DOI:
10.1039/D4CC06382A
(Highlight)
Chem. Commun., 2025,
61, 8969-8983
Recent progress in the decomposition of ammonia as a potential hydrogen-carrier using green technologies
Received
2nd December 2024
, Accepted 3rd April 2025
First published on 4th April 2025
Abstract
To meet the global carbon neutrality target set by the United Nations, finding alternative and cost-effective energy sources has become prominent while enhancing energy conversion methods’ efficiency. The versatile applications of hydrogen (H2) as an energy vector have been highly valued over the past decades due to its significantly lower greenhouse gas emissions compared to conventional fossil fuels. However, challenges related to H2 generation and storage for portable applications have increasingly called attention to ammonia (NH3) decomposition as an effective method for on-site hydrogen production due to its high hydrogen content, high energy density, and affordability. This review highlights recent developments in green decomposition techniques for ammonia, including photocatalysis, electrocatalysis, non-thermal plasma, and other techniques, with a focus on the latest developments in new methods and materials (catalysts, electrodes, and sorbents) employed in these processes. Moreover, technical challenges and recommendations are discussed to assess the future potential of ammonia in the energy sector. The role of machine learning and artificial intelligence in ammonia decomposition is also emphasized, as these tools open up ways of simulating reaction mechanisms for the exploration of a new generation of high-performance catalysts and reducing trial-and-error approaches.
1. Introduction
The demand for global energy has increased due to a highly energy-intensive lifestyle and the continuing growth of the world's population.1 Currently, approximately 733 million people globally lack access to electricity, while 2.4 billion people lack clean fuels and modern cooking technologies.2 According to the Energy Institute Statistical Review of World Energy 2023, fossil fuels, coal, oil, and natural gas are still the main primary energy sources (82%).3 There is a broad consensus that fossil fuel reserves, particularly oil, will be on the verge of depletion and in shortage by the end of this century.4 Due to the continuous reliance on fossil fuels, a substantial amount of greenhouse gases (GHGs) such as CO2, CO, SO2, NOx, and volatile organic compounds (methane (CH4), benzene (C6H6), formaldehyde (CH2O), and ethanol (C2H5OH)) have been emitted into Earth's atmosphere.5–8 CO2 is recognized as the primary driver of global warming, with approximately 80% originating from the combustion of fossil fuels within the industrial sector.9 To respond to global climate change and meet the Paris Agreement's temperature control goals, there is a worldwide consensus on reducing greenhouse gas emissions toward net-zero carbon emissions.9 In this context, China has set a target to reach “peak carbon” by 2030 and “carbon neutrality” by 2060.10
Renewable energy technologies, including solar and wind power, are increasingly achieving cost parity with conventional fossil fuel-based energy sources.11 However, as these energy sources are intermittent and unevenly distributed across the globe, it is still complex to completely replace traditional energy.12 Therefore, hydrogen (H2) has attracted attention as a new energy source without pollution or CO2 emissions.
Hydrogen, with the smallest relative molecular mass, has attracted significant interest as a secondary energy source due to its high gravimetric energy density (∼33 kW h kg−1), which is greater than that of gasoline and diesel, and its capacity for zero-emission output.13,14 The U.S. Department of Energy (DOE) initially set hydrogen storage targets in 2009 for applications such as portable power, onboard light-duty vehicles, and material-handling equipment. The DOE set specific targets for on-board hydrogen storage: 0.030 kg H2 L−1 and 4.5 wt % for volumetric and gravimetric storage capacities by 2020.15 In addition, China is actively working to increase its production of carbon-neutral hydrogen (green hydrogen) to meet its carbon neutrality goals, which involves water splitting (eqn (1) and (2)) to extract hydrogen using electricity generated from renewable sources such as wind and solar energies.9,16 It is expected that till 2025, the specific system targets aim for 1.8 kW h kg−1 system (0.055 kg H2 kg−1 system), 1.3 kW h L−1 system (0.040 kg H2 L−1), and $9 per kW h storage system ($300 per kg stored H2 system).
In acidic solution
| 2H2O → O2 + 4H+ + 4e− E0 = 1.23 V vs. NHE |
| 4H+ + 4e− → 2H2 E0 = 0.00 V vs. NHE |
| | | 2H2O → 2H2 + 2O2 E0 = 1.23 V vs. NHE, ΔG0 = 237.2 kJ mol−1 | (1) |
In alkaline solution
| 4OH− → O2 + 2H2O + 4e− E0 = 0.4 V vs. NHE |
| 4H2O + 4e− → 2H2 + 4OH− E0 = −0.83 V vs. NHE |
| | | 2H2O → 2H2 + 2O2 E0 = 1.23 V vs. NHE, ΔG0 = 237.2 kJ mol−1 | (2) |
Currently, hydrogen can be stored in carbon fiber tanks at high pressures (>35 MPa), achieving a gravimetric H2 capacity of 0.025 kg H2 kg−1 at 350 bar.17 Hydrogen can only be liquefied at an extremely low temperature of −253 °C or pressures above 70 MPa that significantly increase the costs associated with the storage and transportation of H2 energy.9 Therefore, the transportation and storage of hydrogen remains a critical barrier, significantly impeding its industrial applications.14
One potential solution to address hydrogen transport issues involves the utilization of liquid or solid hydrogen energy carriers, from which H2 is chemically extracted upon arrival. The selection of a hydrogen energy carrier focuses on environmental friendliness, efficiency, ease of handling and transport, and a high hydrogen mass and volume percentage. Given this, methanol and ammonia are frequently discussed as feasible carriers.9
Accordingly, ammonia (NH3) possesses high H2 content (17.8 wt%) and a large energy density (3000 W h kg−1). It has greater volumetric hydrogen density than liquid H2 (121 kg H2 m−3) and can be liquefied and stored at room temperature, facilitating its transportation and storage, particularly in the liquid phase, as NH3 gas is liquefied under a pressure of 8.5 MPa at 20 °C.18,19 Notably, hydrogen produced through ammonia decomposition typically contains fewer impurities compared to hydrogen derived from hydrocarbons (like methanol).20
Ammonia decomposition has been investigated since the 19th century.18 In 1904, Perman and Atkinson reported that complete decomposition of ammonia is not achievable below 1100 °C. They also noted that the degree of decomposition depends significantly on the nature of the surface in contact with the ammonia, particularly the catalysts involved.20 To date, various metals, alloys, and their compounds (such as oxides and nitrides) have been extensively studied as active catalysts for NH3 decomposition. Given that ammonia decomposition is the exact reverse of industrial ammonia synthesis from N2 and H2, the microkinetic principle suggests that catalysts effective for NH3 generation should, in theory, facilitate ammonia decomposition. However, the catalytic activity trends differ significantly between the two processes due to their opposing reaction pathways and targeted products.21
Prior to 1990, Fe-based catalysts attracted significant interest; however, in the past decade, research has increasingly shifted toward noble metal catalysts, with growing attention on metal nitrides, carbides, and alloys as active components for the decomposition reaction.22 Various monometallic systems based on non-noble metals have been investigated for hydrogen production from ammonia. Over the last decade, the catalytic decomposition of NH3 over catalysts such as platinum (Pt), palladium (Pd), ruthenium (Ru), and rhodium (Rh) has gained a lot of attention in substitution of iron.18 While these metals show outstanding activities, their large-scale applications significantly increase the cost, which is a substantial drawback.23 To tackle this issue, transition metal carbides (MoCx, VCx, WCx, and FeCx) and nitrides (MoNx, VNx, and WNx), along with zirconium oxynitrides, have been studied. Amidst those, molybdenum nitride and tungsten carbide have received the most attention in ammonia decomposition studies. Notably, these catalysts are generally evaluated under conditions relevant not only to hydrogen production but also to gasification mixture clean-up.24 For bimetallic catalysts, several studies have been explored, including Ni–Pt, Ni/Ru, Pd/Pt/Ru/La, and Fe–MOx (M = Ce, Al, Si, Sr, and Zr). However, a key challenge for bimetallic catalysts remains the structural stability of these catalysts under reaction conditions, particularly concerning metal segregation. This could lead to increased energy consumption, creating potential obstacles for the decomposition of NH3.25 Given that enhanced metal interactions appear to contribute to higher catalytic activity, optimizing preparation methods and selecting appropriate metal salts could offer promising strategies for improving performance.26
Research has shown that alloying strategies can play a crucial role in facilitating the ammonia decomposition reaction to their monometallic counterparts by improving the catalytic performance of catalysts.25,27 In this area, a broad range of alloy systems have been developed, including Co alloys with Ni,28 Re29 and Ce,30 Ni alloys with Co, Fe, and Cu,31 Ru–Ni,32 Cu–Zn,33etc. These findings demonstrate that alloy-based catalysts could provide a more cost-effective alternative while preserving—or even exceeding—the catalytic efficiency of noble metals.34 However, choosing the right elements and appropriate stoichiometric composition remains a significant challenge for NH3 decomposition over alloyed catalysts to ensure both optimal activity and long-term stability.35 Therefore, one of the most significant challenges in the preparation of hydrogen from ammonia decomposition is to customize an effective catalyst that is energy efficient, highly selective, scalable, and affordable, providing a stable rate of decomposition at low temperatures.36
Recent research studies have shifted their focus from noble metals-based catalysts to alternative transition metal (TM) and TM-free catalysts. Among these, alkali amides and imides, particularly lithium (Li)-based compounds such as LiNH2 and Li2NH, have demonstrated a significant reduction in activation barriers by stabilizing M–N bond intermediates. Theoretical studies further highlight the importance of surface disorder dynamics in non-stoichiometric lithium amide compounds (Li2−x(NH2)x(NH)1−x) for TM-free catalysis, suggesting mechanistic differences from TM-based systems. Similar to TM-free NH3 synthesis catalysts, lithium amides/imides exhibit catalytic activity for ammonia decomposition both with and without transition metals. Notably, addition of TM to LiNH2 enhances NH3 conversion at 440 °C from 54% to 86%, in which the catalytic performance was also influenced by ammonia flow rates.37Table 1 provides a summary of the effective heterogeneous catalysts used for the thermal decomposition of ammonia.
