and supramolecular networks that rely on a single bond type by synergistically combining the properties of multiple bond types within one multifunctional material. This approach enhances the overall performance by leveraging the strengths of each bond type, enabling improved mechanical properties, ion transport, and reprocessability. By integrating dynamic covalent bonds with non-covalent interactions or other reversible chemistries, researchers can create materials that exhibit superior self-healing, recyclability, and durability, making them more suitable for advanced applications, such as in solid polymer electrolytes.Disulfide dynamic bonds, known for their excellent interfacial properties, have been effectively combined with other types of dynamic, reversible bonds to tackle the limitations in ion conductivity and self-healing ability. Recently, a poly(ether-urethane)-based SPEs incorporating both disulfide and hydrogen bonds was designed to reduce interfacial resistance and enhance the performance of solid-state Li-metal batteries (Fig. 7C).52 This combination leverages the dynamic nature of disulfide bonds alongside hydrogen bonding to deliver exceptional interfacial self-healing ability, achieving full recovery within 1 hour at 30 °C, while maintaining robust interfacial contact. The optimized composition of the SPE exhibited remarkable mechanical strength, with a breaking strength of 88.3 MPa and an ultimate elongation of 2000%, attributed to the extended hydrogen-bond network and the increased molecular weight facilitated by a chain extender. These properties suggest that the vitrimer network can theoretically act as a mechanical barrier, helping to suppress the growth of lithium dendrites, while maintaining good ion conductivity of 2.4 × 10−4 S cm−1 at 25 °C. When integrated into Li‖Li symmetric cells, the SPE demonstrated stable long-term cycling for over 6000 hours. Additionally, in solid-state Li–S batteries, the electrolyte contributed to a prolonged cycling life, reaching 700 cycles at a 0.3C rate. A intriguing class of shape-memory polyurethane-based solid polymer electrolytes has been developed by combining polycaprolactone (PCL) as the soft segment and urea containing disulfide bonds as the hard segment (Fig. 8A).53 The soft segments primarily control the temporary shape and restoration of the original shape, while the mechanical strength of the electrolyte films can be adjusted by varying the ratio of soft to hard segments (0.63–5.38 MPa). The SPE benefits from abundant disulfide metathesis and hydrogen bonding, imparting self-healing properties when exposed to heat, with a healing efficiency of 91% after 12 hours at 60 °C. However, these vitrimers exhibit relatively low ion conductivity at 30 °C, around 10−7 S cm−1, likely due to the crystallinity of the PCL segment. Upon heating to above the melting point of PCL (60–80 °C), the ionic conductivity significantly improves, reaching values in the range of 10−4 S cm−1. The assembled Li/LiFePO4 battery presents a specific discharge capacity about 160 mA h g−1 at the charge/discharge rate of 0.1C, and the capacity retention rate is close to 92.4% after 100 cycles. Another class of SPE containing disulfide bonds and urea groups has been designed by employing reversible addition–fragmentation chain transfer (RAFT) polymerization of poly(ethylene glycol) methyl ether acrylate (PEGA, Mn = 480 g mol−1) and crosslinker consisting of hydrogen bond and disulfide bond interactions (Fig. 8B).54 The hydrogen bonding between the urea groups and disulfide metathesis reaction endows the SPE with a high level of self-healing without external stimuli at room temperature (30 min) as well as ultrafast self-healing at elevated temperatures (5 min at 60 °C and 1 min at 80 °C, respectively). The completely healed SPE displayed a tensile strength and elongation at break of 92.3 kPa and 62.1%, respectively, with self-healing efficiency of 98.7%. Even after severe damage, the completely healed SPE maintains high self-healing efficiency and shows no significant changes in ionic conductivity or cycling performance when used in a solid-state lithium metal/LiFePO4 cell compared to the pristine state (Fig. 8B). The initial discharge capacity of the SPE battery at a current density of 0.2C is 128.5 mA h g−1, which is very close to the pristine value of 130.3 mA h g−1. Moreover, the specific capacity increases to 131.2 mA h g−1 after 100 cycles. Beyond urea bonds, incorporating ureidopyrimidinone (UPy) groups, which can form quadrupole hydrogen bonds, has demonstrated potential in enhancing the properties of vitrimeric electrolytes. UPy groups introduce strong self-healing functionality within the polymer matrix, contributing to robust mechanical properties. Notably, as demonstrated in our recent studies,55,56 these UPy groups remain stable and maintain their closed and clustered structure even in highly ionic environments. Combining disulfide dynamic bonds and UPy groups, a series of quasi-solid electrolytes was synthesized from poly(arylene ether sulfone) co-grafted with polyureidopyrimidinone and poly(ethylene glycol) (PAES-g-PU/2PEG) and PAES co-grafted with poly(1,2-bis(ureidoethylenemethacrylateethyl)disulfide) and PEG (PAES-g-PUS/2PEG) for long-term stable lithium sulfur battery applications (Fig. 8C).57 The synthesized self-assembled SPEs exhibited a good Li-ion conductivity of 0.712 × 10−3 S cm−1 and tensile strength of 2.2 MPa at room temperature. Their self-healing ability, enabled by hydrogen and/or disulfide bondings, resulted in a high self-healing efficiency of approximately 95.3% within an ultrafast healing time of 5 minutes at room temperature. Moreover, these flexible membranes demonstrated excellent thermal stability (∼200 °C) and interfacial stability without short circuiting after 1000 hours of cycling at 2.0 mA cm−2. When used in a lithium sulfur battery cell, the healed SPE retained 98.7% of its original discharge capacity of 929.8 mA h g−1 and a Coulombic efficiency of 99.5% after 200 cycles at 0.2C-rate. An innovative class of dynamic supramolecular ionic conductive elastomers have been developed via phase-locked strategy (Fig. 8D).58 In this approach, the soft phase polyether backbone is locked in place to facilitate efficient Li-ion transport, while the combination of dynamic disulfide metathesis and robust supramolecular quadruple hydrogen bonds in the hard domains imparts exceptional self-healing capabilities and mechanical flexibility. This multifunctionality approach, enables a tradeoff between ionic conductivity, self-healing capability, and mechanical compatibility of the resulting self-healing electrolytes. The optimized SPE displayed high ionic conductivity reaching 3.77 × 10−3 S cm−1 at 30 °C. Additionally, the material showcases high transparency (92.3%), superior stretchability (up to 2615% elongation), tensile strength (up to 27.8 MPa) and toughness (164.36 MJ m−3). Vitrimeric films exhibited good self-healing capability (∼99% efficiency after 6 h at room temperature), whereas efficient reprocessing could be achieved both from solution (THF, 2 h at room temperature) or via hot press (70 °C, 0.5 MPa, 30 min).
