
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilyl- and germyl-bridged neutral square-planar Ag4 clusters with short Ag–Ag distances exhibiting red emission†
Reon Ishiia,
Yoshimasa Wada *ab and
Yusuke Sunada*ab
*ab and
Yusuke Sunada*ab
aDepartment of Applied Chemistry, School of Engineering, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, 153-8505 Tokyo, Japan
bInstitute of Industrial Science, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, 153-8505 Tokyo, Japan
First published on 18th February 2025
Abstract
We report silyl- and germyl-bridged neutral square-planar Ag4 clusters with very short Ag–Ag distances (2.695 and 2.704 Å), as revealed by single-crystal X-ray diffraction analysis. On the basis of the results of theoretical calculations, we attribute these short distances to attractive Ag–Ag interactions that reduce the optical energy gap, resulting in a red emission reaching wavelengths of up to 700 nm. These wavelengths are among the longest observed for emissions from Ag4 clusters.
Considerable attention has been given to multinuclear transition metal molecular clusters because of their unique electronic states. These states arise from intermetallic interactions between metal centres and distinguish these multinuclear clusters from their mononuclear counterparts. Specific ligand designs can be used to produce multinuclear clusters with distinct electronic states, resulting in unique structural, optical, and reactive properties.1 Group 11 coinage metals (Cu, Ag, and Au) in the d10 monovalent state are known to form multinuclear cluster structures with non-covalent intermetallic interactions.2 Structures with d10 Ag(I) centres form various multinuclear clusters, where Ag4 clusters are among the stable forms frequently observed in silver cluster chemistry.3 The Cambridge structural database (CSD) contains 157 structures determined by X-ray diffraction (XRD) for Ag4 clusters with Ag–Ag interactions (Table S1, ESI†). These clusters have been studied both for their structural diversity and potential applications in chemical reactions and sensing.4
From a photophysical perspective, intermetallic distances within clusters play a crucial role in determining the electronic and optical properties of clusters.5 Short intermetallic distances enhance orbital interactions, modifying the electronic structure and narrowing the energy gap (ΔEg) between high-lying occupied dσ* orbitals and low-lying empty pσ/sσ orbitals.6 Consequently, the excited states and emission properties are changed. Although d10 Cu(I) and Au(I) tetranuclear clusters readily exhibit long-wavelength red emissions,1a Ag4 clusters with emission maxima (λMAX) exceeding 600 nm are rare. This rarity is primarily due to Ag(I) complexes having larger ΔEgs than Cu(I) and Au(I) complexes6,7 because ligand-centred transitions often dominate over transitions involving metal centres.
In this study, we developed two square-planar Ag4 clusters bridged by silyl and germyl ligands. The clusters have exceptionally short Ag–Ag distances and exhibit a deep red emission around 700 nm. We performed a single-crystal XRD (SC-XRD) analysis and theoretical calculations to investigate the intermetallic interactions contributing to these distinctive structural properties and electronic states. Additionally, we examined the origin of these long-wavelength emissions by comparing them with previously reported Ag4 clusters. We present results showing how intermetallic interactions produce red-shifted emissions in this communication.
The Ag4 clusters 1 and 2 were synthesised as shown in Scheme 1. Dipotassium salts (1b and 2b) were prepared, in a manner similar to the foundational procedure developed by Marschner and co-workers,8 by reacting the silyl compound 1a and germyl compound 2a with 2 equiv. of t-BuOK in 1,2-dimethoxyethane (DME), followed by treatment with 2 equiv. of AgOTf in Et2O under −70 °C. The corresponding Ag4 clusters were selectively obtained, as confirmed by 1H NMR. It was necessary to add 4 equiv. of pyridine as an auxiliary ligand to improve the cluster yield (see ESI†); otherwise, only a trace quantity of 2 was obtained. Recrystallisation from Et2O produced both 1 and 2 as orange crystals in yields of 52% and 36%, respectively. Both clusters were fully characterised by SC-XRD and elemental analysis, as well as by 1H, 13C, and 29Si NMR (Fig. S3–S10, ESI†).