Table 1 The effective heterogeneous catalysts utilized for the decomposition of ammonia18
| Catalyst |
Temperature (°C) |
Conversion (%) |
TOF (s−1) |
Ref. |
| Ni/MgAl2O4-LDH |
600 |
88.7 |
2.18 |
38
|
| Co/NC-600 |
500 |
80 |
|
39
|
| Ru/SmCeOx |
400 |
74.9 |
25.81 |
40
|
| 35Co/BHA |
500 |
87.2 |
|
41
|
| 2.5Ru/10C-rGO |
400 |
96 |
75.4 |
42
|
| CoRe1.6 |
500 |
∼90 |
|
29
|
| K+–Fe/C |
470 |
20 |
∼0.5 |
43
|
| Ru/Al2O3 |
580 |
|
6.85 |
44
|
| 20Co–10Ni/Y2O3 |
550 |
71.2 |
|
45
|
| Co-containing CNTs |
700 |
∼100 |
|
46
|
| Ni-10/ATP |
650 |
64.3 |
|
47
|
| α-FeO2O3-50@pSiO2 |
800 |
100 |
|
48
|
| 10%Co/MWCNTs |
600 |
|
8.15 |
49
|
| Ru/La0.33Ce0.67 |
450 |
91.9 |
11.4 |
50
|
| Ni5Co5/SiO2 |
550 |
76.8 |
|
28
|
| 5CMLa-5 |
550 |
82.7 |
|
51
|
| 1%K–Co/SiC |
350 |
33.1 |
9.3 |
52
|
| Pr–Ni/Al2O3 |
550 |
∼90 |
|
53
|
From this perspective, significant progress has been made in recent years in developing alternative methods, with a focus on reducing reaction temperatures. This advancement aims to lower energy consumption while enhancing hydrogen production efficiency.54 Electric currents, electron beams, microwaves, plasma, and solar energy are alternative approaches to provide new feasible solutions for ammonia decomposition reaction (Fig. 1).55 Although numerous reviews have been published on the progress of ammonia decomposition, the literature has overlooked assessing the different approaches for green H2 generation from NH3 and the subsequent technical barriers to achieving a futuristic fuel and sustainable energy vector.
 |
| | Fig. 1 The status of hydrogen generation from ammonia decomposition. This figure was adapted with permission from ref. 56. Copyright 2023, MDPI. | |
Hence, this review aims to summarize and analyze previously reported green ammonia cracking aspects, such as photocatalysis, electrocatalysis, plasma, and other approaches, as well as their mechanisms of catalytic activity. Furthermore, the influences of recent revolution in data science, artificial intelligence (AI), and machine learning (ML) on the discovery of novel catalysts for the NH3-to-H2 reaction are evaluated. Finally, based on the literature and our experience, the current challenges and future perspectives for achieving rapid commercialization of NH3 decomposition and green H2 production are discussed.
2. Photocatalysis
In recent decades, the study of photocatalytic ammonia decomposition technology has garnered significant attention due to their potential applications in energy production, driven by growing concerns over environmental impact and the increasing demand for energy amid dwindling nonrenewable fossil fuel resources.19 The photocatalytic decomposition of NH3 into N2 and H2 presents a viable approach, as it can be conducted at room temperature using recyclable catalysts and allows facile control of light exposure via a switch. Moreover, utilizing sunlight for ammonia decomposition represents an artificial photosynthetic reaction that proceeds under alkaline conditions.56
Fundamentally, photocatalysis involves a redox reaction that utilizes photo-generated electrons (e−) and holes (h+) in semiconductors. The overall process unfolds in three key stages. First, photons excite charge carriers, initiating the reaction. Next, these charges are separated and migrate across the photocatalytic surface. Finally, the photo-induced charge carriers drive catalytic reactions at the surface, facilitating water oxidation and reduction. These steps are elaborated in eqn (3)–(12).57–62
| | | Photocatalyst + hν → h+ + e− | (3) |
| | | O2 + e− → ˙O2− E0 = −0.33 V vs. NHE | (4) |
| | | H2O2 + e− + H+ → ˙OH + H2O E0 = 0.38 V vs. NHE | (5) |
| | | O2 + 2e− + 2H+ → H2O2 E0 = 0.68 V vs. NHE | (6) |
| | | HO2− + H2O + 2e− → 3OH− E0 = 0.87 V vs. NHE | (7) |
| | | 2˙O2− + 2H+ + e− → H2O2 E0 = 0.89 V vs. NHE | (8) |
| | | ˙O2H + H+ + e− → H2O2 E0 = 1.49 V vs. NHE | (9) |
| | | OH˙ + e− → OH− E0 = 1.64 V vs. NHE | (10) |
| | | H2O2 + e− + 2H+ → 2H2O E0 = 1.77 V vs. NHE | (11) |
| | | ˙OH + e− + H+ → H2O E0 = 2.33 V vs. NHE | (12) |
From a thermodynamic perspective, the production of hydrogen through the decomposition of ammonia (eqn (13) and (14)) is more favorable compared to water splitting (eqn (1) and (2)).63,64
| |  | (13) |
| |  | (14) |
Notably, photocatalytic reactions involving ammonia are feasible only when the reduction and oxidation potentials of ammonia fall between the semiconductor's conduction band (CB) and valence band (VB) potentials. During this process, various nitrogen-containing products such as N2, NO2−, NO3−, and NOx can be formed owing to their closely related redox potentials. The specific potentials of these reactions are detailed in eqn (15)–(20)65 and Fig. 2(a). Technically, ammonia decomposition can yield varying quantities of different products depending on several reaction parameters such as pH, temperature, initial ammonia or O2 concentration, and the presence of trapping or sacrificial agents. For an effective photocatalytic cracking of ammonia, the photo-generated electrons and holes on the semiconductor surface must possess appropriate reduction and oxidation capabilities. These enable reactions with species adsorbed on the catalyst surface, such as O2, NH4+, NO2−, and NO3−, facilitating the generation of free radicals or diverse products.
| | | 6H2O(l) + 6e− → 3H2(g) + 6OH−(aq) Cathode | (15) |
| | | 2NH4+(aq) + 8OH−(aq) → N2(g) + 8H2O(l) + 6e− Anode | (16) |
| | | 2NH4+(aq) + 2OH−(aq) → N2(g) + 3H2(g) + 2H2O(l) Full cell, Ecell = 0.27 V vs. SHE | (17) |
| | | 8H2O(l) + 8e− → 4H2(g) + 8OH− Cathode | (18) |
| | | NH4+(aq) + 10OH−(aq) → NO3−(aq) + 7H2O(l) + 8e− Anode | (19) |
| | | NH4+(aq) + 2OH−(aq) + H2O(l) → NO3−(aq) + 4H2(g) Full cell, Ecell = 0.88 V vs. SHE | (20) |
 |
| | Fig. 2 (a) Schematic process of photocatalytic NH3 decomposition. This figure was adapted with permission from ref. 66. Copyright 2020, Royal Society of Chemistry. (b) Proposed reaction mechanism for NH3 decomposition to N2 and H2 on the TiO2 photocatalyst. This figure was adapted with permission ref. 19, Elsevier. (c) TEM image and (d) HAADF-STEM image of SA Co/CeO2. (e) Photograph of SA Co/CeO2 loaded in a novel solar-heating device to drive hydrogen fuel cell under 2 solar irradiation. (f) H2 production rate from NH3 decomposition by SA Co/CeO2. (g) Energy profiles of NH3 decomposed as H2 and N2 on SA Co/CeO2 (111) and Co (111) surfaces. These figures were adapted with permission from ref. 67. Copyright 2023, Elsevier. | |
Furthermore, the photocatalytic decomposition of ammonia typically ceases under acidic conditions, suggesting that H+ ions impede the transformation of ammonia into NH2 free radicals. As the pH increases, ammonia is likely to react with surrounding oxygen, forming NO2−, NO3−, and other nitrogen oxides and adversely affecting hydrogen production. In addition, many photocatalysts currently face significant challenges with carrier recombination and poor light-harvesting efficiency. Consequently, the development of more effective photocatalytic materials remains crucial for the advancement of ammonia treatment through photocatalysis.68
To date, only a group of photocatalysts, such as TiO2, ZnO, ZnS, Mo2N, graphene, and their metal-loaded hybrid materials, have been found effective in decomposing aqueous ammonia solutions.56 However, their hydrogen production rate, capped at 15.56 μmol g−1 min−1, remains insufficient to satisfy practical application requirements.67 To address this shortcoming, Utsunomiya et al.19 investigated the photocatalytic performance of Ni/TiO2 catalysts toward ammonia decomposition and explored the mechanism of NH3 breakdown by proposing three distinct reaction pathways (Fig. 2(b)). These pathways involved the formation of N2 and H2 through intermediates radicals: route 1 entailed the formation of NH radicals via the removal of one hydrogen atom from two NH2 radicals; route 2 involved the direct coupling of adjacent NH2 radicals to form NH2–NH2; and route 2′, where NH2–NH2 formation occurred through the interaction of H2N–NH3. The activation energies for routes 1 and 2 were determined to be 236 kcal mol−1 and 74.8 kcal mol−1, respectively, with route 2 being more energetically favorable. Additionally, the pathways for N2 and H2 formation via NH2–NH2 coupling were further delineated into route 2, which involved the coupling of NH2 radicals to form H2N–NH2, and route 2′, where NH2 interacted with an NH3 molecule in the gas phase.