 | ||
| Fig. 8 (A) Synthesis of shape-memory polyurethane-based solid polymer electrolytes and stress–strain curves of Net-PU4000SS33 with different healing times in the presence of 20 wt% of LiClO4. Reproduced with the permission from ref. 53. © 2022 Elsevier B.V. All rights reserved. (B) RAFT polymerization of poly(ethylene glycol) methyl ether acrylate and crosslinker containing both hydrogen and disulfide bonds, photographs of self-healing of disk-shaped 3PEG-SSH cut into different forms, ionic conductivity of healed 3PEG-SSH after 10 cutting–healing cycles, and cycling of Li|3PEG-SSH|LFP and Li|3PEG-CCH|LFP cells with healed SPEs at 0.2C. All experiments were conducted at 60 °C. Reproduced with the permission from ref. 54. Copyright © 2020, American Chemical Society. (C) Schema of quasi-solid electrolytes synthesized from poly(arylene ether sulfone) co-grafted with polyureidopyrimidinone and poly(ethylene glycol) (PAES-g-PU/2PEG) and PAES co-grafted with poly(1,2-bis(ureidoethylenemethacrylateethyl) disulfide) and PEG (PAES-g-PUS/2PEG), images and stress vs. strain curves before and after self-healing. Reproduced from ref. 57 with permission from the Royal Society of Chemistry. (D) Phase-locked strategy towards vitrimeric electrolytes. Reproduced with the permission from ref. 58. Copyright © 2022, The Author(s). | ||
The concept of dual networks in vitrimeric SPEs can be extended to composite electrolytes, aiming to improve the mechanical integrity of conventional ceramic composite electrolytes and mitigate structural degradation caused by external or internal stress and temperature fluctuations. One such approach involves grafting a self-healing polyurethane–urea disulfide polymer (PUS) onto Li7P3S11via nucleophilic addition (Fig. 9A).59
 | ||
| Fig. 9 (A) Grafting polyurethane–urea disulfide polymer (PUS) onto Li7P3S11 in a vitrimeric electrolyte. Reproduced with the permission from ref. 59. © 2022 Elsevier B.V. All rights reserved. (B) Illustration of vitrimeric polyurethane/LLZTO composite electrolyte and photographs of PBHL@LLZTO@DDB membrane lifting weight after self-healing for 1 h. Reproduced with the permission from ref. 60. © 2024 Elsevier Inc. All rights are reserved, including those for text and data mining, AI training, and similar technologies. (C) Preparation and depolymerisation of polydisulfide-co-polyethylenimine urethane (P(LpC/PEIU)) covalent adaptive network (CAN). Reproduced with the permission from ref. 61. Copyright © 2024, American Chemical Society. (D) Reversibly interlocking polymer network based on differently functionalized PEO blocks, optical images of the (a1) original, (a2) bisected, and (a3) healed specimens, and (a4) the healed specimen stretched to 200% strain and cycling performance of the cells assembled from original and healed RILNs-3/1-LiPF6 at 1C. Reproduced from ref. 62 with permission from the Royal Society of Chemistry. (E) Ceramic–composite polymer electrolyte with interlocked polymer networks. Reproduced with the permission from ref. 63. Copyright © 2024 American Chemical Society. | ||
This method ensures a uniform distribution of Li7P3S11 within the PUS matrix, maintaining close chemical contact between the two components and preventing phase separation during cooling-heating cycles – a common issue in traditional systems. The chemically bonded interface restricts the movement of PUS chains, thereby enhancing the composite's resistance to structural degradation under thermal cycling. This leads to exceptional resilience, with the composite maintaining a stable ionic conductivity of 5 × 10−4 S cm−1, even under temperature fluctuations. Additionally, the dynamic disulfide bonds in PUS enable rapid self-healing, allowing the composite electrolyte to recover nearly 100% of its current capacity within just three minutes after mechanical damage at room temperature.
When tested in a Li|PUS-LPS|LiFePO4 full cell, the composite electrolyte exhibited impressive post-damage performance, with a 95.1% capacity recovery and excellent cycling stability. After 200 cycles, the cell retained 95.4% of its initial capacity (125 mA h g−1), highlighting the potential of this dual-network composite for high-performance, durable solid-state batteries with self-healing capabilities. Another reported composite electrolyte consists of polyurethane as the polymer matrix and Li6.4La3Zr1.4Ta0.6O12 (LLZTO) modified by silane coupling agent as inorganic fillers (Fig. 9B).60 Disulfide bond exchange reactions between polyurethane matrix and modified nanofillers facilitate the creation of a robust, three-dimensional (3D) inorganic/organic hybrid network. This enhanced structure ensures uniform dispersion of LLZTO particles within the matrix, significantly boosting ionic conductivity (1.24 × 10−4 S cm−1 at 30 °C) and expanding the electrochemical stability window to 5.16 V vs. Li+/Li. The synergy between the dynamic disulfide bond exchange and the reversible cleavage/remodeling of hydrogen bonds imparts excellent self-healing properties (1 h at room temperature) to the composite electrolyte. This self-healing capacity ensures that both all-solid-state symmetric and full cells maintain outstanding cycling performance, even after mechanical damage. Notably, the electrochemical properties of the healed composite electrolyte are nearly identical to those of the original, undamaged material. LiFePO4//Li cell assembled with healed composite electrolyte can keep a stable cycling with a discharge capacity of 151.7 mA h g−1 at 0.2C, which is close to the cycle performance of the pristine one (164 mA h g−1 at 0.2C).
In addition to hydrogen bonds, imine-based dynamic bonds can also be successfully combined with disulfide bonds to enhance the overall properties of vitrimeric SPEs. Most recently, a novel recyclable polydisulfide-co-polyethylenimine urethane (P(LpC/PEIU)) covalent adaptive network (CAN), was prepared via a nonisocyanate process involving polyethylenimine (PEI) and a newly designed (2-oxo-1,3dioxolan-4-yl)methyl-5-(1,2-dithiolan-3-yl)pentanoate monomer (LpC) (Fig. 9C).61
The obtained network structure of the P(LpC/PEIU) CANs can undergo dissociation in specific solvents (e.g. DMF or DMSO at 60 °C, or even in DMF/THF mixture) via the self-catalyzed depolymerization of disulfide groups. Subsequently, the network structure can be reconstructed via ring-opening polydisulfide linkage formation from dithiolane pendant groups under UV light at 365 nm. Upon addition of LITFSI salt the films exhibited a maximum ion conductivity of 7.48 × 10−4 S cm−1, attributed to the high concentration of the pendant ethylene carbonate group in the CANs. The recyclability of the network was investigated, revealing dissolution of the network in an appropriate solvent within 1 hour at 60 °C without the need for an additional amine catalyst. Additionally, the investigated films demonstrated good self-healing performance (1 hour at 60 °C, 95% efficiency).
