
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defluorinative thio-functionalization: direct synthesis of methyl-dithioesters from trifluoromethylarenes†
Marcus
Söderström
a,
Esther
Olaniran Håkansson
b and
Luke R.
Odell
 *b
*b
aThe Beijer Laboratory, Department of Medicinal Chemistry, Uppsala University, 751 23 Uppsala, Sweden
bDepartment of Medicinal Chemistry, Uppsala University, 751 23 Uppsala, Sweden. E-mail: luke.odell@ilk.uu.se
First published on 25th November 2024
Abstract
A new functional group transformation allowing the synthesis of methyl-dithioesters from readily available trifluoromethyl arenes via defluorinative functionalization has been developed. This microwave-assisted method is operationally simple, rapid, and eliminates the need for pre-functionalization while accommodating a broad range of functional groups. In addition, it does not rely on highly odorous thiol sources, and utilizes the commercially available reagent BF3SMe2 complex as a multifunctional Lewis acid/sulfur source/defluorination and demethylation agent. Finally, this approach is suitable for late-stage functionalizations, as shown by the transformation of pharmaceuticals leflunomide, flufenamic acid and celecoxib into novel methyl-dithioester derivatives.
Trifluoromethyl (CF3)-substituted arenes are common structural motifs in many biologically active molecules as the CF3 substituent often has a profound influence on their biological, chemical and physical characteristics.1 As a consequence, CF3 groups can be found in a number of drugs such as celecoxib, flufenamic acid and leflunomide.