 | ||
| Scheme 1 Synthesis of the Ag4 clusters 1 and 2. | ||
The molecular structures of the clusters were unambiguously determined by SC-XRD analysis. Both crystals contain two quasi-equivalent molecules; Fig. 1 shows representative molecules for 1 and 2 (Fig. S9 and S10, ESI†). All the Ag atoms lie in the same plane, with two pairs of Si (or Ge) atoms bonded to Ag centres positioned above and below the plane. The X-type silyl or germyl ligands form μ2-bridged structures with two Ag atoms, resulting in 3-centre 2-electron bonds. The bulky –SiMe3 substituents on the terminal Si (or Ge) atoms hinder solvation by other molecules (Fig. S11 and S12, ESI†), leaving the formal Ag(I) centres highly unsaturated and electron-deficient. This structural arrangement results in very short average Ag–Ag distances (dAg–Ag) of 2.695 Å for 1 and 2.704 Å for 2. These distances are among the shortest for the 157 CSD entries of Ag4 clusters (Table S1, ESI†). The dAg–Ag values of the SiH3- and GeH3-bridged Ag4 clusters have been theoretically predicted to be 2.799 Å and 2.822 Å, respectively3a—approximately 0.1 Å larger than those of our clusters. This difference may be attributed to agostic interactions between the H atoms of SiH3 and GeH3 and the electron-deficient Ag centres, which increase the electron density and dAg–Ag. Thus, the presence of silyl or germyl ligands and the formation of highly unsaturated, electron-deficient Ag centres likely contribute to the distinctively short dAg–Ag of our clusters.
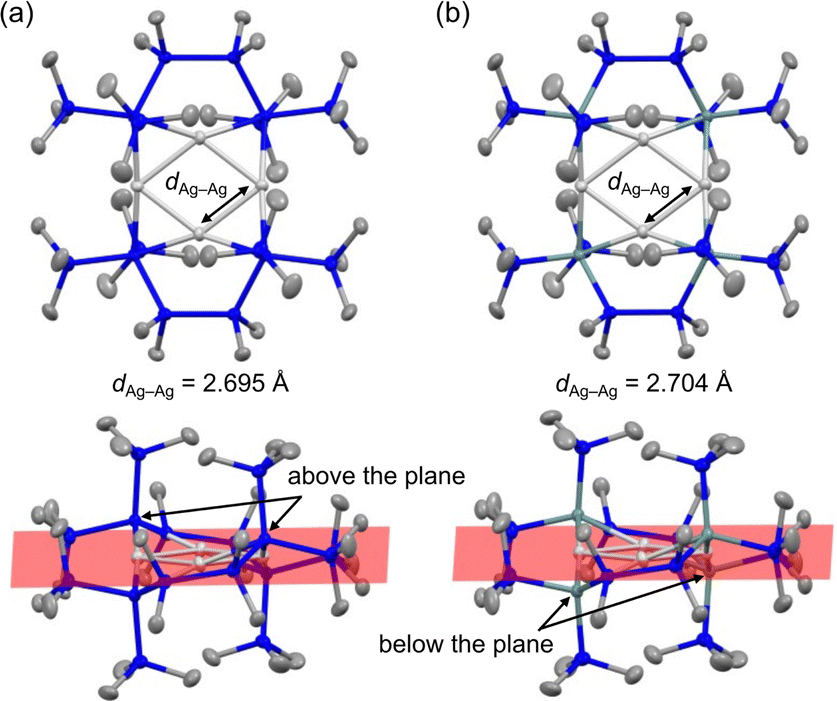 | ||
| Fig. 1 ORTEP drawings (50% probability level) of (a) 1 and (b) 2 from a top-down view. The lower images show the corresponding side profiles with the Ag4 plane highlighted in red. The blue, pale teal, silver, and grey balls represent Si, Ge, Ag, and C atoms, respectively. The H atoms are omitted for clarity. | ||
The electronic states of the clusters were investigated by performing DFT calculations employing Gaussian 16.9 The hybrid functional PBE0 with Grimme's D3BJ dispersion corrections was employed. The Def2-TZVP basis set was applied to the H, C, Si, and Ge atoms, and the SDD basis set was used for the Ag atoms. Although the dAg–Ag values are overestimated by approximately 0.03 Å, the ground-state (S0) optimised structures (Tables S2 and S3, ESI†) are in good accordance with the SC-XRD structures. Fig. 2 shows the HOMO and LUMO of the clusters. For both clusters, the energy of the HOMO−1 is close (within 0.2 eV) to that of the HOMO; therefore, the HOMO−1 is also shown. The HOMO and HOMO−1 are largely localised on the silyl and germyl ligands. The HOMO is partially distributed on two opposing Ag atoms, and the LUMO is predominantly localised on the Ag atoms and their spatial centre. Natural atomic orbital (NAO) analysis revealed that the HOMO and HOMO−1 mainly originate from the four terminal Si or Ge atoms bonded to four Ag atoms and are composed of Si 3p and Ge 4p orbitals (Tables S4–S7, ESI†). The Ag orbitals contribute minimally (2.6%) to the HOMO−1 for both clusters but non-negligibly to the HOMO (approximately 18.4% for cluster 1 and 18.8% for cluster 2), indicating the important role of Ag. Specifically, the contributing Ag orbitals are 5s–4d hybridised orbitals. The contribution from the Ag atoms to the LUMO is 50.1% (cluster 1) and 50.4% (cluster 2) and mainly involves the 5s and 5p orbitals. A Hirshfeld analysis for the LUMO is provided in the ESI,† that validates the NAO analysis.