Alternatively, beyond photocatalysis, solar heating catalysis demonstrates the highest efficiency in sunlight utilization (approaching 100%) and can achieve temperatures up to 400 °C. This facilitates the heating of catalysts for thermocatalysis under natural solar irradiation. In the context of solar-powered ammonia decomposition, cobalt-based catalysts are preferred due to their abundance and effectiveness.67 Yuan et al.67 developed a catalyst by immobilizing single atoms of cobalt on cerium dioxide nanosheets (SA Co/CeO2) (Fig. 2(c) and (d)) for photocatalytic degradation of ammonia in tubular reactor at low temperatures (Fig. 2(e)). As can be seen in Fig. 2(f), integrated with a custom-built TiC/Cu-based solar-heating device, the SA Co/CeO2 demonstrated a stable hydrogen generation rate of 2.7 mmol g−1 min−1 under 2 suns irradiation, which is 572 times more effective than traditional weak sun-light-driven NH3 decomposition. The hydrogen produced was found to be sufficiently pure to power a hydrogen fuel cell without further purification directly. Theoretical calculations revealed that SA Co/CeO2 significantly lowers the energy barrier for nitrogen binding during ammonia decomposition, thereby enhancing the reaction's progress (Fig. 2(g)).
Moreover, Lin et al.69 employed a straightforward nebulization-coating technique to immobilize a wide array of single-atom transition metals (TMs: Co, Mn, Fe, Ni, and Cu) on microporous carbon nitride (MCN) (Fig. 3(a)), creating catalyst panels designed for solar-light-driven photocatalytic gaseous ammonia splitting (Fig. 3(b)). Under ambient conditions, the optimized Ni-MCN demonstrated a hydrogen production rate of 35.6 μmol g−1 h−1, significantly outperforming pure MCN (by approximately 14-fold) and other composite alternatives (Fig. 3(c)). This enhanced photocatalytic activity and photocurrent response (Fig. 3(d)) can be attributed to the presence of Ni–N4 sites, which enhance the optical properties, expedite charge carrier separation/transfer, and improve the kinetics of ammonia splitting on the catalysts. Regarding Fig. 3(e), the Ni site on MCN is the most favorable for NH3 splitting among all these TMs due to its lowest free energy increase in the potential-determining step (PDS).
 |
| | Fig. 3 (a) Schematic of the synthesis procedure of TMs-MCN SACs. (b) Illustration of the reaction system and the nebulization-coating method for fabricating TM-MCN panels. (c) Rates of H2 production by CN, MCN, and Ni-MCNs with different Ni loadings and TMs MCNs (TMs = Mn, Fe, Co, Ni, Cu, with a similar loading percentage of ∼1.4 wt%). (d) Photocurrent responses of MCN and Ni-1.4-MCN (inset: picture of the large-sized cell). (e) Energy profiles for the reaction process. These figures were adapted with permission from ref. 69. Copyright 2023, American Chemical Society. (f) Correlative probe and electron microscopy image of hematene sheet. (g) Decomposition of ammonia over Ru-hematene samples to optimize weight loading of Ru. (h) Schematic mechanism of ammonia decomposition by the Ru-hematene photocatalyst. These figures were adapted with permission from ref. 70. Copyright 2023, Elsevier. | |
In another study, Dzíbelová et al.70 utilized an ultrasound-supported exfoliation technique to anchor ruthenium oxide nanoparticles onto 2D hematene (α-Fe2O3) (Fig. 3(f)) for the decomposition of an aqueous ammonia solution into hydrogen and nitrogen under visible light irradiation. Experimental results demonstrated that with an optimal ruthenium dosage of 0.5 wt%, the Ru-hematite, after 24 hours, achieved a hydrogen yield that was 2.5 times higher than that of pure hematene (Fig. 3(g)), attributed to the enhanced generation of electrons and holes (Fig. 3(h)). Moreover, without any cleaning interventions, the Ru-hematite photocatalyst exhibited only an 11% reduction in photocatalytic activity after five consecutive runs, suggesting its suitability for practical applications.
3. Electrocatalysis
Recently, hydrogen generation through electrochemical reactions, such as ammonia decomposition by electrocatalysis at moderate temperatures, has attracted more attention.71 However, electrocatalytic ammonia decomposition faces challenges such as high overpotentials, unfavorable thermodynamics, and slow reaction kinetics.72 Based on the different existing states of NH3, electrochemical ammonia decomposition can be categorized into the electrolysis of (i) aqueous ammonia solution, or (ii) liquid ammonia. In the electrolysis of alkaline aqueous ammonia solutions, NH3 undergoes oxidation in the existence of OH− ions at the anode (eqn (21)),73 whereas H2O can be reduced at the cathode (eqn (22) and (23)).71,74,75| | | 2NH3 + 6OH− → N2 + 6H2O + 6e− Anode, E0 = −0.77 V vs. SHE | (21) |
| | | 6H2O + 6e− + 3H2 + 6OH− Cathode, E0 = −0.83 V vs. SHE | (22) |
| | | 2NH3 → N2 + 3H2 Overall, Ecell = 0.06 V | (23) |
In 2002, Zisekas et al.76 utilized silver as the reactor electrode to conduct the first tests on hydrogen production via electrocatalytic decomposition of ammonia. These tests demonstrated that NH3 conversion efficiency ranged between 25 and 35% at temperatures of 773–873 K, indicating that reactor temperatures remained relatively high. It is well accepted that the energy density of hydrogen in liquid ammonia [NH3(l), 3.6 kW h L−1] is significantly higher compared to that in alkaline aqueous ammonia solution [NH3(aq), 1.0 M, 0.1 kW h L−1]. Fundamentally, the electrolysis of liquid ammonia differs from that of aqueous ammonia solutions, as both the anodic and cathodic half-reactions in liquid ammonia avoid the excessive oxidation of NH3 that typically occurs in the presence of water.71 Furthermore, the gravimetric H2 density is as low as 6.1 mass% according to its solubility in water, 34.2 mass% at 20 °C.77 Accordingly, Hanada et al.78 evaluated the direct electrolysis of liquid ammonia, where alkaline metal amides (MNH2, M = Li, Na, K) were utilized as the supporting electrolyte. Amides such as LiNH2, NaNH2, KNH2, and N,N-dimethylformamide have enabled the electrolysis of liquid ammonia.79 This process was conducted at exceptionally low temperatures, ranging from −70 to −65 °C, using pure platinum electrodes.80
Moreover, during electrocatalytic reaction, NH3 can be converted aqueous to NO3− and then to products such as HNO2, NO, NH2OH, NH3, N2O, and N2 (eqn (24)–(30)). Generated nitrogen is benign and easily separable, and is the most stable nitrate reduction product with a standard redox potential (E0) of 1.25 V vs. RHE.72
| | | NH3(aq) + 3H2O(l) → NO−3(aq) + 9H+ + 8e− E0 = 0.82 V vs. RHE | (24) |
| | | NO−3(aq) + 2H+ + 2e− → NO−2(aq) + H2O(l) E0 = 0.85 V vs. RHE | (25) |
| | | NO−3(aq) + 3H+ + 2e− → HNO2(aq) + H2O(l) E0 = 0.89 V vs. RHE | (26) |
| | | NO−3(aq) + 4H+ + 3e− → NO(aq) + 2H2O(l) E0 = 0.96 V vs. RHE | (27) |
| | | NO−3(aq) + 7H+ + 6e− → NO2OH(aq) + 2H2O(l) E0 = 0.67 V vs. RHE | (28) |
| | | 2NO−3(aq) + 10H+ + 8e− → N2O(g) + 5H2O(l) E0 = 1.12 V vs. RHE | (29) |
| | | 2NO3−(aq) + 12H+ + 10e− → N2(g) + 6H2O(l) E0 = 1.25 V vs. RHE | (30) |
Technically, the electrocatalytic decomposition of NH3 encompasses two primary phases: (i) the ammonia oxidation reaction (AOR) and (ii) the hydrogen evolution reaction (HER). An effective high-efficiency electrocatalyst should be capable of catalyzing both the AOR and HER at a low potential.36 Despite its potential, the activity level of AOR is insufficient for low-temperature operations. The AOR integrates more easily with fuel cell technologies and provides a more effective method for hydrogen generation than water electrolysis, which represents the second most common hydrogen production method and accounts for approximately 4% of global hydrogen production. In addition, water electrolysis, which is not thermodynamically favored, theoretically necessitates applying a voltage as high as 1.23 V to break down highly stable water molecules, requiring about 180 MJ of energy to produce 1 kg of H2. In contrast, the AOR has a significantly lower energy requirement of approximately 33 MJ per kg of hydrogen generated.81
