
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structurally and electronically driven uniaxial negative thermal expansion in BaIrO3†
Alexander J.
Browne
 ab,
A. Dominic
Fortes
c,
Andreas W.
Rost
*b and
Alexandra S.
Gibbs
ac
ab,
A. Dominic
Fortes
c,
Andreas W.
Rost
*b and
Alexandra S.
Gibbs
ac
aEaStCHEM, School of Chemistry, University of St Andrews, St Andrews, KY16 9ST, UK
bSUPA, School of Physics and Astronomy, University of St Andrews, St Andrews, KY16 9SS, UK. E-mail: a.rost@st-andrews.ac.uk
cISIS Pulsed Neutron and Muon Source, STFC Rutherford Appleton Lab, Harwell Campus, Didcot, OX11 0QX, UK
First published on 17th December 2024
Abstract
Negative thermal expansion is known to exist in a range of structure types but is extremely rare in hexagonal perovskites. Here we demonstrate that BaIrO3 displays negative linear thermal expansion in the direction of its face-shared IrO6 trimers, and apparent zero volume thermal expansion below 100 K. We present evidence that this anomalous thermal expansion is driven by an unusual form of rigid body phonon behaviour governed by the effective trimer valence state and therefore has structural and electronic components to the underlying mechanism.
Negative thermal expansion (NTE) is a fascinating – and highly functional – property.1 But as only a small minority of materials are known to exhibit NTE, discovering new examples, and revealing the mechanisms behind this unusual behaviour, is of great value.1–3 The underlying mechanisms are generally either of structural or electronic origin. The most well-known example for the first type is that of vibrations transverse to the bond direction in flexible framework structures constructed of rigid units as in ReO3.4 Complementary to this are electronically mediated cases driven by thermal changes in the interatomic bonding potential or by magnetostriction.2 Such behaviour generally occurs at phase transitions,5 or gradually evolves in a low-temperature phase as a function of the (magnetic) order parameter.6 NTE can be considered to be strong if the linear thermal expansion coefficient, α, is of order |α| > 10−5 K−1i.e. larger than the α of positive thermal expansion in many everyday materials.7
In hexagonal perovskites (HP), which contain units of face-shared octahedra, very few examples of NTE are known. Ba3BiRu2O9, Ba3BiIr2O9 and Ba4BiIr3O12 all fall in the class of electronically driven NTE coinciding with their magnetic ordering transitions.8–10 [(CH3)2NH2]PbBr3 exhibits uniaxial NTE, as a result of a bonding-related order–disorder phase transition.11 To the best of our knowledge, there are no examples of HPs that exhibit structurally-driven NTE. Simplistically that might be expected – face-sharing octahedra cannot be tilted by transverse phonons in the way vertex-sharing octahedra can. But that does not rule out the face-sharing units themselves being the rigid units that experience vibrational motion. Here, we present evidence for such NTE in a HP, BaIrO3, for the first time. Intriguingly our data indicate that it is closely linked to the electronic structure of the trimer building blocks, hinting at a mixed structural-electronic origin.
The structure of BaIrO3, which is a monoclinically-distorted (space group C2/m) variant of the 9R HP structure is illustrated in Fig. 1a.12 There are four Ir crystallographic sites, each octahedrally coordinated and arranged through face-sharing into linear Ir3O12 trimers oriented approximately parallel to the c axis of the unit cell. In the undistorted 9R structure these trimers are crystallographically equivalent, but the monoclinic distortion present in BaIrO3 breaks this degeneracy. Half the trimers comprise Ir1–Ir2–Ir1 iridium sites, whilst the other half comprise Ir3–Ir4–Ir3 sites. The trimers of one type share vertices exclusively with trimers of the other type (i.e. Ir1–O–Ir3 linkages). This results in a structure comprising alternating ‘layers’ of each trimer type stacking along the c axis. As will become apparent, the inequivalence of the two types of trimer is essential to the occurrence of NTE in BaIrO3.