Zhang and colleagues62 introduced a novel class of SPEs based on a reversibly interlocking polymer network. This type of network was created through the topological reorganization of two distinct poly(ethylene oxide) (PEO) networks, each crosslinked via different dynamic covalent bonds—one network utilizing reversible imine bonds, and the other crosslinked by disulfide bonds (Fig. 9D). The reversible nature of the cross-linkages (both imine and disulfide bonds) allowed for dynamic dissociation and reassociation, promoting segmental motion and intrinsic self-healing capability. As a result, the SPEs demonstrated a commendable ionic conductivity of 6.97 × 10−4 S cm−1, a broad electrochemical stability window exceeding 4.9 V, good mechanical properties with a tensile strength of 0.46 MPa and failure strain of 285.8%, as well as excellent interfacial stability. The assembled Li/LiFePO4 cell showed a specific discharge capacity of 145.4 mA h g−1 (0.1C, 25 °C), and a capacity retention of 88.4% after 300 cycles. Damaged materials could be fully restored within 24 h at room temperature, as evidenced by the recovery of mechanical strength, ionic conductivity, and battery cycling performance (Fig. 9D). The concept of interlocked polymer networks has been further extended to ceramic–composite polymer electrolytes.63 In this approach, Li10GeP2S12 (LGPS) particles are incorporated into poly(ethylene oxide) (PEO)-based reversibly interlocked polymer networks, formed by topologically rearranging two PEO networks cross-linked by reversible imine bonds and disulfide linkages (Fig. 9E). This interlocking structure effectively suppresses PEO crystallization, thereby increasing the amorphous phase conducive to Li+ transport. Additionally, it stabilizes the conductive pathways of the LGPS particles through a unique confinement effect. The LGPS particles synergize with the polymer matrix to create a filler-polymer interfacial phase that further enhances Li+ transport. This phase also strengthens the composite electrolyte via strong intermolecular Li+–O2− interactions. The dynamic nature of the reversible bonds in the system provides the electrolyte with intrinsic self-healing properties. As a result, the composite electrolyte with 15 wt% LGPS and an optimized amount of LiPF6 salt achieves high ionic conductivity (1.06 × 10−3 S cm−1) and a Li+ transference number of ∼0.6 at 25 °C. It also features a wide electrochemical stability window (>4.9 V), good mechanical properties (0.63 MPa tensile strength, 377% elongation), and a stable electrolyte/Li anode interface. The performance of the integrated Li/LGPS composite solid electrolyte (CSE)/LiFePO4 battery was notable, delivering a specific discharge capacity of 110.8 mA h g−1 at 1C (25 °C) and a capacity retention of 76.9% after 200 cycles. Thanks to the material's good self-healing capabilities (restored within 24 hours at room temperature), the damaged electrolyte can regain its structural integrity, ionic conductivity, and overall cycling performance. This work demonstrates an effective strategy for fabricating ceramic composite solid electrolytes that can operate efficiently in lithium metal batteries at ambient temperature. Fang et al.64 introduced a novel soy protein isolate (SPI)-based matrix that incorporates a dynamic imine cross-linked network in conjunction with hydrogen bonding (Fig. 10A). This SPI-based vitrimer system, combined with LiTFSI as an ionic conductive filler, demonstrated high mechanical strength, reproducibility, and non-flammability. The catalyst-free, inexpensive vitrimer matrix exhibited Arrhenius-like ductility when exposed to heat and could be recycled for multiple generations. The activation energy (Ea) for the imine exchange reaction was calculated to be 21.5 kJ mol−1, suggesting that at low temperatures, the vitrimer maintains dimensional stability due to a large relaxation time, while at higher temperatures, it can respond quickly to shape editing and self-healing. The polymer powder was molded at 100 °C under 5 MPa pressure for 30 minutes, showing that the recycled material retained high mechanical strength, albeit slightly reduced from its initial values (Fig. 10A), indicating the recyclability of the catalyst-free dynamic reaction of imine. The stress–strain behavior under varying atmospheric humidity showed that the vitrimer film's flexibility improved with increasing humidity. Under 50% humidity, the vitrimer maintained a high tensile strength of 32 MPa and an elongation at break of over 250%, demonstrating excellent toughness (67.64 MJ m−3). Additionally, the material could be reshaped and recycled in water at ambient temperature, which highlights its potential for environmentally friendly processing. The interaction between the SPI matrix and the TFSI anions facilitated the transport of free Li+ ions, significantly enhancing ionic conductivity. The solid polymer electrolytes exhibited good ionic conductivity of 3.3 × 10−4 S cm−1 at 30 °C and thermal stability up to 240 °C, making them suitable candidates for applications in energy storage devices like lithium batteries. Ionic conductive elastomers were designed by integrating imine-based covalent adaptable networks with supramolecular hydrogen bonding networks.65 In this system, β-amino ester networks, formed via the aza-Michael reaction, were combined with LiTFSI salt to achieve both elasticity and dynamic reprocessability (Fig. 10B). While the dynamic β-amino ester networks were expected to possess certain elasticity and reprocessability, the introduction of lithium salts is expected to enhance the resulting material's mechanical properties and facilitate their dynamic exchange by forming ionic, hydrogen, and coordination bonds with the polymer network. The introduction of LiTFSI imparted the system with both conductivity and improved stretchability (from 67 to 231%) without significantly compromising other properties. Additionally, the dynamic exchange of β-amino ester bonds was accelerated, allowing for reprocessing at milder conditions (120 °C for 20 minutes). By fine-tuning the molar ratio of acrylate to amine and the dosage of lithium salts, the vitrimeric SPE demonstrated remarkable transparency (greater than 94%), a broad operational temperature range (−20 °C to 100 °C), and modest ionic conductivity (5.76 × 10−6 S cm−1 at 20 °C). However, when looking at the electrical properties of the recycled vitrimer, a slight increase of conductivity from 1.38 × 10−6 to 2.00 × 10−6 S cm−1 at 20 °C was observed, indicating some degradation of the vitrimeric network during reprocessing process (Fig. 10B). An innovative strategy presented by Zhao et al.66 involves the development of dual dynamic self-healing SPEs, leveraging the synergistic effects of dynamic hydrogen bonds and imine bonds (Fig. 10C). These SPEs were synthesized through a Schiff base reaction between polyethylene glycol derivatives and terephthalaldehyde at varying molar ratios. The combination of reversible imine bonds and hydrogen bonds within the polymer structure imparts rapid self-healing capabilities, allowing mechanical damage to be repaired within just 5 minutes at room temperature. This dual dynamic bonding not only enables excellent self-repairing properties but also maintains the high stretchability of the SPEs, which is crucial for accommodating the external deformation typical in flexible battery systems. Remarkably, after self-healing, the SPEs retain their original ionic conductivity, ensuring that performance remains unaffected by the repair process. In terms of battery performance, the SPE-based cell exhibits an initial discharge capacity of 149.3 mA h g−1 at a 0.1C current rate. Even after 100 cycles, the capacity remains high at 144.2 mA h g−1, showcasing excellent cycle stability. Furthermore, the SPE demonstrates good interfacial stability and a cycle life exceeding 650 hours at a current density of 0.2 mA cm−2, highlighting its potential for long-term use in high-performance batteries. Another reported self-healing polymer electrolyte was cross-linked by dual network system consisting of dynamic imine and boroxine bonds.67 This vitrimeric SPE was prepared via the reaction of 2-formylphenylboronic acid and poly(ethylene glycol) diamine (Fig. 10D). The dual dynamic network enables the SPE to undergo rapid bond exchange reactions involving both boroxine bonds with B–N coordination and imine bonds. These reactions allow for the self-healing of the electrolyte within 4 hours at room temperature (with a self-healing efficiency of 97%), all while maintaining its ionic conductivity and mechanical properties. The vitrimeric electrolyte exhibits an impressive ionic conductivity of 5.08 × 10−3 S cm−1 at 30 °C. In addition to its high conductivity, it promotes the formation of a robust LiF-rich solid electrolyte interphase and effectively suppresses lithium dendrite growth, which is a significant issue in lithium batteries. When tested in LiFePO4/Li cells, the SPE delivers excellent cycling performance with a capacity retention of 98.6% after 80 cycles, and good rate capability, achieving a specific capacity of 130.5 mA h g−1 at a 2C rate. Moreover, the electrolyte's ability to self-repair within the battery allows it to restore cell performance after damage, enhancing the overall reliability and safety of the battery system (Fig. 10D).