In addition to its interesting properties, the CF3-group has found increasing utility as a latent synthetic handle. Despite the high activation energy of the C–F bond posing a significant synthetic challenge, recent advances have allowed the group to be seen as a precursor for a range of different transformations.2 These approaches include Lewis acids activation,2c,d transition metal catalysis2e and photo/electrochemical acitivation.2f However, the incorporation of sulfur nucleophiles has only rarely been reported3 and, moreover, these methods all require the use of highly odorous thiols or sulfides,3a specialized substrates3b complex reaction conditions3c (Fig. 1), or report very few examples of sulfur transformations.3d–g We have previously disclosed the use of BF3SMe2 complex as a convenient and non-odorous Lewis acid and thiomethyl source.4 Noting that many CF3 functionalization approaches hinge on a Lewis acid/nucleophile combination, we speculated that BF3SMe2 might also promote C–F activation and subsequent thiomethyl incorporation. Given the propensity for the CF3-group to undergo multiple defluorination events,2 a key potential issue was identifying selective reaction conditions.
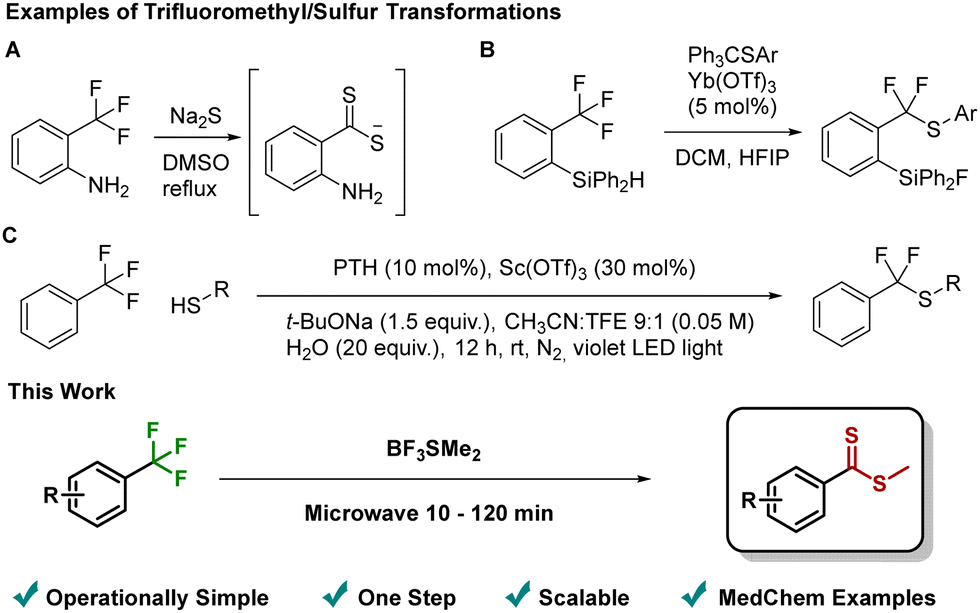 | ||
| Fig. 1 Top: Examples of CF3/sulfur transformations: (A) sulfur insertion into activated CF3-group3a; (B) substrate assisted C–F activation3b; (C) Lewis acid promoted, photo-redox catalyzed C–F activation.3c Bottom: This work. | ||
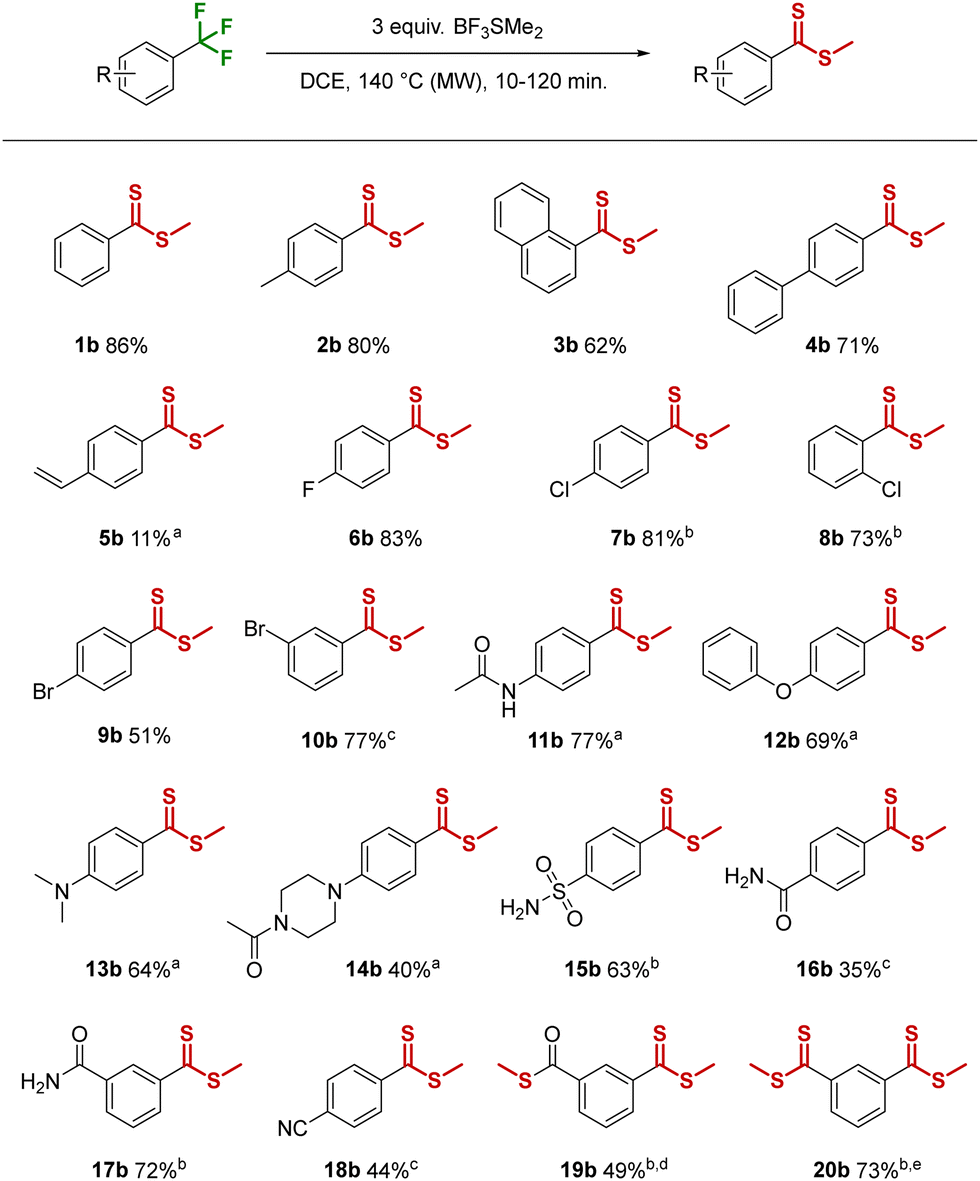
Pleasingly, when trifluorotoluene (1a) was treated with an excess of BF3SMe2 at elevated temperatures, a single main product was formed that was ultimately identified as the methyl-dithioester 1b (Table 1). Importantly, the transformation of an aryltrifluoromethyl group into a dithioester is, to the best of our knowledge, unknown in the literature.