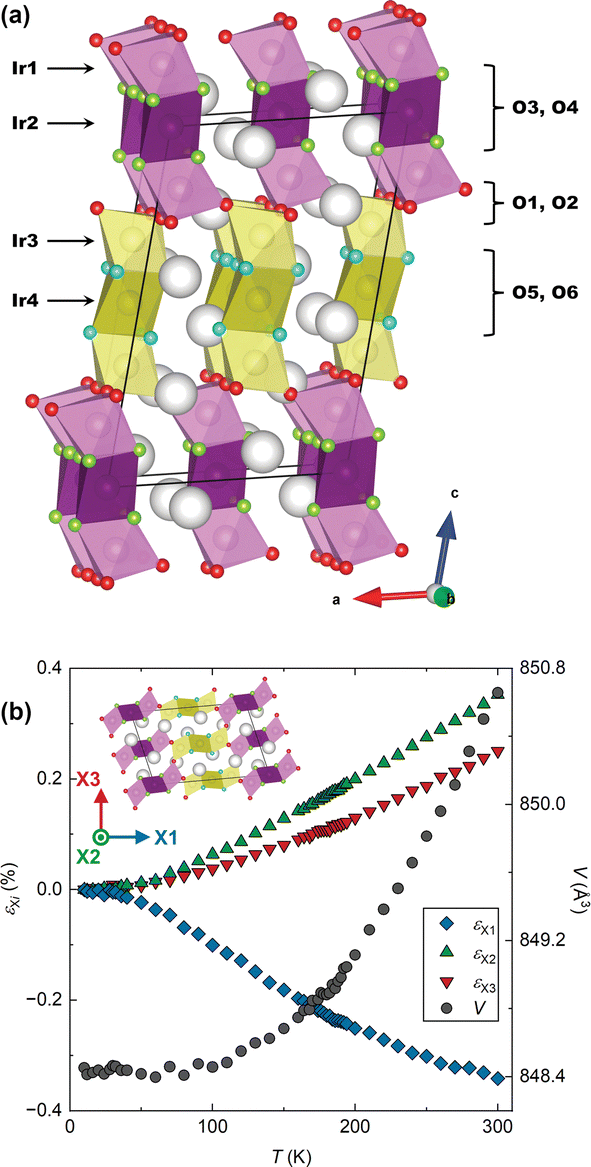 | ||
| Fig. 1 (a) The crystal structure of BaIrO3, with the Ir and O sites labelled. (b) The lattice strains εXi along the principal axes Xi as a percentage of the 10 K values, and the unit cell volume. The inset shows the orientation of the unit cell relative to the three principal axes. | ||
BaIrO3 is metallic, with magnetic transition at TC = 184 K.13 Transport and optical conductivity studies indicate that this is accompanied by a charge density wave,14 with μSR suggesting a small moment consistent with very weak ferromagnetism.15 The details of the transition remain contentious,16–22 and no structural changes have been resolved crystallographically. While additional transitions at 26 K and 80 K have been reported, they were absent in high quality powder samples.23
NTE at low temperatures was first reported based on laboratory X-ray measurements by Cheng et al.18 A recent synchrotron X-ray diffraction study was able to connect this to the distortions of the trimer units, revealing inequivalency of the temperature dependencies of the two symmetry-inequivalent trimers.21 While at the same time being only weakly affected by the 184 K transition. The potentially unusual NTE mechanism in this compound motivated our high-resolution neutron diffraction study reported here.