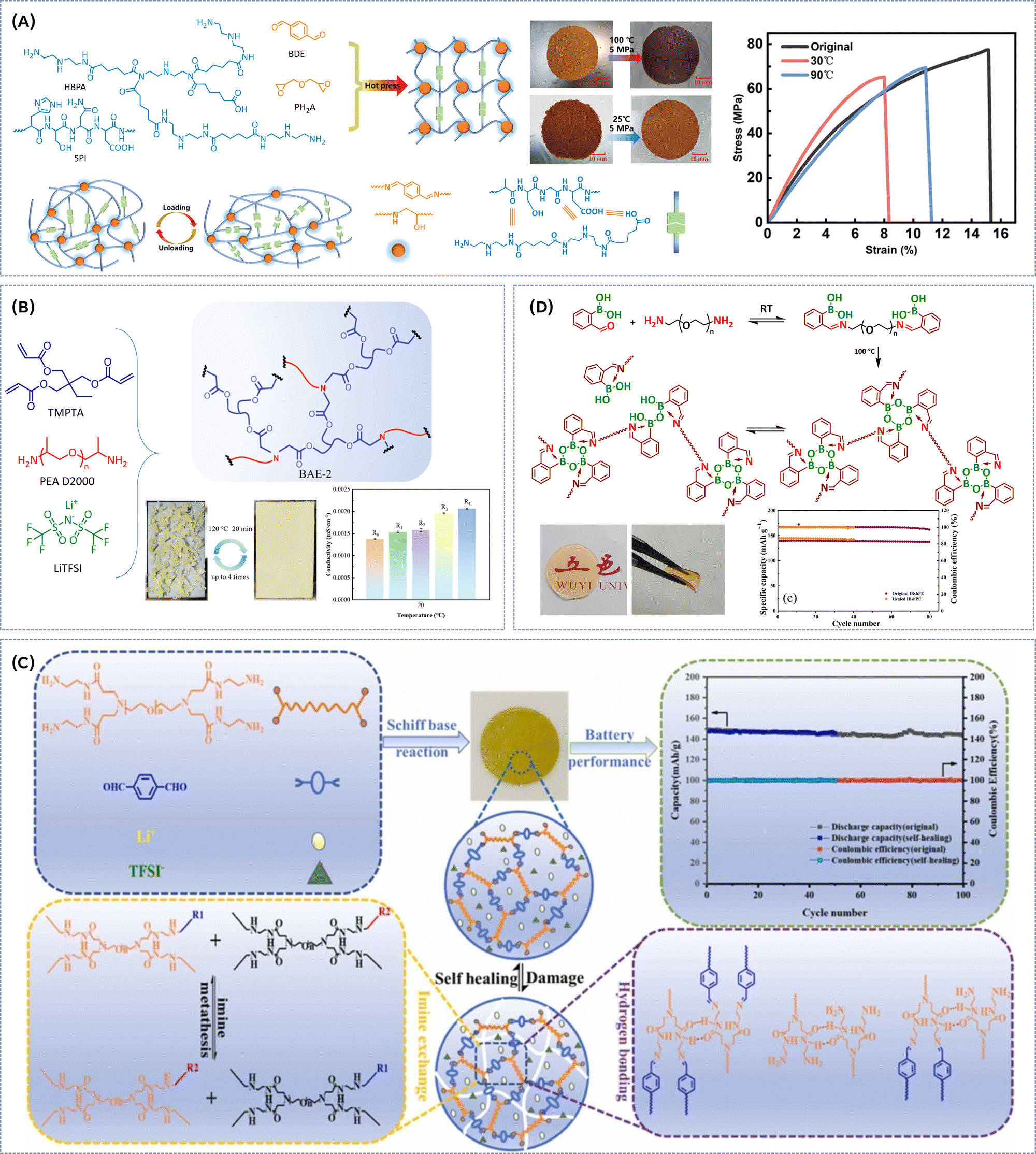 | ||
| Fig. 10 (A) Soy protein isolate (SPI)-based vitrimeric matrix, vitrimer film formed from powder by hot-pressing at 100 °C and 5 MPa for10 min and vitrimer film formed from wet powder, with the weight ratio of dry powder:water of 2:1, by pressing at 25 °C and 5 MPa for 24 h; the strain–stress curves of dry vitrimer film after immersing in warm water (30 °C) and hot water (90 °C). Reproduced with the permission from ref. 64. © 2022 The Authors. Advanced Science published by Wiley-VCH GmbH. (B) β-Amino ester networks, formed via the aza-Michael reaction and combined with LiTFSI, reprocessing and conductivity of the vitrimer after 4 reprocessing cycles. Reproduced with the permission from ref. 65. Copyright © 2024 American Chemical Society. (C) Dual dynamic self-healing SPEs, containing dynamic hydrogen bonds and imine bonds, self-healing process and corresponding battery performance. Reproduced with the permission from ref. 66. © 2023 Elsevier B.V. All rights reserved. (D) Dual network system consisting of dynamic imine and boroxine bonds, digital images of the vitrimer film and cycle performance of LFP/original IBshPE/Li cell and of LFP/healed IBshPE/Li cell at 1C rate. Reproduced with the permission from ref. 67. © 2022 Elsevier B.V. All rights reserved. | ||
The concept of dual synergistic dynamic networks indeed holds great promise for advancing next-generation solid polymer electrolytes. By varying both the polymer matrix and the types of dynamic bonds (such as hydrogen bonds, disulfide linkages, boronic ester, imine bonds etc.), this approach provides a flexible platform to address several critical challenges in battery technology: (a) enhanced safety by improving mechanical robustness and suppressing dendrite formation; (b) the incorporation of dynamic bonding enables the suppression of crystallization and creation of highly amorphous, flexible polymer networks that allow faster ion transport, even at room temperature. In turn enhancing ion conductivity in the range of commercial liquid organic electrolytes; (c) prolonged lifespan; (d) enable room temperature performance in contrast to conventional SPEs; (e) enable materials to be reprocessed or recycled under feasible conditions, making them both technically and economically viable for scalable production.
Vitrimers as binders
Si-based anode binders incorporating self-healing dynamically cross-linked polymers have been a focal point of research due to their ability to address one of the key challenges associated with Si anodes: their significant volume expansion during lithiation and delithiation, which leads to mechanical stress and capacity degradation. This type of binders creates a 3D cross-linked network with Si anodes through molecular interactions, significantly enhancing both the mechanical integrity and electrochemical performance of the electrodes. When mechanical damage occurs due to volume expansion, the self-healing polymer can repair and realign itself, restoring the damaged 3D network. This mechanism reduces performance degradation, a common issue with Si-based materials, and enhances the electrode's long-term cyclability. Additionally, the self-healing process also contributes to the formation of a stable solid electrolyte interphase layer during early-stage operation, minimizing charge and electrolyte losses and increasing the initial capacity. In the meantime, various dynamic self-healing mechanisms can be employed in Si binders, including boronic ester bonding, imine bonding, hydrogen bonding, Diels–Alder and transesterification reactions etc. Recent summaries have highlighted the various self-healing concepts for electrode binders;17,68,69 therefore, we will now shift our focus to the development of vitrimeric-based binders.Recently, cross-linked poly(acrylic acid) (PAA)-based polymer was developed as a new aqueous binder material for Si anodes.70 The binder was prepared by reacting boronic ester-grafted PAA with excess dopamine-grafted PAA, forming a reversible boronic ester bond (Fig. 11A). This dynamic bond exchange allows for self-healing and structural stability of the binder. Dopamine, incorporated into the binder, enhances adhesion with Si particles through its catechol group, improving the interfacial interaction between the Si particles and the binder. The cross-linking of PAA chains into a 3D network effectively inhibits excessive Si volume expansion during charging/discharging, ensuring structural stability. Unreacted dopamine further enhances the binder's mechanical properties and adhesive characteristics. The Lewis acid–base interactions between the boron atoms and Li salt anions contribute to uniform Li ion distribution and improved interfacial stability. Under optimal mixing conditions, a remarkable adhesion force of 3.5 N cm−1 was achieved. An anode using this binder exhibited an initial Coulombic efficiency (ICE) of 74.9% at a 0.5C charge and discharge rate. After 350 cycles, the capacity only decreased to 1755.4 mA h g−1, demonstrating a significant capacity retention of 53.7%. While Diels–Alder reactions have not been extensively explored for electrolyte applications, they offer potential for incorporating self-healing capabilities into binders for Si anodes, thus compensating volume expansion. A study demonstrated the in situ room temperature preparation of a covalently crosslinked three-dimensional hyperbranched biopolymer supramolecular network using a Diels–Alder reaction.71 This network was constructed by a Diels–Alder reaction between furan-modified branched arabinoxylan from corn fiber gum (CFG) and an ionically conductive cross-linker, maleimido-poly(ethylene glycol) (PEG) (Fig. 11B). The maleimide groups in PEG can react spontaneously with the furan groups in CFG at room temperature without any other stimulation, thus forming strong covalent bonds in the network. The cross-linked CFG–PEG binder has demonstrated robust adhesive properties with Si-active materials and the current collector. The branching of CFG and functional groups of PEG contribute to improved lithium-ion conductivity in the silicon anode, leading to enhanced rate performance. The mechanical properties of the binder can be tailored by adjusting the ratio of CFG to PEG (Fig. 11B). The resulting Si–CFG–PEG electrode exhibits a highly stable reversible capacity of 1500 mA h g−1 at a current density of 2000 mA g−1 for 200 cycles. Furthermore, the high Si mass loadings achieve a stable areal capacity at a current density of 0.5 mA cm−2 for 100 cycles.