The methyl-dithioester group is a versatile synthetic intermediate that has been employed in the synthesis of a variety of heterocycles,5 as a heterodieneophile,6 and a key step in the synthesis of a shikimic acid derivative7 and the potassium channel activator aprikalim.8 Despite the versatility of the methyl-dithioester moiety, there are very few convenient procedures for its synthesis. Existing methods all require either the use of complex9 or expensive10 reagents, sensitive organometallic precursors and toxic CS2,11 malodourous H2S12 or laborious multistep syntheses.13 Therefore, the development of a novel, cheap and straightforward synthesis of this important functional group would be a valuable addition to the synthetic toolbox. Furthermore, a route from the relatively inert trifluoromethyl-moiety to the versatile dithioester-group would allow the selective downstream insertion of various functional groups in the late stages of a multistep synthesis.14
Herein we report the discovery and development of the first defluorinative synthesis of methyl-dithioesters from readily available trifluoromethyl arenes. Furthermore, this method was exemplified through the late-stage modification of drug molecules, highlighting its utility in real world applications.
Our initial investigation of the reaction conditions, using 1a as model substrate, quickly revealed that elevated temperatures were advantageous, and heating at 120 °C for 1 h with 3 equivalents of BF3SMe2 afforded 74% of dithioester 1b (Table S1, ESI†). A microwave reactor was employed to ensure rapid heating and pressure control, however conventional heating at lower temperature (80 °C) was also possible, but increased the reaction time significantly and resulted in lower yields. Screening different solvent conditions (neat, i-hexane, toluene) revealed DCE as the solvent of choice (83%), and a screen of temperature and time gave the optimal combination of 140 °C and 30 min. Finally, lowering the amount of Lewis acid (1.5 equiv. BF3SMe2 and 1.5 equiv. SMe2) proved detrimental, resulting in incomplete conversion and only 30% isolated yield. With the optimized conditions in hand (3 equiv. BF3SMe2, DCE, 140 °C, 30 min) we proceeded to investigate the scope and functional group tolerance of the reaction (Table 1).
Toluene, naphthalene and 4-phenyl trifluoro substrates were well tolerated and produced methyl-dithioesters 2b, 3b and 4b in 80%, 62% and 71% yield respectively. The presence of a 4-vinylbenzene group led to significant side-product formation/degradation, although with a 10 min reaction time the desired product 5b could be isolated in 11% yield. 4-Fluoro and 4-chlorobenzotrifluoride were smoothly converted to the methyl-thioesters 6b and 7b, and a chloro-group in the ortho-position resulted in only a slight decrease in the yield of 8b. 4-Bromobenzotrifluoride resulted in a moderate yield of 9b (51%), due to competing thiomethyl substitution of the bromo group (9b′, 7% isolated yield, see Scheme S1, ESI†). However, no traces of bromo-substitution were observed using 3-bromobenzotrifluoride and the expected product 10b was isolated in good yield (77%), although a longer reaction time (2 h) was required.
In contrast, the more electron-rich 4-(trifluoromethyl)acetanilide required only 10 min to reach full conversion, affording 77% of the desired product 11b. The same trend was observed for 1-phenoxy-4-(trifluoromethyl)benzene and N,N-dimethyl-4-(trifluoromethyl)aniline affording 69% and 64% of 12b and 13b respectively after 10 min reaction time. A free NH-group in 1-(4-(trifluoromethyl)phenyl)piperazine was unfortunately not tolerated (see Scheme S2 for unsuccessful substrates, ESI†), resulting in an insoluble black solid. This could be overcome by acetyl-protection and the corresponding target acetamide product 14b was isolated in 40% yield.
Turning to more electron-deficient substrates, 4-(trifluoromethyl)benzenesulfonamide gave the dithioester 15b in a moderate yield of 63%. The reaction of 4-(trifluoromethyl)benzamide was rather sluggish and led to a modest yield of 16b (35%). However, moving the position of the electron withdrawing group with 3-(trifluoromethyl)benzamide changed the reactivity drastically, affording 72% of 17b after 60 min. The presence of a strong electron withdrawing group in 4-(trifluoromethyl)benzonitrile afforded a moderate 44% yield of 18b after 2 h of heating. Electron deficient heteroaromatic pyridines (2-(trifluoromethyl)- and 3-(trifluoromethyl)pyridine) were found to be completely inert under the reaction conditions, returning only unreacted starting material.