We collected high-resolution powder neutron diffraction data for a 193Ir-enriched24 sample of BaIrO3 between 10 K and 300 K. Subsequent Rietveld refinement allowed us to determine the lattice parameters, all atomic positions and atomic displacement parameters Biso over the whole temperature range. Using PASCal25 we calculate the principal axes Xi and the infinitesimal Lagrangian strains εXi along them (Fig. 1b). X1 is effectively parallel to the c axis (it has components 0.9996c and −0.0287a, equating to a 5° offset); X2 is exactly parallel to b; and X3 is orthogonal to X1 and X2 (its components are 0.9869a and 0.1612c). From εXi we extract the principal linear thermal expansion coefficients αXi (see ESI†) as shown in Table 1. Linear temperature dependence is found for all three αXi for T ≥ 80 K.
| α Xi /×10−6 K−1 | 80–182 K | 186–270 K |
|---|---|---|
| α X1 | −16.0(2) | −10.6(2) |
| α X2 | 13.2(1) | 15.1(1) |
| α X3 | 8.7(1) | 11.9(1) |
The phenomenology of this linear NTE is notable. Not only is the magnitude very large (|αX1| > 10−5 K−1),7 but below 100 K εX1 effectively cancels with εX2,3 resulting in an average volumetric thermal expansion coefficient ![[small alpha, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c2.gif) V in the range of 10 to 100 K of V = 1.9 × 10−7 (ESI†), comparable to the best examples in the literature and of potential interest for cryogenic applications.26
V in the range of 10 to 100 K of V = 1.9 × 10−7 (ESI†), comparable to the best examples in the literature and of potential interest for cryogenic applications.26
Surprisingly we found only a small, albeit significant, change in the linear thermal expansion on crossing the magnetic ordering temperature TC = 184 K. This contrasts with the NTE found in the other HPs as introduced previously, which all contract abruptly within the narrow temperature range of a first-order phase transition. Ba3BiRu2O9, Ba3BiIr2O9 and Ba4BiIr3O12 all contract uniaxially along the direction in which their constituent dimers/trimers are oriented – and the contractions are driven by a shortening of the Ir–Ir (or Ru–Ru) distances within those face-sharing units. Thanks to the face-sharing geometry, the metal centres within neighbouring octahedra have both the orientation and proximity for direct d–d overlap to be possible. We do not find such a simple correlation in BaIrO3, not only are the distances within the Ir1–Ir2–Ir1 and Ir3–Ir4–Ir3 trimers different to one another (a consequence of the monoclinic distortion present), but their variation with temperature is different too (see Fig. 2). Above TC = 184 K both the Ir1–Ir2 and Ir3–Ir4 distances lengthen on cooling, though the latter do so at a rate approximately three times greater than the former. But below TC, the Ir1–Ir2 distance switches to a slight contraction on cooling, whilst the Ir3–Ir4 bond not only retains its abnormal behaviour but its rate of extension increases further. In short, the NTE is completely due to the Ir3–Ir4–Ir3 trimer with the Ir1–Ir2–Ir1 trimer effectively invariant.
 | ||
| Fig. 2 Variation of the two face-sharing Ir–Ir distances in BaIrO3, both show a change in slope in crossing TC = 184 K, and the magnetic ordering transition evident in the susceptibility χ = M/H (μ0H = 0.1 T). | ||
To further investigate the mechanism we now turn to the temperature evolution of the remaining atomic parameters, which were inaccessible to previous studies. Most significant is the displacement of the constituent Ir and O atoms, determined via refinement of Biso. Anomalously large atomic displacements are a signature of phonon-driven NTE, as they capture, in the static picture of a diffraction pattern, dynamic displacements of atoms away from their equilibrium sites. Large (and anisotropic) atomic displacements are seen in a number of such materials,7 well known examples including ZrW2O8 and ScF3.27,28 Neutron diffraction is particularly powerful for studying this as it allows the oxygen positions to be captured in the presence of heavy elements and the flat form-factor improves the precision and accuracy of displacement parameter determination. We used a single Biso for the iridium sites (Ir1,2 and Ir3,4) and oxygen sites (O3,4 and O5,6) within each symmetry-inequivalent trimer, as well as an independent Biso for the bridging oxygen sites (O1,2). A comparison of the refined values (Fig. 3) clearly shows that the atoms in the Ir3–Ir4–Ir3 trimer (those Ir sites and the O5,6 pair) have far larger displacement parameters than their analogues in the Ir1–Ir2–Ir1 trimer. And the Biso of the O1,2 pair is the largest of all. The behaviour of the Ir3,4 trimer over the whole temperature regime we studied would suggest that the NTE in BaIrO3 is of structural origin and the result of vibrations in which the Ir3O12 trimers, rather than individual IrO6 octahedra, are the rigid units. Within the unit cell there are 30 atoms and thus 90 possible phonon modes, making it very challenging to determine which specific mode(s) are responsible. However, from a crude analysis one can readily visualise cooperative displacements of the Ir and O sites that would result in NTE. A transverse displacement of the O1 and O2 sites causing a rotation of the trimers in the ac plane, for example, analogous to the tilting of vertex-sharing octahedra in ReO3-type NTE materials, would contract the lattice along c. This is the first time such behaviour has been found in a HP.