Research has demonstrated that the electrocatalytic activity for ammonia oxidation correlates with the surface properties of materials. The catalyst remains active when intermediate NHx species are present but becomes inhibited when strongly binding nitrogen species are formed.82 In this context, the widely accepted mechanism was proposed by Gerischer and Mauerer in 1970, with the fundamental steps detailed in eqn (31)–(35). Briefly, NH3 can be deprotonated in the presence of hydroxyl ions in eqn (31)–(33), producing water molecules while simultaneously releasing an electron at each step. N* adatoms (formed in eqn (33)) are surface poisons because of a typically large kinetic barrier for N–N bond formation and release nitrogen. Thus, adsorbed NHx (and NHy) species can interact with one another to form an N–N bond, subsequently generating an HxNNHy species (eqn (34)). Regarding the Gerischer–Mauerer mechanism, these species are then deprotonated to N2, which desorbs from the surface; however, the identity of the NHx and NHy species that react to form the N–N bond remains in dispute (eqn (35)).83
| | | NH3 + OH− + * → NH2* + H2O + e− | (31) |
| | | NH2* + OH− + * → NH* + H2O + e− | (32) |
| | | NH* + OH− + * → N* + H2O + e− | (33) |
| | | NHx* + NHy* → HxNNHy* + * | (34) |
| | | HxNNHy* + (x + y)OH− → N2 + (x + y)H2O + (x + y)e− + * | (35) |
The catalytic activity can be substantially enhanced when a portion of the metal is utilized as an electrode by applying a current or potential between the catalyst and a counter electrode deposited on the solid electrolyte.36 To date, a diverse range of materials, such as noble metals, metal oxides, and non-metals, have demonstrated high catalytic activity for ammonia electro-oxidation. Notably, Pt has emerged as the most effective electrocatalyst for this reaction. However, most ammonia electro-oxidation systems necessitate strong alkaline media (such as NaOH), leading to rapid deactivation and poisoning of Pt catalysts, as well as oxygen evolution and NOx production. Under strongly alkaline conditions, electrodeposited Pt-based electrodes and nanotubes have effectively oxidized ammonia into hydrogen.80
In this context, Herron et al.83 examined AOR efficiency on various face-centered cubic (fcc) metals (Au, Ag, Cu, Pd, Pt, Ni, Ir, Co, Rh, Ru, Os, and Re) using density functional theory (DFT) calculations. They reported that Pt exhibited the most promising catalytic activity, followed by Ir and Cu, due to its low onset potential (Fig. 4(a) and (b)). It was found that adsorbed NH2 was the dominant intermediate, facilitating the preferred N–N bond formation both kinetically and thermodynamically (Fig. 4(c)). In another study, Zhong et al.82 investigated catalytic electro-oxidation of liquid ammonia using transition metal dimers (Fe2, Co2, Ru2, Rh2, and Ir2) anchored on graphite-carbon nitride monolayers (TM2@g-CN). Their findings reinforce the mechanism proposed by Gerischer and Mauerer, where N–N bond formation is facilitated by hydrogenated NHx species rather than N adatoms (Fig. 4(d)). Importantly, catalytic activity studies demonstrated that Fe, Co, Ru, Rh, and Ir anchored in g-CN monolayers are exceptionally promising AOR catalysts due to their low limiting potentials of −0.47, −0.5, −0.48, −0.52, and −0.48 V, respectively. Among them, Ir2@g-CN, as a bifunctional catalyst for electrocatalytic NH3 decomposition, showed low energy barriers of 0.48 eV and 0.20 eV for the AOR (Fig. 4(e)) and HER (Fig. 4(f)), respectively. It was observed that modulating TM atoms with varying d-electron numbers allows for tuning the d-band center (εd) of TM atoms on TM2@g-CN composites, providing a predictive measure for AOR performance and offering theoretical guidance for designing advanced AOR electrocatalysts.
 |
| | Fig. 4 (a) Estimated onset potential for close-packed facets of transition metals. (b) Activity as predicted by Sabatier analysis for both mechanisms at 0 VRHE. (c) Free-energy diagram for ammonia electro-oxidation on Pt(111) at 0 VRHE. These figures were adapted with permission from ref. 83. Copyright 2015, American Chemical Society. (d) Proposed mechanism for the AOR on TM2@g-CN. (e) The calculation free energy diagram of the AOR through the Gerischer–Mauerer mechanism over Ir2@g-CN at different applied potentials. (f) The calculated free energy diagram of the HER on TM2@g-CN samples. These figures were adapted with permission from ref. 82. Copyright 2023, Elsevier. | |
In Modisha's study,84 decomposition of ammonia using a Pt–Ir electrocatalyst was investigated in a potassium hydroxide (KOH, 5 M) solution (Fig. 5(a)). This research demonstrated that the current density of NH3 electro-oxidation reaction rose at high temperature and ammonia concentration, achieving a peak ammonia conversion of 78% at 2300 ppm (Fig. 5(b)). From Fig. 5(c), the highest hydrogen flow rate recorded was 25 L h−1, with an associated energy consumption of 1.6 W h L−1 H2−1. The purity of hydrogen, as determined by gas chromatography, was found to be 86%. Moreover, Dong et al.85 synthesized five types of electrocatalysts, including Pt-black, Rh, Pt–Ir, Rh–Pt, and Rh–Pt–Ir alloys, aimed at reducing the overpotential of the anode reaction for ammonia cracking (Fig. 5(d)). These alloys were electrochemically evaluated for their efficacy in ammonia decomposition in the presence of NH4Cl. The trimetallic Rh–Pt–Ir and the bimetallic Pt–Ir, as well as the Rh-contained alloy electrodes, demonstrated enhanced activity and reduced deactivation. Notably, the Rh–Pt–Ir alloy anode (Fig. 5(e)) exhibited the highest electrocatalytic performance, achieving the lowest minimum potential (Emin) of approximately 0.47 V and the highest current density of 46.9 mA cm−2 at 2.0 V (Fig. 5(f)). The results of this research reflect that the energy required for hydrogen generation from the electrocatalysis of liquid NH3 can be significantly lowered through strategic selection and compositional optimization of alloy catalysts.
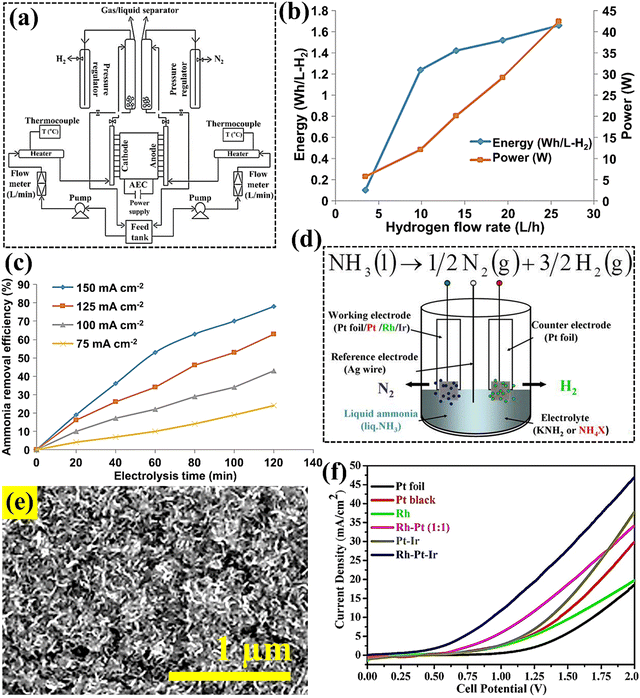 |
| | Fig. 5 (a) Schematic representation of the ammonia electrolysis process for hydrogen production. (b) Volumetric hydrogen generation rates and the required energy and power input (cell retention time (Rt): was 12.5 min, 2000 ppm NH3 in 5 M KOH at 55 °C). (c) Ammonia decomposition efficiency as a function of time and current density. These figures were adapted with permission from ref. 84. Copyright 2016, Elsevier. (d) Schematic electrolysis of liquid ammonia using different ammonium salt electrolytes with the reference electrode. (e) SEM image of the freshly prepared Rh–Pt–Ir electrocatalyst. (f) Cyclic voltammetry curves of NH3 (l) with 1 M NH4Cl in a two-electrode system. These figures were adapted with permission from ref. 85. Copyright 2016, Elsevier. | |
4. Plasma
As mentioned earlier, significant research efforts have been directed toward developing alternative energy supply methods for ammonia decomposition. These methods include the use of electric currents,86 electron beams,87 microwaves,88 and plasma,89 which offer higher conversion rates at lower temperatures compared to traditional thermocatalysis. Among them, there has been a notable shift towards exploring non-thermal plasma (NTP) for catalytic ammonia decomposition at low temperatures. This approach potentially enhances the response time and modularity of ammonia-based hydrogen production systems, thereby improving their applicability in sectors like transportation.90
Plasma is generally defined as a state of gas where the atoms are partially or fully ionized, maintaining overall electrical neutrality, containing a large number of highly energetic electrons and reactive species (e.g., excited molecules, atoms, ions, and radicles) (Fig. 6(a)–(c)).9,91,92 It was found that integrating non-thermal plasma with a catalyst can significantly modify the catalytic reaction pathway, leading to enhanced selectivity or reaction rates through the interaction of plasma, reactant, and catalyst.91 Notably, the H2 energy yield from the plasma catalysis method is nearly five times greater than that achieved through thermocatalysis. This indicates that plasma catalysis substantially enhances the economic efficiency of hydrogen production from ammonia.9 In 2006, research on the impact of dielectric barrier discharge plasma on ammonia decomposition revealed that using a commercially available bulk Fe-based catalyst significantly increased NH3 conversion rates. Specifically, NH3 conversion escalated from 7.4% to 99.9% when the Fe-based catalyst was positioned within a plasma zone at 410 °C.93 In this context, the researchers further optimized the ammonia decomposition process by utilizing low-temperature plasma, either using high-performance catalysts or by identifying optimal reaction conditions.