3-(Trifluoromethyl)benzoic acid, in addition to methyl-dithioester formation, also resulted in OH-substitution to form the thioester 19b in 26% yield, which could be improved to 49% by increasing the amount of BF3SMe2. Finally, 1,3-bis(trifluoromethyl)benzene was also successfully converted into the 1,3-dithioester 20b in good yield (73%).
Potential halide selectivity was explored by reacting 1-(trichloromethyl)-3-(trifluoromethyl)benzene under the conditions. Interestingly, both functional groups were transformed into methyl dithioesters and the 1,3-bis methyl dithioester 20b was isolated in 82% yield. This demonstrates that the trichloromethyl moiety is also a viable substrate for direct methyl-dithioester synthesis.
The scalability of this method was also explored, and the conditions were applied on a 5 mmol scale using trifluorotoluene (1a). Pleasingly, no reduction in yield was observed with the methyl-dithioester 1b isolated in 85% yield, highlighting the utility of this method on a preparative scale.
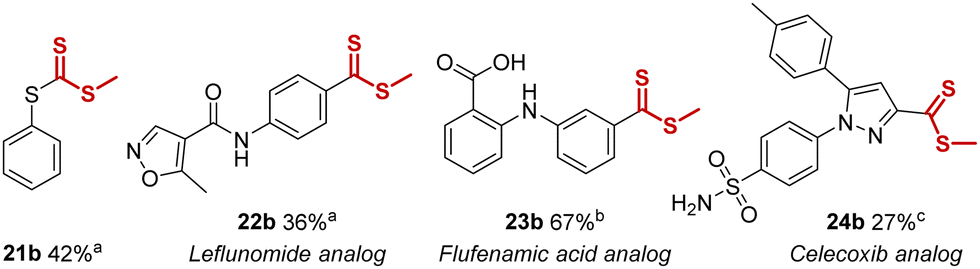
During our exploration of non-aromatic trifluoromethyl-groups, we found (trifluoromethoxy)benzene and (2,2,2-trifluoroethyl)benzene to be unsuitable for this method due to competing side-reactions (e.g. demethylation). In contrast, trifluoromethylthiobenzene was found to be a productive substrate, leading to formation of the rarely encountered trithiocarbonate functional group, and 21b was isolated in 42% yield (Table 2). Notably, this is the first trithiocarbonate synthesis that does not require odorous sulfides or activated thiocarbonyl substrates. Furthermore, the method could also be applied to the late-stage functionalization of the antirheumatic drug leflunomide and the nonsteroidal anti-inflammatory drugs flufenamic acid and celecoxib. All three substrates were smoothly converted into novel methyl-dithioester analogs 22b, 23b and 24b in useful yields of 36%, 67% and 27% respectively, representing interesting late-stage transformations (Table 2).
The mechanism of the defluorination/thioester synthesis is thought to proceed via an initial defluorination of the trifluoromethyl group by BF3, followed by attack from SMe2, resulting in intermediate I (Scheme 1). The higher reactivity of electron-rich substrates suggest that this occurs via an SN1-type mechanism in accordance with other Lewis acid mediated defluorination processes.15 Following this, demethylation of the thionium ion occurs to give II, followed by abstraction of a second fluorine, resulting in intermediate III. Intermediate III should be highly electrophilic, and react readily with a second SMe2 molecule, which after demethylation results in the fluoro-thioacetal intermediate IV. Alternatively, intermediate IV can also be formed by an SN2-type attack by SMe2 on intermediate II followed by demethylation (Scheme 1, dotted line). Intermediate IV is then converted into Vvia a third defluorination and this is followed by a final demethylation to form the methyl-dithioester product, compound 1b.
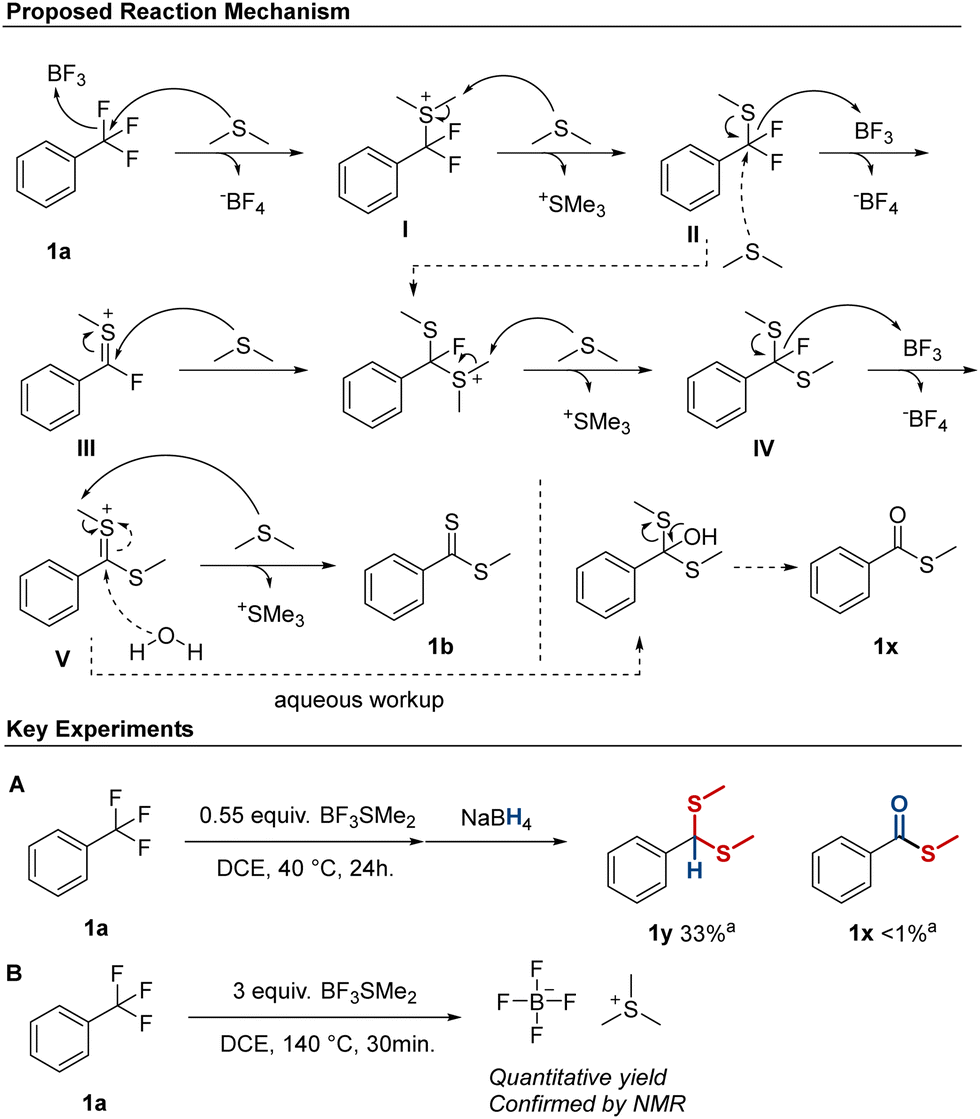 | ||
Scheme 1 Proposed mechanism for the transformation of the trifluoromethyl group into the methyl-dithioester using BF3SMe2 and key experiments supporting intermediate V (A), and SMe2 as demethylating reagent (B). a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Yields determined by 1H-NMR. Yields determined by 1H-NMR. | ||
To further delineate the reaction mechanism a number of control experiments were performed (ESI,† Scheme S3). In an attempt to form intermediate I, 1a was reacted with 0.55 equiv. of BF3SMe2 at 40 °C (ESI,† Experiment 1). This failed to produce intermediate I, and aqueous workup instead revealed S-methyl carbothioate (thioester) 1x as the major product (22%, NMR) along with a minor amount of methyl-dithioacetal 1y (9%). Thioester formation was also observed during the previous scope investigation of some trifluoromethyl substrates, and we had reasoned that the formation could be due to the presence of moisture in the reaction mixture.
To investigate this, a small amount of water (1.1 equiv.) was added to our standard conditions. This was surprisingly well-tolerated, and 64% of dithioester 1b was formed. Notably, there were only traces of 1x detected, indicating that water was not the main reason for thioester formation and we were therefore curious to further probe the origins of 1x. The presence of thioacetal 1y supported the formation of intermediate V, and we therefore treated the crude reaction mixture from experiment 1 with NaBH4 instead of performing an aqueous work up (Scheme 1A). This resulted in formation of significant amounts of dithioacetal 1y, and only traces of 1x were detected. We therefore suggest that 1x is formed from intermediate V during aqueous workup rather than during the reaction (Scheme 1).
Finally, we investigated our hypothesis of SMe2 acting as the demethylating species, as there is also the possibility that the demethylation occurs via a halide (fluoro) nucleophile. This pathway was suggested by Ikeada et al.16 using BBr3 as C–F activator and methanol as nucleophile forming carbonyls from trifluoromethyl groups. By treating a crude reaction mixture with i-hexane, a residue was formed, which upon further purification resulted in an off-white solid (Scheme 1B). This was analyzed by NMR and the data were consistent with the formation of trimethyl sulfonium tetrafluoroborate (1H-, 13C- and 19F NMR). This, together with the amount of trimethyl sulfonium tetrafluoroborate recovered (quant.), suggests that SMe2 is the predominant demethylating reagent under these conditions, which is in line with the known demethylating properties of BF3SMe2.
In conclusion, we have developed a new strategy for the activation of the trifluoromethyl group and its transformation into methyl-dithioesters. The methyl-dithioester is a versatile and useful group in synthetic chemistry and this method offers not only a valuable alternative entry point but also arguably the most straight-forward route to this functional group. We have also reported a rare example of a trithiocarbonate synthesis, as well as applications on active pharmaceutical scaffolds. We hope that these efforts will encourage the further investigation into the application of this unusual synthetic handle, and applications of this method towards other transformations are currently underway in our laboratory.
The authors would like to thank Dr Lisa Haigh (Imperial College London, U.K.) and Dr Reza Shariatgorji (Uppsala University, Sweden) for assistance with high-resolution mass determination. The authors would also like to thank the Kjell and Märta Beijer Foundation. The research was supported by Uppsala University and the Swedish Research Council (Vetenskapsrådet 2018-05133, 2022-04831).
Data availability
Data for this article, including details on experimental procedures, mechanistic experiments and characterization data of all compounds are available as part of the ESI.†17
Conflicts of interest
There are no conflicts to declare.Notes and references
- A. S. Nair, A. K. Singh, A. Kumar, S. Kumar, S. Sukumaran, V. P. Koyiparambath, L. K. Pappachen, T. M. Rangarajan, H. Kim and B. Mathew, Processes, 2022, 10, 2054 CrossRef CAS.
- (a) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (b) G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915–3942 RSC; (c) T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578–1587 CrossRef CAS; (d) K. Lye and R. D. Young, Chem. Sci., 2024, 15, 2712–2724 RSC; (e) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef CAS; (f) Z. Wang, Y. Sun, L. Y. Shen, W. C. Yang, F. Meng and P. Li, Org. Chem. Front., 2022, 9, 853–873 RSC.
- (a) G. P. Jourdan and B. A. Dreikorn, J. Org. Chem., 1982, 47, 5255–5257 CrossRef CAS; (b) Y. Kim, K. Kanemoto, K. Shimomori, T. Hosoya and S. Yoshida, Chem. – Eur. J., 2020, 26, 6136–6140 CrossRef CAS PubMed; (c) J. Xu, J.-W. Liu, R. Wang, J. Yang, K.-K. Zhao and H.-J. Xu, ACS Catal., 2023, 13, 7339–7346 CrossRef CAS; (d) D. Mandal, R. Gupta, A. K. Jaiswal and R. D. Young, J. Am. Chem. Soc., 2020, 142, 2572–2578 CrossRef CAS; (e) R. Idogawa, Y. Kim, K. Shimomori, T. Hosoya and S. Yoshida, Org. Lett., 2020, 22, 9292–9297 CrossRef CAS PubMed; (f) L. Yuan, Z. Wang, W. Zhuang, X. Li, C. Shi, X. Li and D. Shi, Org. Lett., 2024, 26, 7066–7071 CrossRef CAS PubMed; (g) K. Muta, K. Okamoto, H. Nakayama, S. Wada and A. Nagaki, ChemRxiv, 2024, preprint DOI:10.26434/chemrxiv-2024-5ml66-v3.
- (a) M. Söderström, E. Zamaratski and L. R. Odell, Eur. J. Org. Chem., 2019, 5402–5408 CrossRef; (b) M. Söderström, C. Matt and L. R. Odell, ACS Omega, 2023, 8, 4320–4330 CrossRef PubMed.