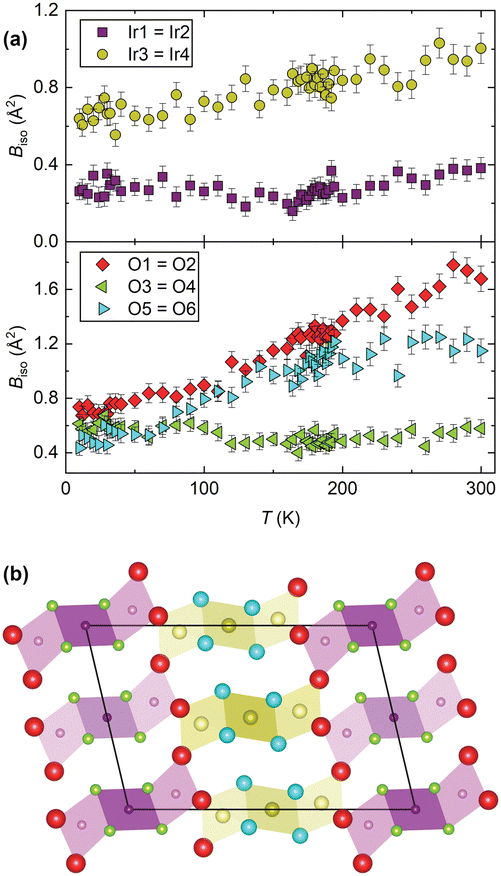 | ||
Fig. 3 (a) Refined Biso values for the Ir and O sites. (b) Projection of the structure along b with the atomic radii proportional to the 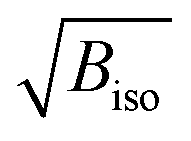 values at 300 K (Ba sites omitted for clarity). values at 300 K (Ba sites omitted for clarity). | ||
An essential comparison can be made to BaRuO3, with an undistorted R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m 9R HP structure, and an entirely conventional positive thermal expansion (and no anomalies in its atomic displacement parameters).29 All of its Ru3O12 trimers are equivalent – in other words, whichever effect is responsible for the NTE in BaIrO3 is not available in its Ru-analogue. Comparison also rules out the electronic effect of strengthening metal–metal bonding being in itself responsible for the NTE, because it is similarly present in both of them. At 300 K, the Ir1–Ir2 distance in BaIrO3 is 2.63 Å whilst the Ir3–Ir4 distance is 2.58 Å. Both of these are shorter than the 2.69 Å nearest-neighbour separation in elemental Ir at the same temperature. Comparably, the 300 K Ru–Ru distance in BaRuO3 is 2.55 Å,29 compared to 2.65 Å in elemental Ru.
m 9R HP structure, and an entirely conventional positive thermal expansion (and no anomalies in its atomic displacement parameters).29 All of its Ru3O12 trimers are equivalent – in other words, whichever effect is responsible for the NTE in BaIrO3 is not available in its Ru-analogue. Comparison also rules out the electronic effect of strengthening metal–metal bonding being in itself responsible for the NTE, because it is similarly present in both of them. At 300 K, the Ir1–Ir2 distance in BaIrO3 is 2.63 Å whilst the Ir3–Ir4 distance is 2.58 Å. Both of these are shorter than the 2.69 Å nearest-neighbour separation in elemental Ir at the same temperature. Comparably, the 300 K Ru–Ru distance in BaRuO3 is 2.55 Å,29 compared to 2.65 Å in elemental Ru.