 |
| | Fig. 6 (a) Schematic representation of the plasma-catalytic NH3 decomposition. This figure was adapted with permission from ref. 94. Copyright 2024, American Chemical Society. In situ plasma-assisted catalytic NH3 conversion system (b) under NTP conditions and (c) glow discharge reactor. These figures were adapted with permission from ref. 95. Copyright 2022, MDPI. (d) TEM micrographs of Fe, Co, Ni, and Cu catalysts supported on fumed SiO2 (reduced in H2 plasma). (e) NH3 conversions on Co catalysts on various supports as a function of relative dielectric constants of supports in plasma + catalyst mode (NH3 feed 40 mL min−1, temperature 450 °C, supported catalyst 0.88 g, discharge gap 3 mm, discharge frequency 12 kHz). (f) Influence of metals on NH3 conversion in plasma + catalyst, plasma, and catalyst modes (similar condition for 3b). These figures were adapted with permission from ref. 94. Copyright 2024, American Chemical Society. | |
In 2015, Wang et al.93 conducted a comparative study on the efficiencies of hydrogen generation from ammonia using both thermal and plasma catalysis methods over different low-cost metal catalysts (Fe, Co, Ni, Cu) on a fumed SiO2 support (Fig. 6(d)). The results indicated that the NH3 conversion strongly depends on the metal–N bond strengths and on the relative dielectric constant (εd) of the support in the plasma reaction. It was observed that the moderately strong Co–N bonds can be expected to improve the plasma–catalyst synergy, thus leading to higher NH3 conversions (Fig. 6(e)). Notably, the relative εd of the support can efficiently contribute to plasma-catalytic NH3 decomposition performance. As shown in Fig. 6(f), a support with a small εd facilitates plasma-catalytic NH3 decomposition. Technically, plasma gas discharge can rapidly heat the reaction/catalyst, enhancing the energy efficiency of hydrogen production. Additionally, plasma has been shown to facilitate the rate-limiting step by accelerating the recombinative desorption of Nad from the catalyst bulk structure.91 In another study, the effects of varying discharge zone lengths on the efficiency of plasma-catalytic ammonia decomposition at a set discharge frequency were investigated. The research revealed that doubling the discharge zone length from 3.0 cm to 3.5 cm at a frequency of 10 kHz resulted in a twofold increase in NH3 conversion efficiency.9 Consequently, these findings indicate that non-thermal plasma and catalysts can synergistically interact to efficiently convert NH3 to H2 under mild conditions. However, despite the potential of plasma-catalytic processes, the number of catalysts tested and evaluated remains limited compared to those used in thermocatalysis.
Tables 2–4 summarize the literature on hydrogen generation from ammonia cracking through photocatalytic, electrocatalytic, and plasma processes, respectively.
Table 2 Performance comparison of different catalysts toward photocatalytic NH3 decomposition
| Photocatalyst |
Light source |
Time |
Initial NH3 concentration |
Maximum decomposition ability |
Ref. |
| Pt–TiO2 (0.5 wt% Pt) |
450 W high pressure Hg lamp |
24 h |
5 mM |
Over 95% |
96
|
| Ni/TiO2 (0.5 wt% Ni) |
500 W Xe lamp |
3 h |
5 mL, 0.59 mol L−1 |
131.7 μmol H2 per g-catalyst |
19
|
| Ce-doped TiO2 (1.4 wt% Ce) |
8 W Hg pen-ray lamp |
10 h |
100 mL, 0.8274 g L−1 |
1010 mmol H2 per g-catalyst |
97
|
| N–C@TiO2 |
25 W UV lamp |
5 min |
100 μL aqueous ammonia (30%) |
100% |
98
|
| MoS2@TiO2 |
25 W UV lamp |
7 min |
100 μL aqueous ammonia (30%) |
91% |
99
|
| MoS2/N-doped graphene |
300 W UV-visible lamp |
8 h |
100.0 mg L−1 |
99.6% |
100
|
| Nitrogen-doped rGO/TiO2 |
8 W Hg pen-ray lamp |
12 h |
0.883 g L−1 |
208 μmol h−1 g−1 |
80
|
| GQDs (graphene quantum dots)/CN (g-C3N4) |
150 W Xe arc lamp |
7 h |
1.5 mg L−1 |
90% |
101
|
| ZnO/Ag |
300 W Xe lamp |
2.5 h |
1.5 mg L−1 |
Circa 90% |
102
|
Table 3 Performance comparison of different catalysts toward electrocatalytic NH3 decomposition
| Catalyst |
Catalyst loading (mg cm−2) |
Electrolyte |
Onset potential |
Current density (mA cm−2) |
Scan rate (mV s−2) |
Ref. |
| PtIr/C |
2.00 |
1.0 M NH3 |
0.470 VRHE |
— |
10 |
103
|
| RGO/Pt–Ir |
— |
1.0 M NH4OH |
−0.400 VAg/AgCl |
20.00 at 0 VAg/AgCl |
10 |
104
|
| Pt90Ru10/C |
1.00 |
1.0 M NH4OH + 1.0 M KOH |
— |
0.920 at −0.210 VHg/HgO |
20 |
105
|
| PtxIr100−x/MgO |
— |
0.1 M NH3 + 0.2 M NaOH |
0.530 VRHE |
1.00 at 0.710 VRHE |
20 |
106
|
| SnO2–Pt/C |
0.028 |
0.1 M NH3 + 0.1 M KOH |
0.450 VRHE |
1.62 at 0.690 VRHE |
20 |
53
|
Table 4 Performance comparison of different catalysts toward plasma-assisted catalytic NH3 decomposition
| Catalyst |
Power |
Reactor configuration |
Temperature (°C) |
Pressure (bar) |
Catalyst amount (g) |
NH3 flow rate (L min−1) |
NH3 conversion rate (%) |
Ref. |
|
Note: dielectric barrier discharge.
|
| FeO |
12 kHz |
DBDa reactor |
410 |
— |
10 |
0.04 |
>99.9 |
107
|
| 26 W |
| Ni–Al2O3 |
23.8 kHz |
Non-thermal arc reactor |
400–843 |
— |
200 |
30.00 |
— |
108
|
| 0–700 W |
| Ru–Al2O3 |
10 kHz |
DBD reactor |
— |
1 |
— |
0.10–1.00 |
85.7 |
109
|
| 12–20 kV |
| Metal–MgAl2O4 |
1.0–3.5 kHz |
DBD reactor |
— |
1 |
— |
1.00 |
82.0 |
110
|
| 10–25 W |
| No catalyst |
10 kHz |
DBD reactor |
— |
— |
— |
0.50 |
13.0 |
111
|
| 3.5–22 kV |
5. Other approaches
It is well accepted that the alternative methods for ammonia decomposition, such as electrocatalysis and photocatalysis, often depend on complex catalysts that include costly precious metals. While non-precious metals are more affordable, they tend to exhibit lower catalytic efficiencies and reduced stabilities.112 To tackle the challenges in conversion performance, recently, ammonia–water has been recognized as a promising liquid hydrogen carrier with the potential for widespread use in hydrogen generation. However, the hydrogen derived from ammonia–water still poses major challenges. Despite advancements in various methods to enhance the efficiency of H2 production from ammonia–water, these techniques have yet to reach a level that is suitable for practical industrial applications.112 In this context, Yan et al.112 developed a novel, eco-friendly, and ultrafast method for extracting hydrogen from ammonia–water without a catalyst and under ambient conditions using the laser bubbling in liquids (LBL) approach. This technique is entirely different from conventional catalytic methods for hydrogen extraction from ammonia–water. The LBL involves a focused pulsed laser directly beneath the surface of the liquid. When the pulsed laser is applied to ammonia–water, the molecules can be rapidly excited and ionized. This process generates cavitation bubbles at the focus point that achieve transient high temperatures, creating an optimal microspace for efficient hydrogen extraction. It was reported that the real adequate time of laser action on ammonia–water was just 0.36 ms per hour and the actual hydrogen yield reached 93.6 mol h−1 at laser “light-on” time, reflecting the acceptable efficiency of the LBL process.
Another attempt to develop a new approach toward ammonia decomposition was performed by McLennan and Greenwood,113 discovering that electric discharge in a cathode ray tube can rapidly decompose ammonia. By eliminating the electric current, they focused on the decomposition using only high-speed electron beams, achieving up to 30% decomposition efficiency with pure ammonia. They also examined the effects of the presence of high-speed electrons (electron spark) on the reaction rate, noting ammonia dilution with N2 enhanced the reaction while H2 inhibited it. Similarly, Hirabayashi and Ichihashi114 explored ammonia decomposition using ion beams on various catalysts, identifying vanadium and niobium nitrides (VnNm+ and NbnNm+ (n = 3–6; m = n, n − 1)) as promising for hydrogen production.
6. Predicting NH3 decomposition efficiency by machine learning
It is well recognized that the broad temperature range of the ammonia decomposition process in practical applications makes it difficult to monitor catalyst changes during the reaction, which is also a major barrier to its practical implementation.115 In addition, discovery of catalysts for NH3 decomposition is a crucial aspect and has traditionally relied on trial-and-error experiments.115–117 Therefore, utilizing DFT and numerical modeling techniques can significantly accelerate research by validating the fundamental mechanisms of ammonia decomposition.34
In recent decades, machine learning (ML) technology has emerged as a powerful tool in designing novel catalysts, understanding composition-structure–property relationships, and analyzing complex data patterns. This efficient computational approach streamlines thermocatalytic ammonia decomposition while minimizing the need for extensive human and material resources in catalyst design.115 The strength of ML algorithms lies in their capacity to learn from historical data without the need for explicit programming. This approach is anticipated to demonstrate high fidelity in identifying optimal operating conditions, not only for NH3 cracking in the gas phase but also for optimizing CO2 capture, hydrocracking, and dimethyl ether synthesis.118
To date, several studies have utilized ML models to identify and optimize catalysts for ammonia decomposition. For instance, Williams et al.119 assessed the integration of ML with high-throughput experimentation to optimize catalyst compositions with low ruthenium content for NH3 decomposition in a 16-channel parallel reactor system. They developed a model training in three progressive stages, utilizing datasets of 3, 22, and 28 catalysts. By analyzing the chemical properties of the secondary metal and reaction temperature, the model effectively predicted ammonia decomposition efficiency. It was found that by employing the random forest algorithm the catalyst performance was predicted with a mean absolute error of less than 0.16, demonstrating the approach's accuracy and reliability. In another study, Guo et al.115 utilized ML using random forest regression, support vector machines, and gradient boost regression approaches to statistically analyze ammonia decomposition as a function of catalyst properties and reaction conditions. Their findings revealed a strong positive correlation between the catalytic efficiency and reaction temperature, with the gas hourly space velocity (GHSV) emerging as a key factor influencing both NH3 conversion and H2 production rates. Notably, optimal decomposition and hydrogen formation were achieved with a total metal loading below 20%wt. It was concluded that among the models tested, the gradient boost regression tree demonstrated strong predictive accuracy, achieving a coefficient of determination (R2) greater than 0.85, a root mean square error (RMSE) below 13.24, and a mean absolute error (MAE) under 10.31.