 | ||
| Fig. 2 (a) Molecular orbitals and their composition determined by natural atomic orbital analysis. RDG isosurface map plotted at an isosurface value of 0.3 with four BCPs (shown by orange spheres) observed along the Ag–Ag bond in clusters (b) 1 and (c) 2. | ||
The noncovalent d10 Ag intermetallic interactions in our clusters were characterised by performing a reduced density gradient (RDG) analysis10 and Bader's quantum theory of atoms in molecules (QTAIM) analysis11 using Multiwfn software.12 Electronic wavefunctions were generated based on the aforementioned structures optimised using DFT. The same level of theory was employed as in a recent study on metal–metal (M–M) interactions in d10 coinage-metal clusters, including Ag4 clusters.13 In the referenced study, red RDG isosurface maps were reported between two metal centres, indicating repulsive interactions. In our case, as shown in Fig. 2 and Fig. S13 (ESI†), we observed blue RDG isosurface maps, indicating that the interactions between the Ag centres are as strong as hydrogen bonds, while red regions were observed at the centre of the Ag–Si (or Ge)–Ag triangles and the cluster centre. There are bond critical points (BCPs)11b within the blue region, suggesting an attractive interaction. Table 1 shows representative values of the topological parameters of the electron density ρ(r) at the BCPs. These values are consistent for the four BCPs in each cluster. The ratio of the Lagrangian kinetic energy (GCP) to the absolute potential energy density (|VCP|) is often used as a reliable indicator for characterising the type of interaction.14 Our clusters have |VCP|/GCP values of ∼1.25, indicating that the Ag–Ag interaction is not purely closed-shell but possesses some electron-shared (covalent) character. We used a previously reported method15 to estimate the strength of argentophilic interactions (Eint) on the basis of the ρ values at a BCP as −23.7 kcal mol−1 (cluster 1) and −22.9 kcal mol−1 (cluster 2). The ρ values for our clusters are larger than the range reported in the referenced study, which may reduce the accuracy of the estimated Eint. However, the larger ρ and Eint values calculated for our clusters indicate strong argentophilic interactions in our systems. The underlying reasons for this enhanced interaction strength will be investigated in future studies.
| Cluster 1 | Cluster 2 | |
|---|---|---|
| Density of all electrons: ρ | 0.041 | 0.040 |
| Laplacian of electron density: ∇2ρ | 0.090 | 0.091 |
| Lagrangian kinetic energy: GCP | 0.030 | 0.030 |
| Potential energy density: VCP | −0.038 | −0.038 |
| Energy density: HCP | −0.008 | −0.008 |
| |VCP|/GCP | 1.254 | 1.250 |
To investigate the effect of the short dAg–Ags of 1 and 2 on the photophysical performance of these clusters, a series of photophysical measurements was performed on solutions of 1 and 2 in methylcyclohexane. Fig. 3 shows the corresponding UV-vis absorption spectra. The spectra of clusters 1 and 2 contain broad absorption bands around 450 nm, with peak maxima (λabs) at 446 nm and 439 nm, respectively. We performed time-dependent (TD)-DFT calculations at the same level of theory as the structural optimisations. The results revealed that these absorptions are mainly attributable to the HOMO → LUMO-based S0 → lowest singlet excited state (S1) transition, with a smaller contribution from the HOMO−1 → LUMO-based S0 → second lowest singlet excited state (S2) transition. This result is obtained because the oscillator strength (f) for the S0 → S1 transition (0.0698 for 1 and 0.0862 for 2) is considerably larger than that for the S0 → S2 transition (0.0090 for 1 and 0.0119 for 2) (see the ESI† for details), even though the S2 energies (2.99 eV for 1 and 2.97 eV for 2) are close to those of S1 (2.78 eV for 1 and 2.85 eV for 2). Neither cluster exhibits noticeable emissions at room temperature but both clusters exhibit bright red emissions in glassy solutions at the temperature of liquid nitrogen. Fig. 3 shows the PL spectra of both solutions containing broad, single-peaked emissions with λMAX at approximately 690 nm (1) and 700 nm (2), which are among the longest wavelengths recorded for emissions from Ag4 clusters to date. The PL quantum yields (ΦPL) are 0.63 (1) and 0.50 (2), and the PL lifetimes (τ) are 102 μs (1) and 58 μs (2) (Fig. S13, ESI†). Given the large Stokes shift, even in frozen matrices, and the relatively long τ, these emissions can be assigned to phosphorescence. The phosphorescence rate constants (kTrs) are 6.1 × 103 s−1 (1) and 8.7 × 103 s−1 (2), whereas the non-radiative rate constants (kTnrs) are 3.6 × 103 s−1 (1) and 8.7 × 103 s−1 (2). Interestingly, both kTr and kTnr are larger for the Ge-containing cluster 2 than for the Si-based cluster 1. Considering that kTr and kTnr are rate constants for spin-forbidden transitions, the higher values of these constants for 2 than 1 can be attributed to the so-called heavy atom effect of Ge. A plausible reason for the relatively high ΦPLs (resulting from the small kTnr) is the presence of bulky –SiMe3 ligands densely packed around the clusters (Fig. S11 and S12, ESI†), which may contribute to restricting large-scale structural rearrangements.