- (a) G. Lingaraju, T. Swaroop, A. Vinayaka, K. Sharath Kumar, M. Sadashiva and K. Rangappa, Synthesis, 2012, 1373–1379 CAS; (b) T. A. Jenifer Vijay, K. N. Nandeesh, G. M. Raghavendra, K. S. Rangappa and K. Mantelingu, Tetrahedron Lett., 2013, 54, 6533–6537 CrossRef CAS; (c) A. B. Ramesha, N. C. Sandhya, C. S. Pavan Kumar, M. Hiremath, K. Mantelingu and K. S. Rangappa, New J. Chem., 2016, 40, 7637–7642 RSC; (d) Kemparajegowda, H. A. Swarup, N. C. Sandhya, S. Rangappa, K. Mantelingu and K. S. Rangappa, ChemistrySelect, 2019, 4, 4611–4614 CrossRef CAS.
- D. M. Vyas and G. W. Hay, Can. J. Chem., 1971, 49, 3755–3758 CrossRef CAS.
- M. Heras, M. Gulea and S. Masson, Chem. Commun., 2001, 611–612 RSC.
- R. Bastin, H. Albadri, A.-C. Gaumont and M. Gulea, Org. Lett., 2006, 8, 1033–1036 CrossRef CAS.
- G. A. Olah, M. R. Bruce and F. L. Clouet, J. Org. Chem., 1981, 46, 438–442 CrossRef CAS.
- H. Davy, J. Chem. Soc., Chem. Commun., 1982, 457–458 RSC.
- (a) H. D. Verkruijsse and L. Brandsma, J. Organomet. Chem., 1987, 332, 95–98 CrossRef CAS; (b) J. Meijer, P. Vermeer and L. Brandsma, Recl. Trav. Chim. Pays-Bas, 1973, 92, 601–604 CrossRef CAS.
- C. S. Marvel, P. de Radzitzky and J. J. Brader, J. Am. Chem. Soc., 1955, 77, 5997–5999 CrossRef CAS.
- (a) S. R. Ramadas and P. S. Srinivasan, J. Chem. Soc., Chem. Commun., 1972, 345–346 RSC; (b) I. Abrunhosa, M. Gulea and S. Masson, Synthesis, 2004, 928–934 CAS.
- L. V. Hooker and J. S. Bandar, Angew. Chem., Int. Ed., 2023, 62, e202308880 CrossRef CAS.
- (a) R. Gupta, D. Mandal, A. K. Jaiswal and R. D. Young, Org. Lett., 2021, 23, 1915–1920 CrossRef CAS PubMed; (b) R. Gupta, D. Csókás, K. Lye and R. D. Young, Chem. Sci., 2023, 14, 1291–1300 RSC.
- M. Ikeda, T. Matsuzawa, T. Morita, T. Hosoya and S. Yoshida, Chem. – Eur. J., 2020, 26, 12333–12337 CrossRef CAS PubMed.
- References found in ESI†: (a) S. T. Keaveney and F. Schoenebeck, Angew. Chem., Int. Ed., 2018, 57, 4073–4077 CrossRef CAS PubMed; (b) R. Devine, M. Kelada, S. Leonard, D. S. D. Martin, J. M. D. Walsh, C. J. Breen, R. B. Driver, G. K. Kinsella, J. B. C. Findlay and J. C. Stephens, Eur. J. Med. Chem., 2020, 202, 112416 CrossRef CAS PubMed; (c) S. Kato, N. Kitaoka, O. Niyomura, Y. Kitoh, T. Kanda and M. Ebihara, Inorg. Chem., 1999, 38, 496–506 CrossRef CAS PubMed; (d) S. Furuta, M. Kuroboshi and T. Hiyama, Bull. Chem. Soc. Jpn., 1999, 72, 805–819 CrossRef CAS; (e) T. Roberie, N. S. Bhacca, D. Lankin and J. Selbin, Can. J. Chem., 1980, 58, 2314–2317 CrossRef CAS; (f) R. Takayama, A. Yamada, K. Fuchibe and J. Ichikawa, Org. Lett., 2017, 19, 5050–5053 CrossRef CAS PubMed; (g) A. Acharya, S. Vijay Kumar, B. Saraiah and H. Ila, J. Org. Chem., 2015, 80, 2884–2892 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc05540c |
| This journal is © The Royal Society of Chemistry 2025 |