Although ML has shown promising results, developed models often tend to be case-specific, limiting their generalizability across different catalyst systems. In addition, traditional ML-guided catalyst screening can be challenging to obtain new catalytic systems, specifically molecular catalysts for AOR electrocatalysts.120 To address these theoretical limitations, recent studies have aimed to develop an ML model specifically designed to accurately predict the conversion of NH3 to H2. By compiling data from published literature, an extensive experimental database was established, and the relationship between independent variables and dependent responses was thoroughly evaluated using statistical analyses and mechanistic insights.115
Moreover, integrating reaction kinetics into the ML framework presents a promising research direction. In this context, understanding the role of reaction kinetics in predicting H2 formation rates during NH3 decomposition can provide valuable insights into catalyst behavior and improve predictive accuracy. Additionally, incorporating hydrogen inhibition effects into the ML model will enable researchers to better capture catalyst dynamics, ultimately enhancing performance predictions.115 Notably, the exploration of advanced ML techniques can offer new insights into developing the connections among the characteristics of substances and their catalytic activity, selectivity, and stability of the complex catalytic systems.121 In the future, with further research and widespread application of artificial intelligence technologies, ammonia decomposition processes are expected to become more efficient and environmentally sustainable, offering robust support for sustainable energy development.
7. Further challenges for ammonia decomposition
Extensive research has focused on developing highly active and durable catalysts for ammonia decomposition at minimal temperatures, intending to further lower the temperature to improve efficiency and promote environmentally sustainable processes. It is widely recognized that support materials for NH3 decomposition catalysts should exhibit high basicity alongside high conductivity, low concentrations of electron-withdrawing groups, and extensive surface area. Increased basicity can enhance the dispersion of the active metal, thereby facilitating the dehydrogenation of ammonia and the recombinative desorption of surface nitrogen atoms, which are likely the rate-limiting steps of the reaction. Additionally, the electron-donating characteristics of the catalyst can indirectly interact with the support to promote stronger basicity. Consequently, adjusting the basicity of the supports is essential for developing efficient catalysts toward NH3 decomposition. Beyond Ru and Ni catalysts, nitrides, carbides, and perovskites have also gained popularity for optimizing active components in catalytic processes. Looking ahead, it will be valuable to explore methods for separating and purifying hydrogen derived from ammonia decomposition in a cost-effective and highly efficient manner. Additionally, microwave and plasma-based decomposition of ammonia merits further investigation.
Moreover, the economic evaluation of a large-scale NH3 decomposition plant has revealed a significant reliance on the cost of the green ammonia industry. It was demonstrated that lower costs of renewable energy and green ammonia production lead to reduced hydrogen production costs via NH3 decomposition. With a baseline price of green ammonia set at 450 € per ton, the estimated levelized cost of hydrogen (LCOH) was approximately 4.82 € kg−1 (in 2019). However, this estimate is influenced by several uncertainties, including the accuracy of total investment cost estimation (±30%), the ammonia decomposition kinetics that affects cracker size and consumption, and the use of ammonia/hydrogen blends as fuel for endothermic reactions in conventional burners. In summary, if green ammonia becomes cost-competitive with fossil-based ammonia, with an estimated price range of 210–215 € ton−1, the cost of producing pressurized hydrogen through ammonia decomposition would be approximately 3.00 € per kg, excluding any potential regulatory or financial incentives.122
8. Conclusions and perspectives
Ammonia, as an ideal hydrogen storage material, is expected to be used to address the challenges of hydrogen storage and transportation in the development of the hydrogen energy industry and overcome the safety problems related to hydrogen utilization. Although many ammonia-related studies have contributed to the promotion of ammonia as a favorable alternative renewable resource, further improvements are required in ammonia decomposition to make it a practical H2 carrier option for on-site generation.
Currently, the great potential of direct ammonia fuel cells for electricity generation in vehicles, hydrogen refilling stations, and ammonia combustion for power underscores the need for ammonia decomposition under mild conditions. Although progress toward the commercialization of direct ammonia fuel cells is ongoing, in the future, within both distributed and grid power supply systems, a carbon-neutral energy system that combines green ammonia synthesis with ammonia fuel cell technology could become a reality.
In conclusion, combining ML with computational modeling or experiments opens up new possibilities for rapid screening of the catalysts, identifying the performance descriptors, and assisting in catalyst manufacturing. However, further steps are required to couple experimental and theoretical techniques to gain a fundamental understanding, which will inspire researchers to design advanced catalysts for mild-condition ammonia synthesis and decomposition.
Data availability
No primary research results, software or code, have been included and no new data was generated or analyzed as part of this review.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge funding from the US National Science Foundation (NSF) grant (IIP 1939876). The NSF supported this study (Award numbers: 2315268—the Great Lakes Water Innovation Engine and 2314720).
References
- M. El-Shafie, S. Kambara, S. P. Katikaneni, S. N. Paglieri and K. Lee, Int. J. Hydrogen Energy, 2024, 65, 126–141 CAS.
- X. Niu, C. Li, X. Li and Y. Zhang, Heliyon, 2024, 10, e25087 Search PubMed.
- Z. Bao, D. Li, Z. Wang, Y. Wen, L. Jin and H. Hu, Fuel, 2024, 373, 132346 CAS.
- F. Schüth, R. Palkovits, R. Schlögl and D. S. Su, Energy Environ. Sci., 2012, 5, 6278–6289 Search PubMed.
- J. Li, S. Lai, D. Chen, R. Wu, N. Kobayashi, L. Deng and H. Huang, Front. Energy Res., 2021, 9, 760356 Search PubMed.
- C. Qin, S. Ruan, C. He and L. Zhang, Colloids Surf., A, 2024, 691, 133898 CAS.
- N. Li, C. Zhang, D. Li, W. Jiang and F. Zhou, Chem. Eng. J., 2024, 495, 153125 CAS.
- M. Ghasemi, J. Choi, S. M. Ghoreishian, Y. S. Huh and H. Ju, J. Electrochem. Soc., 2023, 170, 074501 CAS.
- N. Zhu, Y. Hong, F. Qian and J. Liang, Int. J. Hydrogen Energy, 2024, 59, 791–807 CAS.
- Z. Bao, D. Li, Y. Wu, L. Jin and H. Hu, Int. J. Hydrogen Energy, 2024, 53, 848–858 CrossRef CAS.
- S. S. Rathore, S. Biswas, D. Fini, A. P. Kulkarni and S. Giddey, Int. J. Hydrogen Energy, 2021, 46, 35365–35384 CrossRef CAS.
- G. Glenk and S. Reichelstein, Nat. Energy, 2019, 4, 216–222 CrossRef CAS.
- Y. Gu, Y. Ma, Z. Long, S. Zhao, Y. Wang and W. Zhang, Int. J. Hydrogen Energy, 2021, 46, 4045–4054 CrossRef CAS.
- K. Grubel, H. Jeong, C. W. Yoon and T. Autrey, J. Energy Chem., 2020, 41, 216–224 CrossRef.
- I. Cabria, Int. J. Hydrogen Energy, 2024, 50, 160–177 CrossRef CAS.
- Z. Yan, H. Liu, Z. Hao, M. Yu, X. Chen and J. Chen, Chem. Sci., 2020, 11, 10614–10625 RSC.
- C. Chen, K. Wu, H. Ren, C. Zhou, Y. Luo, L. Lin, C. Au and L. Jiang, Energy Fuels, 2021, 35, 11693–11706 CrossRef CAS.
- P. Adamou, S. Bellomi, S. Hafeez, E. Harkou, S. M. Al-Salem, A. Villa, N. Dimitratos, G. Manos and A. Constantinou, Catal. Today, 2023, 423, 114022 CrossRef CAS.
- A. Utsunomiya, A. Okemoto, Y. Nishino, K. Kitagawa, H. Kobayashi, K. Taniya, Y. Ichihashi and S. Nishiyama, Appl. Catal., B, 2017, 206, 378–383 CrossRef CAS.
- S. Mukherjee, S. V. Devaguptapu, A. Sviripa, C. R. F. Lund and G. Wu, Appl. Catal., B, 2018, 226, 162–181 CrossRef CAS.
- S. Sun, Q. Jiang, D. Zhao, T. Cao, H. Sha, C. Zhang, H. Song and Z. Da, Renewable Sustainable Energy Rev., 2022, 169, 112918 CrossRef CAS.
- S. F. Yin, B. Q. Xu, X. P. Zhou and C. T. Au, Appl. Catal., A, 2004, 277, 1–9 CrossRef CAS.
- S. M. Ghoreishian, K. Shariati, Y. S. Huh and J. Lauterbach, Chem. Eng. J., 2023, 467, 143533 CrossRef CAS.
- T. E. Bell and L. Torrente-Murciano, Top. Catal., 2016, 59, 1438–1457 CrossRef CAS.
- Y. Sun, W. Zeng, Y. Yang, Q. Wu and C. Zou, Chem. Eng. J., 2024, 502, 158043 CrossRef CAS.
- N. Morlanés, S. P. Katikaneni, S. N. Paglieri, A. Harale, B. Solami, S. M. Sarathy and J. Gascon, Chem. Eng. J., 2021, 408, 127310 CrossRef.
- N. Zecher-Freeman, H. Zong, P. Xie and C. Wang, Curr. Opin. Green Sustainable Chem., 2023, 44, 100860 CrossRef CAS.
- Z.-W. Wu, X. Li, Y.-H. Qin, L. Deng, C.-W. Wang and X. Jiang, Int. J. Hydrogen Energy, 2020, 45, 15263–15269 CrossRef CAS.
- K. G. Kirste, K. McAulay, T. E. Bell, D. Stoian, S. Laassiri, A. Daisley, J. S. J. Hargreaves, K. Mathisen and L. Torrente-Murciano, Appl. Catal., B, 2021, 280, 119405 CrossRef CAS.
- N. Morlanés, S. Sayas, G. Shterk, S. P. Katikaneni, A. Harale, B. Solami and J. Gascon, Catal. Sci. Technol., 2021, 11, 3014–3024 Search PubMed.
- E. Fu, Y. Qiu, H. Lu, S. Wang, L. Liu, H. Feng, Y. Yang, Z. Wu, Y. Xie, F. Gong and R. Xiao, Fuel Process. Technol., 2021, 221, 106945 CrossRef CAS.
- I. Lucentini, A. Casanovas and J. Llorca, Int. J. Hydrogen Energy, 2019, 44, 12693–12707 CrossRef CAS.
- V. D. B. C. Dasireddy, Š. Hajduk, F. Ruiz-Zepeda, J. Kovač, B. Likozar and Z. C. Orel, Fuel Process. Technol., 2021, 215, 106752 CAS.
- T. Han, L. Wei, S. Xie, Y. Liu, H. Dai and J. Deng, Ind. Chem. Mater., 2025 Search PubMed.
- Z. Zhao, Z. Liu, M. Li, Y. Yang, L. Deng, Y. Zhao, B. Dou and F. Bin, Sep. Purif. Technol., 2025, 360, 131144 CAS.
- D. Liang, C. Feng, L. Xu, D. Wang, Y. Liu, X. Li and Z. Wang, Catal. Sci. Technol., 2023, 13, 3614–3628 CAS.
- J. C. Verschoor, P. E. de Jongh and P. Ngene, Curr. Opin. Green Sustainable Chem., 2024, 50, 100965 CAS.
- Y. Qiu, E. Fu, F. Gong and R. Xiao, Int. J. Hydrogen Energy, 2022, 47, 5044–5052 Search PubMed.
- G. Li, H. Zhang, X. Yu, Z. Lei, F. Yin and X. He, Int. J. Hydrogen Energy, 2022, 47, 12882–12892 CAS.
- H. Tang, Y. Wang, W. Zhang, Z. Liu, L. Li, W. Han and Y. Li, J. Solid State Chem., 2021, 295, 121946 CAS.
- G. Li, X. Yu, F. Yin, Z. Lei, H. Zhang and X. He, Catal. Today, 2022, 402, 45–51 CrossRef CAS.
- M. Pinzón, O. Avilés-García, A. R. de la Osa, A. de Lucas-Consuegra, P. Sánchez and A. Romero, Sustainable Chem. Pharm., 2022, 25, 100615 CrossRef.
- A. Jedynak, Z. Kowalczyk, D. Szmigiel, W. Raróg and J. Zieliński, Appl. Catal., A, 2002, 237, 223–226 CrossRef CAS.
- J. C. Ganley, F. Thomas, E. Seebauer and R. I. Masel, Catal. Lett., 2004, 96, 117–122 CrossRef CAS.
- H. Li, L. Guo, J. Qu, X. Fang, Y. Fu, J. Duan, W. Wang and C. Li, Int. J. Hydrogen Energy, 2023, 48, 8985–8996 CrossRef CAS.
- J. Zhang, M. Comotti, F. Schüth, R. Schlögl and D. S. Su, Chem. Commun., 2007, 1916–1918 RSC.
- L. Li, F. Chen, J. Shao, Y. Dai, J. Ding and Z. Tang, Int. J. Hydrogen Energy, 2016, 41, 21157–21165 CrossRef CAS.
- M. Feyen, C. Weidenthaler, R. Güttel, K. Schlichte, U. Holle, A.-H. Lu and F. Schüth, Chem. – Eur. J., 2011, 17, 598–605 CrossRef CAS PubMed.
- H. Zhang, Y. A. Alhamed, A. Al-Zahrani, M. Daous, H. Inokawa, Y. Kojima and L. A. Petrov, Int. J. Hydrogen Energy, 2014, 39, 17573–17582 CrossRef CAS.
- T. A. Le, Y. Kim, H. W. Kim, S.-U. Lee, J.-R. Kim, T.-W. Kim, Y.-J. Lee and H.-J. Chae, Appl. Catal., B, 2021, 285, 119831 CrossRef CAS.
- S. Podila, Y. A. Alhamed, A. A. AlZahrani and L. A. Petrov, Int. J. Hydrogen Energy, 2015, 40, 15411–15422 CrossRef CAS.
- M. Pinzón, A. Romero, A. de Lucas-Consuegra, A. R. de la Osa and P. Sánchez, Catal. Today, 2022, 390–391, 34–47 CrossRef.
- K. Okura, T. Okanishi, H. Muroyama, T. Matsui and K. Eguchi, Appl. Catal., A, 2015, 505, 77–85 CrossRef CAS.
- N. Zhu, F. Yang, Y. Hong and J. Liang, Int. J. Hydrogen Energy, 2025, 98, 1243–1261 CrossRef CAS.
- I. Lucentini, X. Garcia, X. Vendrell and J. Llorca, Ind. Eng. Chem. Res., 2021, 60, 18560–18611 CrossRef CAS.
- X. Huang, K. Lei, Y. Mi, W. Fang and X. Li, Molecules, 2023, 28, 5245 CrossRef CAS PubMed.
- S. Mirsadeghi, S. M. Ghoreishian, H. Zandavar, R. Behjatmanesh-Ardakani, E. Naghian, M. Ghoreishian, A. Mehrani, N. Abdolhoseinpoor, M. R. Ganjali, Y. S. Huh and S. M. Pourmortazavi, J. Environ. Chem. Eng., 2023, 11, 109106 CrossRef CAS.
- S. M. Ghoreishian, K. S. Ranjith, B. Park, S.-K. Hwang, R. Hosseini, R. Behjatmanesh-Ardakani, S. M. Pourmortazavi, H. U. Lee, B. Son, S. Mirsadeghi, Y.-K. Han and Y. S. Huh, Chem. Eng. J., 2021, 419, 129530 CrossRef CAS.
- S. M. Ghoreishian, K. S. Ranjith, M. Ghasemi, B. Park, S.-K. Hwang, N. Irannejad, M. Norouzi, S. Y. Park, R. Behjatmanesh-Ardakani, S. M. Pourmortazavi, S. Mirsadeghi, Y.-K. Han and Y. S. Huh, Chem. Eng. J., 2023, 452, 139435 CrossRef CAS.
- H. Martin, C. Ruck, M. Schmidt, S. Sell, S. Beutner, B. Mayer and R. Walsh, Pure Appl. Chem., 1999, 71, 2253–2262 CrossRef CAS.
-
B. G. Petri, R. J. Watts, A. L. Teel, S. G. Huling and R. A. Brown, In situ chemical oxidation for groundwater remediation, 2011, pp. 33–88 Search PubMed.
- Z. Yong and T. Ma, Angew. Chem., Int. Ed., 2023, 62, 202308980 CrossRef PubMed.
- M. Reli, M. Edelmannová, M. Šihor, P. Praus, L. Svoboda, K. K. Mamulová, H. Otoupalíková, L. Čapek, A. Hospodková, L. Obalová and K. Kočí, Int. J. Hydrogen Energy, 2015, 40, 8530–8538 CrossRef CAS.
- H. Kominami, H. Nishimune, Y. Ohta, Y. Arakawa and T. Inaba, Appl. Catal., B, 2012, 111–112, 297–302 CrossRef CAS.
- C. Jo, S. Surendran, M.-C. Kim, T.-Y. An, Y. Lim, H. Choi, G. Janani, S. Cyril Jesudass, D. Jun Moon, J. Kim, J. Young Kim, C. Hyuck Choi, M. Kim, J. Kyu Kim and U. Sim, Chem. Eng. J., 2023, 463, 142314 CrossRef CAS.
- S. Zhang, Z. He, X. Li, J. Zhang, Q. Zang and S. Wang, Nanoscale Adv., 2020, 2, 3610–3623 RSC.
- D. Yuan, F. Xie, K. Li, Q. Guan, J. Hou, S. Yang, G. Han, X. San, J. Hao and Y. Li, Appl. Surf. Sci., 2023, 613, 155973 CrossRef CAS.
- H. Shi, C. Li, L. Wang, W. Wang, J. Bian and X. Meng, J. Alloys Compd., 2023, 933, 167815 CrossRef CAS.
- J. Lin, Y. Wang, W. Tian, H. Zhang, H. Sun and S. Wang, ACS Catal., 2023, 13, 11711–11722 CrossRef CAS.
- J. Dzíbelová, S. M. H. Hejazi, V. Šedajová, D. Panáček, P. Jakubec, Z. Baďura, O. Malina, J. Kašlík, J. Filip, Š. Kment, M. Otyepka and R. Zbořil, Appl. Mater. Today, 2023, 34, 101881 CrossRef.