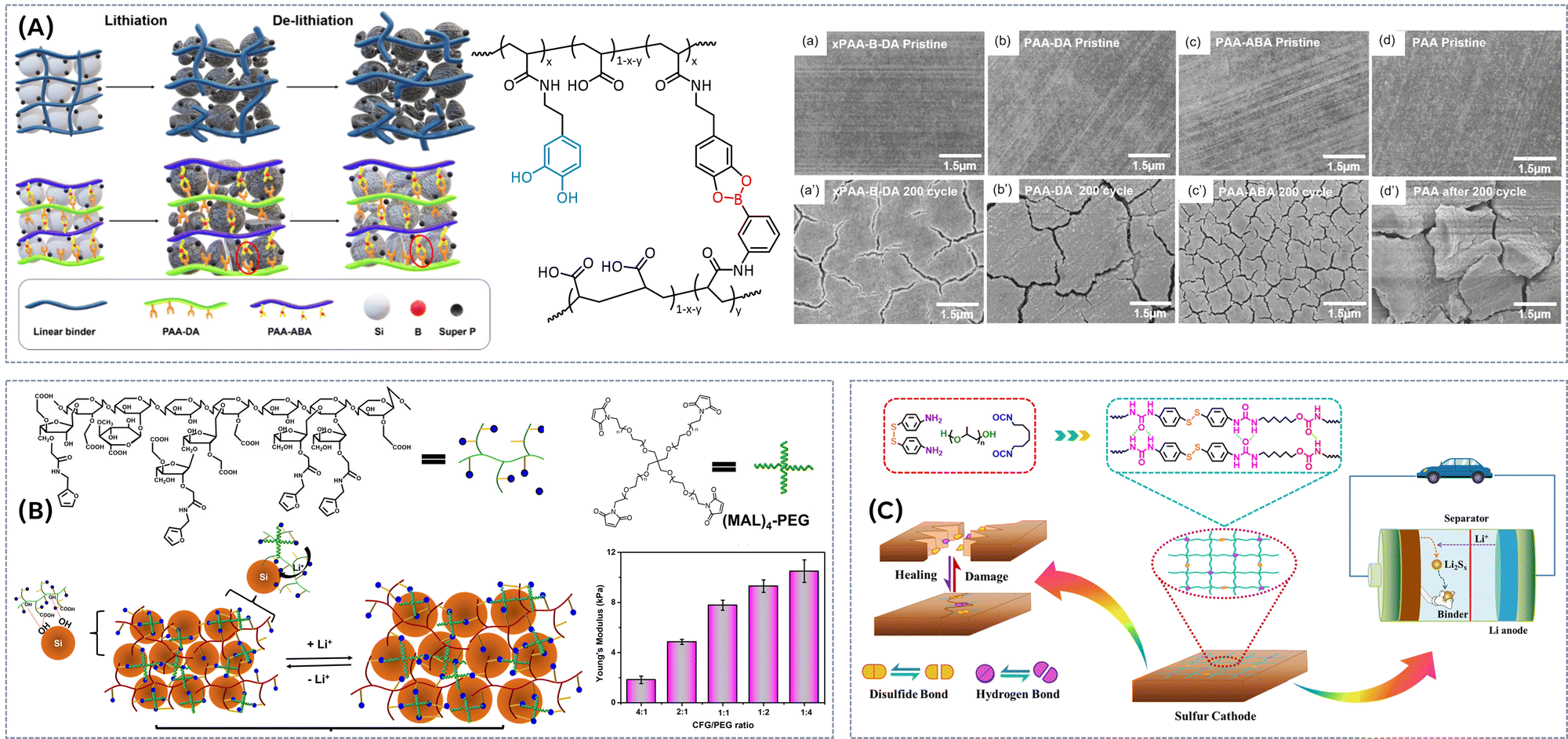 | ||
| Fig. 11 (A) Poly(acrylic acid) grafted with a boronic ester and dopamine as aqueous binder and SEM surface images of Si electrodes: (a) and (d) Si@xPAA-B-DA, Si@PAA-DA, Si@PAA-ABA, and Si@PAA, in the pristine state, and (a′)–(d′) Si@xPAA-B-DA, Si@PAA-DA, Si@PAA-ABA, and Si@PAA, after 200 cycles. Reproduced with the permission from ref. 70. Copyright © 2024 American Chemical Society. (B) Hyperbranched biopolymer supramolecular network using a Diels–Alder reaction and its use as binder for Si-anode and Young's modulus of the hydrogels with different CFG–PEG ratios. Reproduced with the permission from ref. 71. Copyright © 2021 American Chemical Society. (C) Self-healable polyurethane binder featuring a dual synergetic network containing aromatic disulfide bonds and hydrogen bonds. Reproduced with the permission from ref. 72. Copyright © 2021 American Chemical Society. | ||
Vitrimeric binders are not limited to applications in Si-anodes; they have also shown promise in other battery systems, such as Li–sulfur (Li–S) batteries. An example of this is a self-healable polyurethane binder featuring a dual synergetic network containing aromatic disulfide bonds and hydrogen bonds, which has been successfully applied to sulfur cathodes in Li–S batteries (Fig. 11C).72 The binder's self-healing properties allow it to autonomously repair microcracks caused by volume changes during battery operation, ensuring a structurally integrated sulfur cathode. Additionally, the presence of urethane, disulfide, and urea groups effectively adsorbs polysulfides, mitigating the “shuttling effect”, which arises due to the uncontrollable diffusion of soluble intermediate lithium polysulfides in the organic electrolyte, and improving the cycle stability of Li–S batteries. Li–S batteries using this vitrimeric binder exhibit a high electrochemical capacity of 1269.7 mA h g−1 and a low fading rate of 0.04% per cycle at 2C for 500 cycles. This issue of “shuttle effect” can be significantly mitigated by using an adaptive polymer layer (rGO–putty), composed of reduced graphene oxide nanosheets and silly putty (SP)—a polysilicone lightly crosslinked by dynamic boronic ester bonds (Fig. 12A).73 The dynamic network within this polymer layer not only suppresses the “shuttle effect”, but also spontaneously repairs mechanical and structural damages within the cathode during cycling. Specifically, the diffusion of polysulfides is effectively reduced when rGO–putty is applied, thanks to its adaptable structure (Fig. 12A). Over the course of several cycles, the rGO–putty gradually infuses into the gaps between active material particles, tightly adhering to them. This results in the formation of a thin, conductive, and flexible protective layer that seals cracks and maintains the cathode's structural integrity. The viscoelasticity of rGO–putty is key to this process, as it enables the formation of a robust, continuous layer that preserves the multidirectional electric pathways crucial for battery performance. Thanks to this innovative design, the sulfur cathode maintains a compact structure with an ultrathin conductive rGO–putty coating, which stabilizes the electrode and enhances electrochemical performance. The use of this rGO–putty layer in Li–S batteries has demonstrated impressive results, with a capacity of 586 mA h g−1 after 1000 cycles at 1C, and a remarkable cycling stability with only a 0.03% capacity decay per cycle.