 | ||
| Fig. 3 UV-vis absorption and PL spectra of clusters 1 and 2. | ||
Importantly, both complexes exhibit red emissions that are comparable to the longest-wavelength emissions recorded for Ag4 clusters. To explore the reason for this result, we compared our results with those for previously reported emissive clusters. Among the 157 CSD entries, 32 Ag4 clusters have been reported to exhibit emissions, as summarized in Table S1 (ESI†). Most of the emissions of these Ag4 clusters originate from ππ* transitions of the ligands, indicating a large ΔEg for Ag centres (as mentioned in the Introduction), even for those forming tetra-nuclear clusters. For some clusters with an excited state involving a metal centre, such as the states involved in ligand-to-metal charge transfer16 and metal–metal-to-ligand charge-transfer,17 emissions with longer wavelengths that those associated with ππ* transitions have been reported. However, clusters with red emissions approaching wavelengths of 700 nm are scarce. In the solid state, where molecules are aggregated, six reported complexes have been reported with λMAXs beyond 600 nm—five at cryogenic temperatures, one at 298 K, and another at both 298 K and 77 K.17,18 Among these clusters, the longest λMAX of 707 nm (77 K) was reported for a heterometallic Pt2Ag4 cluster.18a By contrast, in solution, where molecules are isolated, only three clusters have been reported to exhibit λMAXs beyond 600 nm (at 298 K or ambient temperature), with the longest-wavelength emissions reaching 628 nm at most.16,18c The observed emission wavelengths of our clusters are only shorter than those of the Pt-containing cluster and correspond to the longest λMAX recorded in an isolated state. On the basis of our DFT calculations, this long-wavelength phosphorescence emission is attributed to a LUMO → HOMO-based transition with considerable involvement of Ag orbitals, particularly because of the cluster-centred σ-type LUMO. We confirmed that, in such cases, the LUMO energy decreases as dAg–Ag decreases in a square-planar Ag4 cluster (Fig. S14, ESI†). Although the long-wavelength emission results primarily from phosphorescence (rather than fluorescence), which induces a large Stokes shift as previously discussed, the low-energy LUMO of our clusters, realised by the short dAg–Ag, likely contributes to the further red-shift of the emission wavelength.
To broaden the scope of this study, we performed theoretical calculations on cluster 1 in which the Ag atoms were replaced with the congeners Cu and Au. The calculated Cu–Cu and Au–Au distances are short (2.49 Å and 2.75 Å, respectively), and the corresponding phosphorescence energies are 1.00 eV (for the Cu4 cluster) and 1.06 eV (for the Au4 cluster), reaching the near-infrared (NIR) region. These values are notably lower than that of the Ag4 cluster 1 (1.68 eV). These theoretical results provide further evidence of the versatility of our molecular design concept, demonstrating that short M–M distances effectively induce red-shifted emission by enhancing M–M interactions.
In conclusion, we developed Ag4 clusters with short Ag–Ag distances that exhibit red emission reaching an λMAX of 700 nm. Our experimental and theoretical investigations underscore the role of both Ag–Ag distances and interactions in influencing the electronic structure and photophysical properties of Ag4 clusters. Subsequent theoretical calculations suggest that employing congeners, such as Cu or Au, could yield even longer-wavelength emissions reaching the NIR region. Therefore, this study deepens our fundamental understanding of Ag clusters and broadens our insight into the molecular design of coinage-metal-based emitters.
We acknowledge financial support from JSPS KAKENHI (Grant No. 23K13757 and 24K01495 (Japan)), JST PRESTO (Grant No. JPMJPR20A9 (Japan)), and JST-ALCA-Next Program (Grant No. JPMJAN23B1 (Japan)). Computational time was provided by the supercomputer facility of ACCMS at Kyoto University. We would like to thank Dr S. Takizawa (The University of Tokyo) for assistance with photophysical measurements.
Data availability
Crystallographic data for clusters 1 and 2 have been deposited at the CCDC under No. 2400625 and 2400626, respectively. Data supporting the results presented in this article are presented in ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) V. W.-W. Yam, et al., Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed; (b) Y. Du, et al., Chem. Rev., 2020, 120, 526 Search PubMed.
- (a) P. K. Mehrotra and R. Hoffmann, Inorg. Chem., 1978, 17, 2187 CrossRef CAS; (b) P. Pyykkö, Chem. Rev., 1997, 97, 597 CrossRef PubMed; (c) S. Sculfort and P. Braunstein, Chem. Soc. Rev., 2011, 40, 2741 Search PubMed; (d) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC; (e) H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746 Search PubMed; (f) N. V. S. Harisomayajula, et al., Chem. – Eur. J., 2019, 25, 8936 Search PubMed.
- (a) E. E. Karagiannis and C. A. Tsipis, Organometallics, 2010, 29, 847 CrossRef CAS; (b) A. Kytsya, et al., Mol. Cryst. Liq. Cryst., 2021, 720, 17 Search PubMed.
- (a) Y. Kikukawa, et al., Angew. Chem., Int. Ed., 2012, 51, 2434 CrossRef CAS PubMed; (b) T. Hu, et al., CrystEngComm, 2017, 19, 3445 RSC; (c) D. K. Jangid, et al., Inorg. Chem., 2022, 61, 13342 Search PubMed; (d) L. Chen, et al., Dalton Trans., 2023, 52, 9441 Search PubMed; (e) C. Ren, et al., Catalysts, 2023, 13, 644 Search PubMed; (f) Y. Xiao, et al., Inorg. Chem., 2024, 63, 8958 Search PubMed; (g) V. J. Catalano, et al., Angew. Chem., Int. Ed., 1999, 38, 1979 Search PubMed.
- V. W.-W. Yam and K. K.-W. Lo, Chem. Soc. Rev., 1999, 28, 323 RSC.
- K. Tsuge, Bull. Jpn. Soc. Coord. Chem., 2010, 56, 24 Search PubMed.
- T. Tsubomura, et al., Bull. Jpn. Soc. Coord. Chem., 2008, 52, 29 CrossRef.
- (a) C. Kayser, et al., Angew. Chem., Int. Ed., 2002, 41, 989 Search PubMed; (b) R. Fischer, et al., Organometallics, 2003, 22, 3723 Search PubMed; (c) J. Fischer, et al., Organometallics, 2005, 24, 1263 Search PubMed.
- M. J. Frisch, et al., Gaussian 16 Rev. C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- E. R. Johnson, et al., J. Am. Chem. Soc., 2010, 132, 6498 Search PubMed.
- (a) R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9 Search PubMed; (b) R. F. W. Bader, Chem. Rev., 1991, 91, 893 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 Search PubMed.
- S. Xu, et al., J. Phys. Chem. Lett., 2024, 15, 2193 CrossRef CAS PubMed.
- E. Espinosa, et al., J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
- S. Burguera, et al., Phys. Chem. Chem. Phys., 2024, 26, 16550 Search PubMed.
- V. W.-W. Yam, et al., Inorg. Chem., 1996, 35, 5116 Search PubMed.
- A. M. Polgar, et al., J. Am. Chem. Soc., 2017, 139, 14045 CrossRef CAS PubMed.
- (a) B. Gil, et al., Inorg. Chem., 2006, 45, 7788 CrossRef CAS PubMed; (b) A. M. Polgar, et al., Inorg. Chem., 2019, 58, 3338 CrossRef CAS PubMed; (c) M. M. Zhang, et al., J. Am. Chem. Soc., 2023, 145, 22310 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of CSD search results for previously reported Ag4 clusters and details of cluster synthesis, theoretical calculations, and photophysical measurements. CCDC 2400625 and 2400626. For crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06105e |
| This journal is © The Royal Society of Chemistry 2025 |