- S. Zhang, L. Yan, H. Jiang, L. Yang, Y. Zhao, X. Yang, Y. Wang, J. Shen and X. Zhao, ACS Appl. Mater. Interfaces, 2022, 14, 9036–9045 CrossRef CAS PubMed.
- Z. Wang, S. D. Young, B. R. Goldsmith and N. Singh, J. Catal., 2021, 395, 143–154 CrossRef CAS.
- Y. Tian, Z. Mao, L. Wang and J. Liang, Small Struct., 2023, 4, 2200266 CrossRef CAS.
- R. Sen, S. Das, A. Nath, P. Maharana, P. Kar, F. Verpoort, P. Liang and S. Roy, Front. Chem., 2022, 10, 861604 CrossRef CAS PubMed.
- A. AlZaabi, F. AlMarzooqi and D. Choi, Int. J. Hydrogen Energy, 2024, 94, 23–52 CrossRef CAS.
- S. Zisekas, G. Karagiannakis, G. Marnellos and M. Stoukides, Ionics, 2002, 8, 118–122 CrossRef CAS.
- K. Goshome, T. Yamada, H. Miyaoka, T. Ichikawa and Y. Kojima, Int. J. Hydrogen Energy, 2016, 41, 14529–14534 CrossRef CAS.
- N. Hanada, S. Hino, T. Ichikawa, H. Suzuki, K. Takai and Y. Kojima, Chem. Commun., 2010, 46, 7775–7777 RSC.
- D. J. Little, M. R. Smith, III and T. W. Hamann, Energy Environ. Sci., 2015, 8, 2775–2781 RSC.
- Z. Wu, N. Ambrožová, E. Eftekhari, N. Aravindakshan, W. Wang, Q. Wang, S. Zhang, K. Kočí and Q. Li, Emergent Mater., 2019, 2, 303–311 CrossRef CAS.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, J. Electrochem. Soc., 2018, 165, J3130 CrossRef CAS.
- J.-J. Zhong, S.-P. Huang, J.-F. Gu, Y. Li, K.-N. Ding, Y.-F. Zhang, W. Lin and W.-K. Chen, Appl. Surf. Sci., 2023, 609, 155280 CrossRef CAS.
- J. A. Herron, P. Ferrin and M. Mavrikakis, J. Phys. Chem. C, 2015, 119, 14692–14701 CrossRef CAS.
- P. Modisha and D. Bessarabov, Int. J. Electrochem. Sci., 2016, 11, 6627–6635 CrossRef CAS.
- B.-X. Dong, H. Tian, Y.-C. Wu, F.-Y. Bu, W.-L. Liu, Y.-L. Teng and G.-W. Diao, Int. J. Hydrogen Energy, 2016, 41, 14507–14518 CrossRef CAS.
- M. Idamakanti, E. B. Ledesma, R. R. Ratnakar, M. P. Harold, V. Balakotaiah and P. Bollini, ACS Eng. Au, 2023, 4, 71–90 CrossRef.
- Y.-S. Son, K.-H. Kim, K.-J. Kim and J.-C. Kim, Plasma Chem. Plasma Process., 2013, 33, 617–629 CrossRef CAS.
- D. Varisli, C. Korkusuz and T. Dogu, Appl. Catal., B, 2017, 201, 370–380 CrossRef CAS.
- M. Akiyama, K. Aihara, T. Sawaguchi, M. Matsukata and M. Iwamoto, Int. J. Hydrogen Energy, 2018, 43, 14493–14497 CrossRef CAS.
- P. Peng, J. Su and H. Breunig, Energy Convers. Manage., 2023, 288, 117166 CrossRef CAS.
- Z. Wang, H. Zhang, Z. Ye, G. He, C. Liao, J. Deng, G. Lei, G. Zheng, K. Zhang, F. Gou and X. Mao, Int. J. Hydrogen Energy, 2024, 49, 1375–1385 CrossRef CAS.
- K. H. R. Rouwenhorst, Y. Engelmann, K. van’t Veer, R. S. Postma, A. Bogaerts and L. Lefferts, Green Chem., 2020, 22, 6258–6287 RSC.
- L. Wang, Y. Yi, Y. Zhao, R. Zhang, J. Zhang and H. Guo, ACS Catal., 2015, 5, 4167–4174 CrossRef CAS.
- N. Wang, H. O. Otor, G. Rivera-Castro and J. C. Hicks, ACS Catal., 2024, 14, 6749–6798 CrossRef CAS PubMed.
- J. Moszczyńska, X. Liu and M. Wiśniewski, Int. J. Mol. Sci., 2022, 23, 9638 CrossRef PubMed.
- S. Shibuya, Y. Sekine and I. Mikami, Appl. Catal., A, 2015, 496, 73–78 CrossRef CAS.
- M. Reli, N. Ambrožová, M. Šihor, L. Matějová, L. Čapek, L. Obalová, Z. Matěj, A. Kotarba and K. Kočí, Appl. Catal., B, 2015, 178, 108–116 CrossRef CAS.
- Y.-N. Li, Z.-Y. Chen, S.-J. Bao, M.-Q. Wang, C.-L. Song, S. Pu and D. Long, Chem. Eng. J., 2018, 331, 383–388 CrossRef CAS.
- S. Pu, D. Long, M.-Q. Wang, S.-J. Bao, Z. Liu, F. Yang, H. Wang and Y. Zeng, Mater. Lett., 2017, 209, 56–59 CrossRef CAS.
- H. Zhang, Q.-Q. Gu, Y.-W. Zhou, S.-Q. Liu, W.-X. Liu, L. Luo and Z.-D. Meng, Appl. Surf. Sci., 2020, 504, 144065 CrossRef CAS.
- R. Wang, T. Xie, Z. Sun, T. Pu, W. Li and J.-P. Ao, RSC Adv., 2017, 7, 51687–51694 RSC.
- W. Guo and H. Liu, Chem. Res. Chin. Univ., 2017, 33, 129–134 CrossRef CAS.
- B. Achrai, Y. Zhao, T. Wang, G. Tamir, R. Abbasi, B. P. Setzler, M. Page, Y. Yan and S. Gottesfeld, J. Electrochem. Soc., 2020, 167, 134518 CrossRef CAS.
- L. Cunci, C. V. Rao, C. Velez, Y. Ishikawa and C. R. Cabrera, Electrocatalysis, 2013, 4, 61–69 CrossRef CAS.
- J. C. M. Silva, S. Ntais, É. Teixeira-Neto, E. V. Spinacé, X. Cui, A. O. Neto and E. A. Baranova, Int. J. Hydrogen Energy, 2017, 42, 193–201 CrossRef CAS.
- N. Sacré, M. Duca, S. Garbarino, R. Imbeault, A. Wang, A. Hadj Youssef, J. Galipaud, G. Hufnagel, A. Ruediger and L. Roue, ACS Catal., 2018, 8, 2508–2518 CrossRef.
- L. Wang, Y. Zhao, C. Liu, W. Gong and H. Guo, Chem. Commun., 2013, 49, 3787–3789 RSC.
- Q. F. Lin, Y. M. Jiang, C. Z. Liu, L. W. Chen, W. J. Zhang, J. Ding and J. G. Li, Energy Rep., 2021, 7, 4064–4070 CrossRef.
- M. El-Shafie, S. Kambara and Y. Hayakawa, J. Energy Inst., 2021, 99, 145–153 CrossRef CAS.
- J. A. Andersen, K. van’t Veer, J. M. Christensen, M. Østberg, A. Bogaerts and A. D. Jensen, Chem. Eng. Sci., 2023, 271, 118550 CrossRef CAS.
- Y. Hayakawa, S. Kambara and T. Miura, Int. J. Hydrogen Energy, 2020, 45, 32082–32088 CrossRef CAS.
- B. Yan, Y. Li, W. Cao, Z. Zeng, P. Liu, Z. Ke and G. Yang, J. Am. Chem. Soc., 2024, 146, 4864–4871 CrossRef CAS PubMed.
- J. C. McLennan and G. Greenwood, Proc. R. Soc. London, 1928, 120, 283–295 CAS.
- S. Hirabayashi and M. Ichihashi, Int. J. Mass Spectrom., 2016, 407, 86–91 CrossRef CAS.
- W. Guo, A. Shafizadeh, H. Shahbeik, S. Rafiee, S. Motamedi, S. A. Ghafarian Nia, M. H. Nadian, F. Li, J. Pan, M. Tabatabaei and M. Aghbashlo, J. Energy Storage, 2024, 89, 111688 CrossRef.
- E. Soltanmohammadi and N. Hikmet, J. Data Anal. Inform. Process., 2024, 12, 544–565 Search PubMed.
- E. Soltanmohammadi, A. Dilek and N. Hikmet, J. Data Anals Inform. Process., 2024, 13, 46–65 Search PubMed.
- D.-N. Vo, J. Hun Chang, S.-H. Hong and C.-H. Lee, Chem. Eng. J., 2023, 475, 146195 CrossRef CAS.
- T. Williams, K. McCullough and J. A. Lauterbach, Chem. Mater., 2019, 32, 157–165 CrossRef.
- Z.-H. Lyu, J. Fu, T. Tang, J. Zhang and J.-S. Hu, EnergyChem, 2023, 5, 100093 CrossRef CAS.
- H. Mashhadimoslem, M. Safarzadeh Khosrowshahi, M. Delpisheh, C. Convery, M. Rezakazemi, T. M. Aminabhavi, M. Kamkar and A. Elkamel, Chem. Eng. J., 2023, 474, 145661 CrossRef CAS.
- C. Makhloufi and N. Kezibri, Int. J. Hydrogen Energy, 2021, 46, 34777–34787 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a