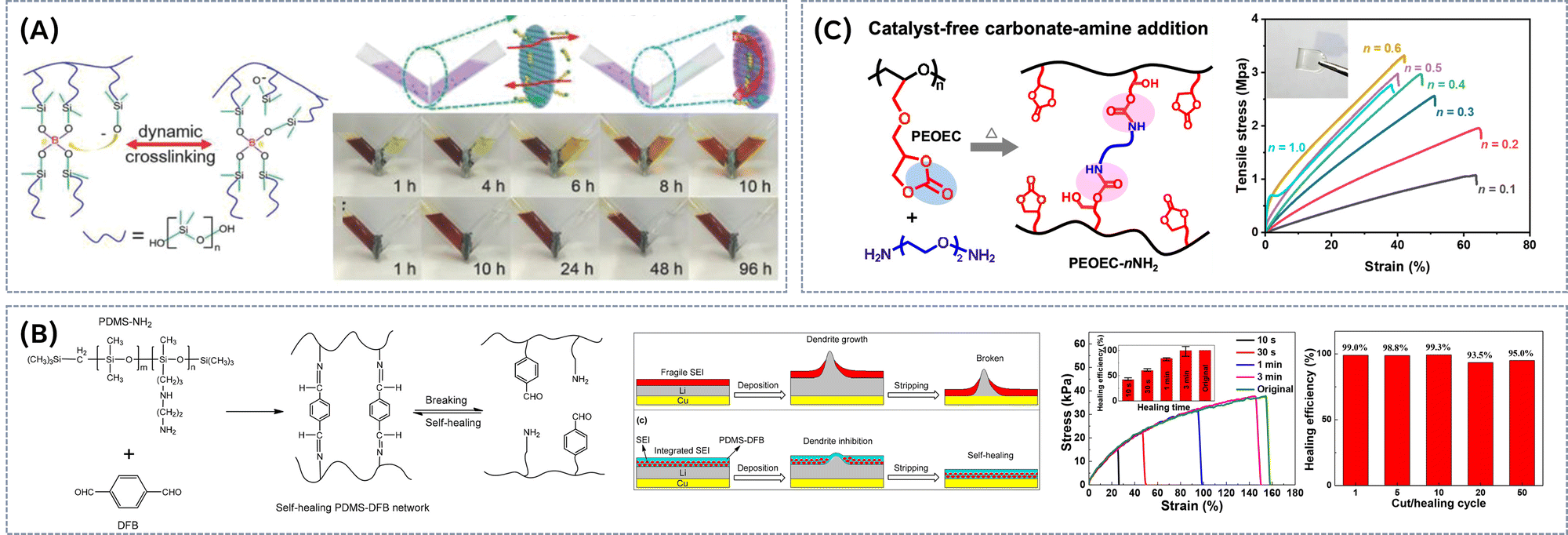 | ||
| Fig. 12 (A) Molecular structure of silly-putty nanocomposite and the photos of the polysulfides diffusion experiment of the PP and rGO–putty separators. Reproduced with the permission from ref. 73. © 2018 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. (B) Integration of the SEI layer into a self-healing polydimethylsiloxane (PDMS) network, cross-linked via dynamic imine bonding, (b) fragile SEI layer and (c) integrated SEI layer during the Li deposition/stripping process, stress–strain curves of the elastomer at different healing stages, inset is the mechanical healing efficiency calculated from the curves and mechanical healing efficiency of the elastomer after multiple cut/healing cycles. Reproduced with the permission from ref. 74. Copyright © 2018 American Chemical Society. (C) Polymer-in-salt electrolytes, which consist of a polyether backbone with pendant cyclic carbonates synthesized through a carbonate–amine reaction and stress–strain curves of the PEOEC-nNH2 samples with different cross-linking densities. The inset is the photograph of the flexible PEOEC-0.2NH2 film. Reproduced with the permission from ref. 75. Copyright © 2024 American Chemical Society. | ||
The primary drawback of Li-metal anodes, limiting their practical application, is their poor stability due to fragile solid electrolyte interphase layers. These layers are not adaptable to the dynamic volume changes that occur during cycling, nor are they self-healing after damage, which leads to performance degradation. Vitrimeric materials can be applied to stabilize interface layer, while adapting to and/or compensating volume changes. One example of this approach involves the in situ integration of the SEI layer into a self-healing polydimethylsiloxane (PDMS) network, cross-linked via dynamic imine bonding (Fig. 12B).74 This self-healing network enables the SEI layer to adapt to the volumetric fluctuations of the Li-metal anode and repair itself after mechanical damage. Remarkably, the elastomer can restore over 84% of its original tensile stress and strain within just 1 minute, with a maximum mechanical healing efficiency of 99.53% achieved after three minutes. This fast and efficient restoration of mechanical properties was sustained over at least 50 cycles, with an average healing efficiency exceeding 95% (Fig. 12B). As a result, detrimental electrolyte decomposition and nonuniform Li deposition is significantly depressed for the resulting Li anode, leading to excellent cycling stability and a dendrite-free morphology. In a Li/LiFePO4 full cell, this strategy leads to capacity retention up to 99% and a Coulombic efficiency >99.5% after 300 cycles. This investigation highlights a novel self-healing approach for developing stable Li-metal anodes, with the potential to significantly enhance the performance and lifespan of high energy-density batteries. Another approach to stabilizing lithium metal surfaces involves the use of elastomeric polymer-in-salt electrolytes, which consist of a polyether backbone with pendant cyclic carbonates synthesized through a carbonate–amine reaction (Fig. 12C).75 The pendant cyclic carbonate moieties, analogous to the classic carbonate liquid electrolytes, can host lithium salts to a high concentration (up to 70%) on the one hand; on the other hand, they can function as reactive sites toward diamine cross-linkers. This simple and additive-free method enables the in situ integration of the polymer electrolyte with the cathode, where the vitrimeric polymer also acts as an ion-conductive binder. The resulting in-built flexible elastomers exhibit enhanced mechanical stability (up to 3.5 MPa), favorable interfacial compatibility, and a high lithium-ion transference number of up to 0.72. Lithium ions are found to interact primarily with the ether segments of the polymer at lower salt concentrations, while at higher salt concentrations, nearly all cyclic carbonates and TFSI counterions participate in lithium-ion coordination. Notably, as the salt concentration increases, the proportion of the cis-conformation of TFSI− rises compared to the trans-conformation. This shift in ion coordination enhances both the ionic conductivity and lithium-ion transference number in the polymer-in-salt system. This integrated composite cathode/electrolyte strategy enables a compact interfacial contact and thus facilitates ion conduction across the boundary, eventually leading to all-solid-state LiFePO4/Li cells with a prolonged cycling stability and a high cathode areal capacity at ambient temperature. The all-solid-state Li/LFP cell has demonstrated a potential of stable operation at 1C for more than 2000 cycles at room temperature, as well as a high capacity beyond 1.0 mA h cm−2.
Although research on the application of vitrimers for stabilizing electrode materials is still in its early stages, adaptive vitrimeric polymers hold significant potential to revolutionize the field. By stabilizing electrode surfaces and compensating for volume changes during battery operation, these materials could pave the way for the development of long-lasting, high-performance, and recyclable battery systems. Their unique ability to undergo dynamic bond exchange offers a promising approach to enhancing both the durability and sustainability of next-generation energy storage devices.