However, if the NTE were of purely structural origin, one would naively expect both trimers to show similar behaviour given their superficially small structural differences. But, as shown in Fig. 2, they are fundamentally different. Hence a purely structural mechanism cannot be sufficient and electronic effects have to be important in BaIrO3: like many iridates, it hosts finely-balanced states.30 Through being able to accurately refine the oxygen site positions we are able to calculate the bond valence sum (BVS)31 for each Ir centre (Fig. 4a). We find that at all temperatures they are significantly different between the two trimers, indicating fundamentally different electronic characteristics. For example, Ir2 and Ir4 differ in their BVS/oxidation state by 0.5 at room temperature. This difference of electron density distribution must be one of the drivers of the different trimer expansivities. Furthermore, whilst all four BVS are temperature-independent above TC, three of them vary significantly below it (Fig. 4a). Ir1 (Ir4.2+ to Ir4.0+) and Ir2 (Ir3.9+ to Ir3.5+) both lose valence, whilst Ir4 (Ir3.4+ to Ir3.8+) gains it, and the Ir3 site is stable Ir3.9+. In other words, at TC there is a transfer of charge from the Ir3–Ir4–Ir3 trimers to the Ir1–Ir2–Ir1 ones. Between 300 K and 10 K this equates to a transfer of 1.0(1) charge. This change in the BVS at TC is a likely source for the evolution of the NTE across the transition, and is further evidence of an electronic component to its mechanism. We cannot say precisely how without more diverse experimental input – spectroscopy, for example, may show a corresponding change in phonon frequencies if the charge transfer stiffens the Ir3–Ir4 interaction and weakens the Ir1–Ir2 one – but there is clearly a change in the electronic structure associated with the magnetic ordering which affects the NTE. The very small ordered moment makes a magnetostrictive origin of the evolution of the NTE below the transition rather improbable. A magnetostrictive scenario would furthermore require significantly different magnetic moments on the two trimers – something not seen by μSR.
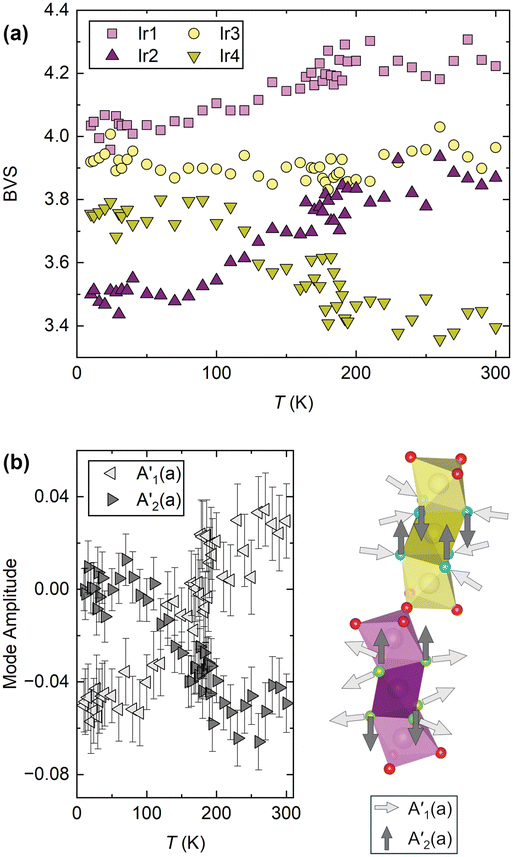 | ||
| Fig. 4 (a) The calculated bond valence sums for the four Ir sites, which show that inter-trimer charge transfer occurs below TC. (b) Symmetry mode analysis reveals a simultaneous structural distortion, by which the Ir2O6 octahedron gains volume on cooling and the Ir4O6 octahedron shrinks by an equivalent amount. Arrows on the crystal structure section indicate the corresponding atomic displacements. | ||
We cannot currently explain the charge transfer occurring at TC = 184 K. The electronic structure of BaIrO3 remains under discussion, though it is reassuring that a recent reflectivity study is consistent with the inter-trimer charge transfer observed by us.22 Further insight is provided by symmetry-mode analysis (Fig. 4b), which reveals an anti-correlation in two modes of the T1+ irrep (which is activated by the monoclinic lattice distortion) that corresponds to a gain in volume by the central Ir2O6 octahedron of one of the trimers and an equivalent loss in volume by the Ir4O6 octahedron of the other trimer (Fig. 4b). These subtle but detectable structural features, which have never been reported before, may help to reconcile the different mechanisms that have been put forward to describe the ordering transition in BaIrO3.