Conclusions and outlook
In summary this article highlights the strategies and potential of incorporating vitrimeric features into solid polymer electrolytes. While research in this field is still in its early stages, the integration of dynamic covalent bonds shows great promise for creating self-healing, reprocessable, and recyclable materials with enhanced properties. These advancements could significantly improve battery performance, address safety concerns, and extend the lifespan of commercial energy storage systems. Future developments in this area are expected to lead to more efficient and environmentally sustainable energy storage solutions, tackling both recyclability and pollution challenges. However, to make these materials commercially viable and suitable for large-scale industrial applications, several key challenges must still be overcome (Fig. 13): | ||
| Fig. 13 Schematically displayed challenges, opportunities and perspectives of incorporation vitrimeric materials into Li-ion batteries. | ||
(a) Dynamic network behavior: the covalent network dynamics must enable self-healing at room temperature or, at a minimum, within the operational range of lithium-ion batteries (approximately 60 °C). Achieving effective self-healing at these temperatures will be critical to ensuring the practicality and durability of these materials.
(b) Balancing mechanical integrity and conductivity: a focus on the trade-off between mechanical strength and ionic conductivity is essential. These materials must provide high ion transport while also stabilizing the solid electrolyte interface and accommodating volume changes. Dual dynamic networks, which combine different bonding mechanisms, offer a promising approach to achieve this balance.
(c) Real-life applicability: systematic investigation of the mechanical integrity, self-healing ability, and reprocessing properties under practical conditions is needed. It is crucial to evaluate how these materials perform in real-world applications to ensure their durability and reliability.
Vitrimeric polymer electrolytes and binders represent a promising technology for next-generation, high-performance, sustainable battery systems. However, not all proposed chemistries are suitable for real-life applications, making it necessary for researchers to continue exploring the synergistic relationship between vitrimeric materials and battery components.
Author contributions
Conceptualization, A. M., W. H. B. and Z. K.; methodology, A. M., W. H. B. and Z. K.; writing – original draft preparation, A. M. and Z. K.; writing – review and editing, W. H. B., A. M. and Z. K.; supervision, project administration, and funding acquisition, W. H. B. and A. M. All authors have read and agreed to the published version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful to the DFG project INST 271/444-1 FUGG for financial support; the DFG-Project BI1337/18-1; BI1337/17-1; BI1337/16-1; BI 1337/14-1 and the GRK 2670, W69000789, ProjectNr 436494874, TP B=1. W. H. B. and Z. K. thank the European Center of Just Transition Research and Impact-Driven Transfer (JTC).Notes and references
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Electrochem. Energy Rev., 2019, 2, 1–28 CrossRef CAS.
- A. Masias, J. Marcicki and W. A. Paxton, ACS Energy Lett., 2021, 6, 621–630 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- Y. Tian, G. Zeng, A. Rutt, T. Shi, H. Kim, J. Wang, J. Koettgen, Y. Sun, B. Ouyang, T. Chen, Z. Lun, Z. Rong, K. Persson and G. Ceder, Chem. Rev., 2021, 121, 1623–1669 CrossRef CAS PubMed.
- E. Fan, L. Li, Z. Wang, J. Lin, Y. Huang, Y. Yao, R. Chen and F. Wu, Chem. Rev., 2020, 120, 7020–7063 CrossRef CAS PubMed.
- Y. Chen, Y. Kang, Y. Zhao, L. Wang, J. Liu, Y. Li, Z. Liang, X. He, X. Li, N. Tavajohi and B. Li, J. Energy Chem., 2021, 59, 83–99 CrossRef CAS.
- X. Feng, M. Ouyang, X. Liu, L. Lu, Y. Xia and X. He, Energy Storage Mater., 2018, 10, 246–267 CrossRef.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- B. Zhu, X. Wang, P. Yao, J. Li and J. Zhu, Chem. Sci., 2019, 10, 7132–7148 RSC.
- L. Sun, Y. Liu, R. Shao, J. Wu, R. Jiang and Z. Jin, Energy Storage Mater., 2022, 46, 482–502 CrossRef.
- H. Adenusi, G. A. Chass, S. Passerini, K. V. Tian and G. Chen, Adv. Energy Mater., 2023, 13, 2203307 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- H. Bae and Y. Kim, Mater. Adv., 2021, 2, 3234–3250 RSC.
- Q. Wang, L. Jiang, Y. Yu and J. Sun, Nano Energy, 2019, 55, 93–114 CrossRef CAS.
- M. Li, C. Wang, Z. Chen, K. Xu and J. Lu, Chem. Rev., 2020, 120, 6783–6819 CrossRef CAS PubMed.
- A. Marinow, Z. Katcharava and W. H. Binder, Polymers, 2023, 15, 1145 CrossRef CAS PubMed.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- A. Campanella, D. Döhler and W. H. Binder, Macromol. Rapid Commun., 2018, 39, 1700739 CrossRef PubMed.
- R. Narayan, C. Laberty-Robert, J. Pelta, J.-M. Tarascon and R. Dominko, Adv. Energy Mater., 2022, 12, 2102652 CrossRef CAS.
- W. Mai, Q. Yu, C. Han, F. Kang and B. Li, Adv. Funct. Mater., 2020, 30, 1909912 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- B. Krishnakumar, R. V. S. P. Sanka, W. H. Binder, V. Parthasarthy, S. Rana and N. Karak, J. Chem. Eng., 2020, 385, 123820 CrossRef CAS.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- J. Zheng, Z. M. Png, S. H. Ng, G. X. Tham, E. Ye, S. S. Goh, X. J. Loh and Z. Li, Mater. Today, 2021, 51, 586–625 CrossRef CAS.
- V. Schenk, K. Labastie, M. Destarac, P. Olivier and M. Guerre, Mater. Adv., 2022, 3, 8012–8029 RSC.
- J. Luo, Z. Demchuk, X. Zhao, T. Saito, M. Tian, A. P. Sokolov and P.-F. Cao, Matter, 2022, 5, 1391–1422 CrossRef CAS.
- B. B. Jing and C. M. Evans, J. Am. Chem. Soc., 2019, 141, 18932–18937 CrossRef CAS PubMed.
- S. Li, C. Zuo, Y. Zhang, J. Wang, H. Gan, S. Li, L. Yu, B. Zhou and Z. Xue, Polym. Chem., 2020, 11, 5893–5902 RSC.
- Z. Katcharava, X. Zhou, R. Bhandary, R. Sattler, H. Huth, M. Beiner, A. Marinow and W. H. Binder, RSC Adv., 2023, 13, 14435–14442 RSC.
- B. Zhou, T. Deng, C. Yang, M. Wang, H. Yan, Z. Yang, Z. Wang and Z. Xue, Adv. Funct. Mater., 2023, 33, 2212005 CrossRef CAS.
- S. Zhou, X. Wang, Z. Xu, T. Guan, D. Mo and K. Deng, J. Energy Storage, 2024, 75, 109712 CrossRef.
- F. Li, G. T. M. Nguyen, C. Vancaeyzeele, F. Vidal and C. Plesse, ACS Appl. Polym. Mater., 2023, 5, 529–541 CrossRef CAS PubMed.
- L. Zhang, P. Zhang, C. Chang, W. Guo, Z. H. Guo and X. Pu, ACS Appl. Mater. Interfaces, 2021, 13, 46794–46802 CrossRef CAS PubMed.
- X. Xue, X. Cao, L. Wan, Y. Tong, T. Li and Y. Xie, Polym. Int., 2022, 71, 1201–1209 CrossRef CAS.
- K. Deng, S. Zhou, Z. Xu, M. Xiao and Y. Meng, J. Chem. Eng., 2022, 428, 131224 CrossRef CAS.