In conclusion, we have quantified the uniaxial NTE in BaIrO3 for the first time, and unveiled an effective volume ZTE below 100 K. While the evolution of Biso in the Ir3,4 trimer suggests a structural origin for the NTE it seems irreconcilable with the more conventional behaviour of the structurally similar Ir1,2 trimer. Irrespective of this, it is clear that the origin of the NTE is fundamentally different from that of the other HP NTE materials. Intriguingly, we uncovered significant differences in the oxidation states of the two trimers of up to 0.5, indicating an electronic origin of the disparities in their thermal behaviour. Furthermore, the NTE response changes upon crossing TC = 184 K, and we have found concurrent changes in the trimer bond valences, which appear to be related to inter-trimer charge transfer at this phase transition. These observations clearly mark the NTE as not easily classifiable into purely structural or purely electronic origin, an unprecedented scenario clearly motivating further studies.
We acknowledge funding from the Engineering and Physical Sciences Research Council [grant numbers EP/T011130/1 and EP/P024564/1]. Experiments at the ISIS Neutron and Muon Source were supported by a beamtime allocation RB1910632 from the Science and Technology Facilities Council: https://doi.org/10.5286/ISIS.E.RB1910632-1.
Data availability
Data for this article have been included as part of the ESI.† Neutron diffraction data are available from: https://doi.org/10.5286/ISIS.E.RB1910632-1.Conflicts of interest
There are no conflicts to declare.References
- K. Takenaka, Sci. Technol. Adv. Mater., 2012, 13, 013001 CrossRef PubMed.
- J. P. Attfield, Front. Chem., 2018, 6, 371 CrossRef PubMed.
- Q. Li, K. Lin, Z. Liu, L. Hu, Y. Cao, J. Chen and X. Xing, Chem. Rev., 2022, 122, 8438–8486 CrossRef CAS PubMed.
- T. Chatterji, P. F. Henry, R. Mittal and S. L. Chaplot, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134105 CrossRef.
- Y. W. Long, N. Hayashi, T. Saito, M. Azuma, S. Muranaka and Y. Shimakawa, Nature, 2009, 458, 60–63 CrossRef CAS PubMed.
- B. Dabrowski, M. Avdeev, O. Chmaissem, S. Kolesnik, P. W. Klamut, M. Maxwell and J. D. Jorgensen, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 104411 CrossRef.
- N. Shi, Y. Song, X. Xing and J. Chen, Coord. Chem. Rev., 2021, 449, 214204 CrossRef CAS.
- W. Miiller, M. Avdeev, Q. Zhou, A. J. Studer, B. J. Kennedy, G. J. Kearley and C. D. Ling, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 220406(R) CrossRef.
- W. Miiller, M. Avdeev, Q. Zhou, B. J. Kennedy, N. Sharma, R. Kutteh, G. J. Kearley, S. Schmid, K. S. Knight, P. E. R. Blanchard and C. D. Ling, J. Am. Chem. Soc., 2012, 134, 3265–3270 CrossRef CAS PubMed.
- W. Miiller, M. T. Dunstan, Z. Huang, Z. Mohamed, B. J. Kennedy, M. Avdeev and C. D. Ling, Inorg. Chem., 2013, 52, 12461–12467 CrossRef CAS PubMed.
- J. S. Rodríguez-Hernández, M. A. P. Gómez, D. S. Abreu, A. Nonato, R. X. da Silva, A. García-Fernández, M. A. Señarís-Rodríguez, M. Sánchez-Andújar, A. P. Ayala and C. W. A. Paschoal, J. Mater. Chem. C, 2022, 10, 17567–17576 RSC.
- T. Siegrist and B. L. Chamberland, J. Less-Common Met., 1991, 170, 93–99 CrossRef CAS.
- A. V. Powell and P. D. Battle, J. Alloys Compd., 1993, 191, 313–318 CrossRef CAS.
- G. Cao, J. Crow, R. Guertin, P. Henning, C. Homes, M. Strongin, D. Basov and E. Lochnere, Solid State Commun., 2000, 113, 657–662 CrossRef CAS.
- M. L. Brooks, S. J. Blundell, T. Lancaster, W. Hayes, F. L. Pratt, P. P. C. Frampton and P. D. Battle, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 220411(R) CrossRef.
- M.-H. Whangbo and H.-J. Koo, Solid State Commun., 2001, 118, 491–495 CrossRef CAS.
- K. Maiti, R. S. Singh, V. R. R. Medicherla, S. Rayaprol and E. V. Sampathkumaran, Phys. Rev. Lett., 2005, 95, 016404 CrossRef PubMed.
- J.-G. Cheng, J.-S. Zhou, J. A. Alonso, J. B. Goodenough, Y. Sui, K. Matsubayashi and Y. Uwatoko, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 104430 CrossRef.
- M. A. Laguna-Marco, D. Haskel, N. Souza-Neto, J. C. Lang, V. V. Krishnamurthy, S. Chikara, G. Cao and M. van Veenendaal, Phys. Rev. Lett., 2010, 105, 216407 CrossRef CAS PubMed.
- W. Ju, G.-Q. Liu and Z. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 075112 CrossRef.
- I. Terasaki, S. Ito, T. Igarashi, S. Asai, H. Taniguchi, R. Okazaki, Y. Yasui, K. Kobayashi, R. Kumai, H. Nakao and Y. Murakami, Crystals, 2016, 6, 27 CrossRef.
- R. Okazaki, S. Ito, K. Tanabe, H. Taniguchi, Y. Ikemoto, T. Moriwaki and I. Terasaki, Phys. Rev. B, 2018, 98, 205131 CrossRef CAS.
- N. Kini, A. Bentien, S. Ramakrishnan and C. Geibel, Phys. B, 2005, 359–361, 1264–1266 CrossRef CAS.
- A. C. Hannon, A. S. Gibbs and H. Takagi, J. Appl. Crystallogr., 2018, 51, 854–866 CrossRef CAS.
- M. Lertkiattrakul, M. L. Evans and M. J. Cliffe, J. Open Source Softw., 2023, 8, 5556 CrossRef.
- T. H. K. Barron, J. G. Collins and G. K. White, Adv. Phys., 1980, 29, 609–730 CrossRef CAS.
- J. S. O. Evans, T. A. Mary, T. Vogt, M. A. Subramanian and A. W. Sleight, Chem. Mater., 1996, 8, 2809–2823 CrossRef CAS.
- M. T. Dove, J. Du, Z. Wei, D. A. Keen, M. G. Tucker and A. E. Phillips, Phys. Rev. B, 2020, 102, 094105 CrossRef CAS.
- P. Kayser, S. Injac, B. J. Kennedy, A. L. M. de Oliveira, Y. Shirako and M. Hasegawa, Dalton Trans., 2017, 46, 2974–2980 RSC.
- A. J. Browne, A. Krajewska and A. S. Gibbs, J. Mater. Chem. C, 2021, 9, 11640–11654 RSC.
- O. C. Gagné and F. C. Hawthorne, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 562–578 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc06174h |
| This journal is © The Royal Society of Chemistry 2025 |