- G. Lopez, L. Granado, G. Coquil, A. Lárez-Sosa, N. Louvain and B. Améduri, Macromolecules, 2019, 52, 2148–2155 CrossRef CAS.
- X. Wang, M. Wang, H. Chen, Y. Zhang, B. Niu, L. Tian and D. Long, J. Chem. Eng., 2024, 496, 153830 CrossRef CAS.
- Y. Wang, Z. Wang, B. Jin, D. Ye, W. Fan and X. Ye, Eur. Polym. J., 2023, 193, 112098 CrossRef CAS.
- J. Liu, J.-J. Li, Z.-H. Luo and Y.-N. Zhou, J. Chem. Eng., 2023, 452, 139452 CrossRef CAS.
- S. Jang, E. I. Hernandez Alvarez, C. Chen, B. B. Jing, C. Shen, P. V. Braun, A. Schleife, C. M. Schroeder and C. M. Evans, Chem. Mater., 2023, 35, 8039–8049 CrossRef CAS.
- Y. Lin, Y. Chen, Z. Yu, Z. Huang, J.-C. Lai, J. B. H. Tok, Y. Cui and Z. Bao, Chem. Mater., 2022, 34, 2393–2399 CrossRef CAS.
- S. Yang, S. Park, S. Kim and S.-K. Kim, Mater. Today Energy, 2024, 45, 101690 CrossRef CAS.
- Z. Sun, J. Wu, H. Yuan, J. Lan, Y. Yu, Y. Zhu and X. Yang, Mater. Today Energy, 2022, 24, 100939 CrossRef CAS.
- R. Kato, P. Mirmira, A. Sookezian, G. L. Grocke, S. N. Patel and S. J. Rowan, ACS Macro Lett., 2020, 9, 500–506 CrossRef CAS PubMed.
- F. Li, G. T. M. Nguyen, C. Vancaeyzeele, F. Vidal and C. Plesse, RSC Adv., 2023, 13, 6656–6667 RSC.
- S. Ullah, H. Wang, G. Hang, T. Zhang, L. Li and S. Zheng, Polymer, 2023, 284, 126318 CrossRef CAS.
- X. Ma, J. Yu, Y. Hu, J. Texter and F. Yan, Ind. Chem. Mater., 2023, 1, 39–59 RSC.
- Z. Katcharava, T. E. Orlamünde, L. T. Tema, H. Hong, M. Beiner, B. Iliev, A. Marinow and W. H. Binder, Adv. Funct. Mater., 2024, 34, 2403487 CrossRef CAS.
- B. Kim, Y. J. Cho, D. G. Kim and J. H. Seo, Mater. Today Chem., 2023, 30, 101583 CrossRef CAS.
- X. Zhou, C. Li, R. Bhandary, Z. Katcharava, F. Du, R. Androsch, A. Marinow and W. H. Binder, ACS Appl. Eng. Mater., 2023, 1, 1997–2003 CrossRef CAS.
- F. Pei, L. Wu, Y. Zhang, Y. Liao, Q. Kang, Y. Han, H. Zhang, Y. Shen, H. Xu, Z. Li and Y. Huang, Nat. Commun., 2024, 15, 351 CrossRef CAS PubMed.
- Y. Huang, Z. Shi, H. Wang, J. Wang and Z. Xue, Energy Storage Mater., 2022, 51, 1–10 CrossRef.
- Y. H. Jo, S. Li, C. Zuo, Y. Zhang, H. Gan, S. Li, L. Yu, D. He, X. Xie and Z. Xue, Macromolecules, 2020, 53, 1024–1032 CrossRef CAS.
- C. Li, R. Bhandary, A. Marinow, D. Ivanov, M. Du, R. Androsch and W. H. Binder, Polymers, 2022, 14, 4090 CrossRef CAS PubMed.
- C. Li, R. Bhandary, A. Marinow, S. Bachmann, A.-C. Pöppler and W. H. Binder, Macromol. Rapid Commun., 2024, 45, 2300464 CrossRef CAS PubMed.
- A. Le Mong and D. Kim, J. Mater. Chem. A, 2023, 11, 6503–6521 RSC.
- J. Chen, Y. Gao, L. Shi, W. Yu, Z. Sun, Y. Zhou, S. Liu, H. Mao, D. Zhang, T. Lu, Q. Chen, D. Yu and S. Ding, Nat. Commun., 2022, 13, 4868 CrossRef CAS PubMed.
- W. Lei, X. Jiao, S. Yang, F. B. Ajdari, M. Salavati-Niasari, Y. Feng, J. Yin, G. Ungar and J. Song, Energy Storage Mater., 2022, 49, 502–508 CrossRef.
- Y. Jiang, K. Chen, J. He, Y. Sun, X. Zhang, X. Yang, H. Xie and J. Liu, J. Colloid Interface Sci., 2025, 678, 200–209 CrossRef CAS PubMed.
- J. Lyu, G. Song, H. Jung, Y. I. Park, S.-H. Lee, J.-E. Jeong and J. C. Kim, ACS Appl. Mater. Interfaces, 2024, 16, 1511–1520 CrossRef CAS PubMed.
- Z. X. Huang, Z. H. Xie, Z. P. Zhang, T. Zhang, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2022, 10, 18895–18906 RSC.
- Z. X. Huang, T. Zhang, Z. P. Zhang, M. Z. Rong and M. Q. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 42736–42747 CrossRef CAS PubMed.
- W. Gu, F. Li, T. Liu, S. Gong, Q. Gao, J. Li and Z. Fang, Adv. Sci., 2022, 9, 2103623 CrossRef CAS PubMed.
- Y. Aierken, Y. Xu, S. Xiang, W. Peng, X. Zhang, M. Hakkarainen and P. Ma, ACS Appl. Mater. Interfaces, 2024, 16, 25374–25384 CrossRef CAS PubMed.
- L. Wan, X. Xue, X. Du, X. Tan, Y. Tong, Y. Qin, H. Huang, D. Zhou, Y. Xie and J. Zhao, Colloids Surf., A, 2023, 675, 132046 CrossRef CAS.
- S. Zhou, K. Deng, Z. Xu, M. Xiao and Y. Meng, J. Chem. Eng., 2022, 442, 136083 CrossRef CAS.
- F. Boorboor Ajdari, F. Abbasi, A. Molaei Aghdam, F. Ghorbani Chehel Khaneh, A. Ghaedi Arjenaki, V. Farzaneh, A. Abbasi and S. Ramakrishna, Mater. Sci. Eng., R, 2024, 160, 100830 Search PubMed.
- J. Xu, C. Ding, P. Chen, L. Tan, C. Chen and J. Fu, Appl. Phys. Rev., 2020, 7, 031304 CAS.
- Y. Kang, S. Kim, J. Han, T. Yim and T.-H. Kim, ACS Appl. Energy Mater., 2024, 7, 2436–2450 CrossRef CAS.
- Z. Cai, S. Hu, Y. Wei, T. Huang, A. Yu and H. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 56095–56108 CrossRef CAS PubMed.
- X. Zhang, P. Chen, Y. Zhao, M. Liu and Z. Xiao, Ind. Eng. Chem. Res., 2021, 60, 12011–12020 CrossRef CAS.
- Z. Chang, Y. He, H. Deng, X. Li, S. Wu, Y. Qiao, P. Wang and H. Zhou, Adv. Funct. Mater., 2018, 28, 1804777 CrossRef.
- X. Cui, Y. Chu, L. Qin and Q. Pan, ACS Sustainable Chem. Eng., 2018, 6, 11097–11104 CrossRef CAS.
- D. You, M. A. Farooq, Z. Lai, W. Wei and H. Xiong, Macromolecules, 2024, 57, 4471–4483 CrossRef CAS.
Footnote |
| † Z. K. and A. M. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |