
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular dynamics simulation to predict assembly structures of bowl-shaped π-conjugated molecules†
Shunsuke Sato a,
Barun Dharab,
Dan Hec,
Daigo Miyajima*d and
Go Watanabe*aef
a,
Barun Dharab,
Dan Hec,
Daigo Miyajima*d and
Go Watanabe*aef
aDepartment of Physics, School of Science, Kitasato University, 1-15-1 Kitazato, Minami-ku, Sagamihara, Kanagawa 252-0373, Japan. E-mail: go0325@kitasato-u.ac.jp
bRIKEN Center for Emergent Matter Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
cCollege of Chemistry and Chemical Engineering, Central South University, Changsha 410083, P. R. China
dSchool of Science and Engineering, the Chinese University of Hong Kong, Shenzhen 518172, P. R. China. E-mail: dmiyajima@cuhk.edu.cn
eKanagawa Institute of Industrial Science and Technology (KISTEC), 705-1 Shimoimaizumi, Ebina, Kanagawa 243-0435, Japan
fDepartment of Data Science, School of Frontier Engineering, Kitasato University, 1-15-1 Kitazato, Minami-ku, Sagamihara, Kanagawa 252-0373, Japan
First published on 6th February 2025
Abstract
The proposed computational method using molecular dynamics simulation investigating the structural stability and dynamics of the molecular assembly could predict bulk crystal structures for the rationally designed bowl-shaped π-conjugated molecules. In addition, the process of the formation of the columnar assemblies was reproduced by our simulated annealing simulation.
Novel materials with high functionality and low environmental load are needed to develop innovative next-generation devices and realize a sustainable society. For example, organic crystals are attracting much attention as metal-free carbon-based materials with unique properties such as flexibility, ferroelectricity, the bulk photovoltaic effect (BPVE), and the chirality-induced spin selectivity (CISS).1–4 In organic crystals, the crystal structure and the physical properties are significantly influenced by the molecular structure. Thus, the synthesis of new molecules and X-ray crystallography are generally required for the development of new organic materials, and these experimental processes need time and effort. Therefore, the utilization of computational approaches is necessary for the efficient design of novel functional molecules.
Computational methods for crystal structure prediction (CSP) have been developed by many researchers5,6 and have improved significantly in recent years.7–9 The goal of CSP is to correctly predict an organic molecule's crystal structure from its chemical structure, including its polymorphism. Dispersion corrected density functional theory (DFT) calculation is widely used in CSP because it can provide the energies of candidate crystal structures for a molecule with high accuracy.9–11 However, it is still difficult to establish a versatile accurate CSP method due to molecular vibration, solvent effects, thermodynamic effects considering energy barriers for phase transitions, and so on.12 In the case of organic materials, the properties of an organic molecule in the crystalline state are important, but the methods for evaluating the structural stability of the crystalline state are still in their infancy.
Molecular dynamics (MD) simulations can analyze the structural stability of the molecular assembly under constant temperature and pressure and investigate the dynamics at the atomic or molecular level. However, a limited number of studies13,14 have reported on the CSP method using MD simulations thus far. Our previous studies15,16 showed that MD simulations can be used to investigate molecular thermal fluctuations and structural stability in the crystal structure of organic semiconductors.
This study proposes a novel method to investigate the stability of the bulk structures for bowl-shaped π-conjugated molecules and to predict the crystal structures using all-atom MD simulation. Moreover, we present the MD simulation model and method to reproduce the assembly structure from randomly dispersed molecules.
The simulated molecules were bowl-shaped π-conjugated molecules, subphthalocyanine (SubPc) derivatives.17,18 SubPc derivatives with fluorine substituted for the axial ligand and halogen atoms substituted at three of the α-positions tend to stack unidirectionally to form one-dimensional stacked columns.19 As these SubPc derivatives are chiral, there can be an enantiopure system and a racemic system in the crystal structure of the SubPc derivatives (Fig. 1). Recently, it has been reported that the SubPc derivatives show different crystal structures in which the arrangements of neighboring columns are parallel or anti-parallel depending on the type of halogen that is introduced and the chirality of the system (Table S1, ESI†).20 On the other hand, the SubPc derivative that is unsubstituted in the α-positions shows herringbone-type assemblies with no columnar structures.21 From the above, the crystal structures of the SubPc derivatives significantly change depending on slight differences in their molecular structure. Therefore, it is of great importance to gain insight into the crystal structure of the SubPc derivatives, as they are promising for optoelectronic applications and attract research interest as organic polar materials.22
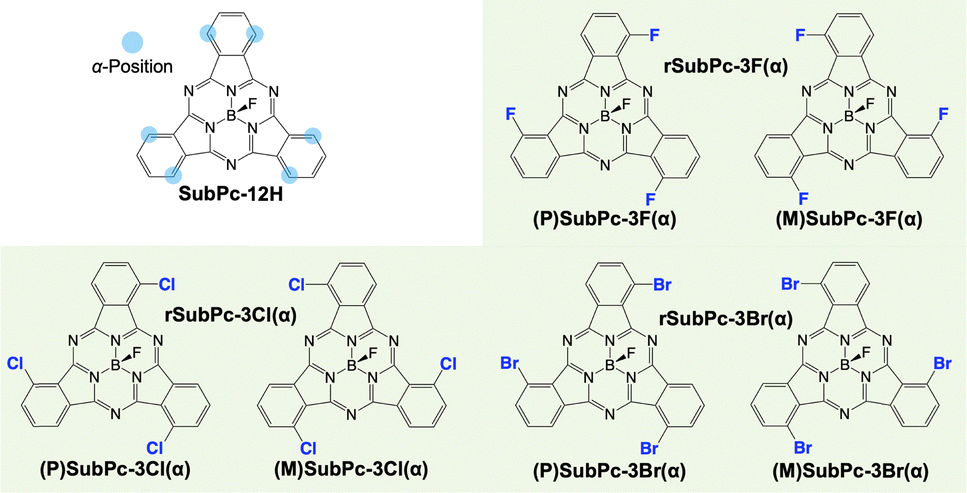 | ||
| Fig. 1 Chemical structures of chiral subphthalocyanine (SubPc) derivatives with a schematic illustration of the α-positions of SubPc. | ||
Firstly, MD simulations of SubPc-12H without halogen substituents were performed for both bulk structures obtained experimentally and those generated virtually: herringbone, parallel columnar and anti-parallel columnar arrangements. All-atom MD simulations were performed using the MD program GROMACS 2020.5. The simulations for each system consisting of nearly 600 molecules were run for 50 ns at several different temperatures between 100 K and 400 K. The details of the MD simulation are described in the ESI.† The initial structures for the MD simulation were created using the experimental crystal structures.19–21 To quantitatively compare the structural stability for the simulated systems, we analyzed the energy landscapes from MD simulation results in accordance with previous studies5,9 (Fig. 2a and Table S10, ESI†). Since the molecule exhibits the assembly structure at the low potential energy and the high density of the energy landscape, Fig. 2a indicates that the herringbone structure was suitable for SubPc-12H at each temperature and it corresponds the experimental results. Actually, the results of MD simulations suggest that the difference in the van der Waals interaction energies for herringbone, parallel columnar, and anti-parallel columnar structures contribute to the energy gain in the three simulated systems (Table S2, ESI†).
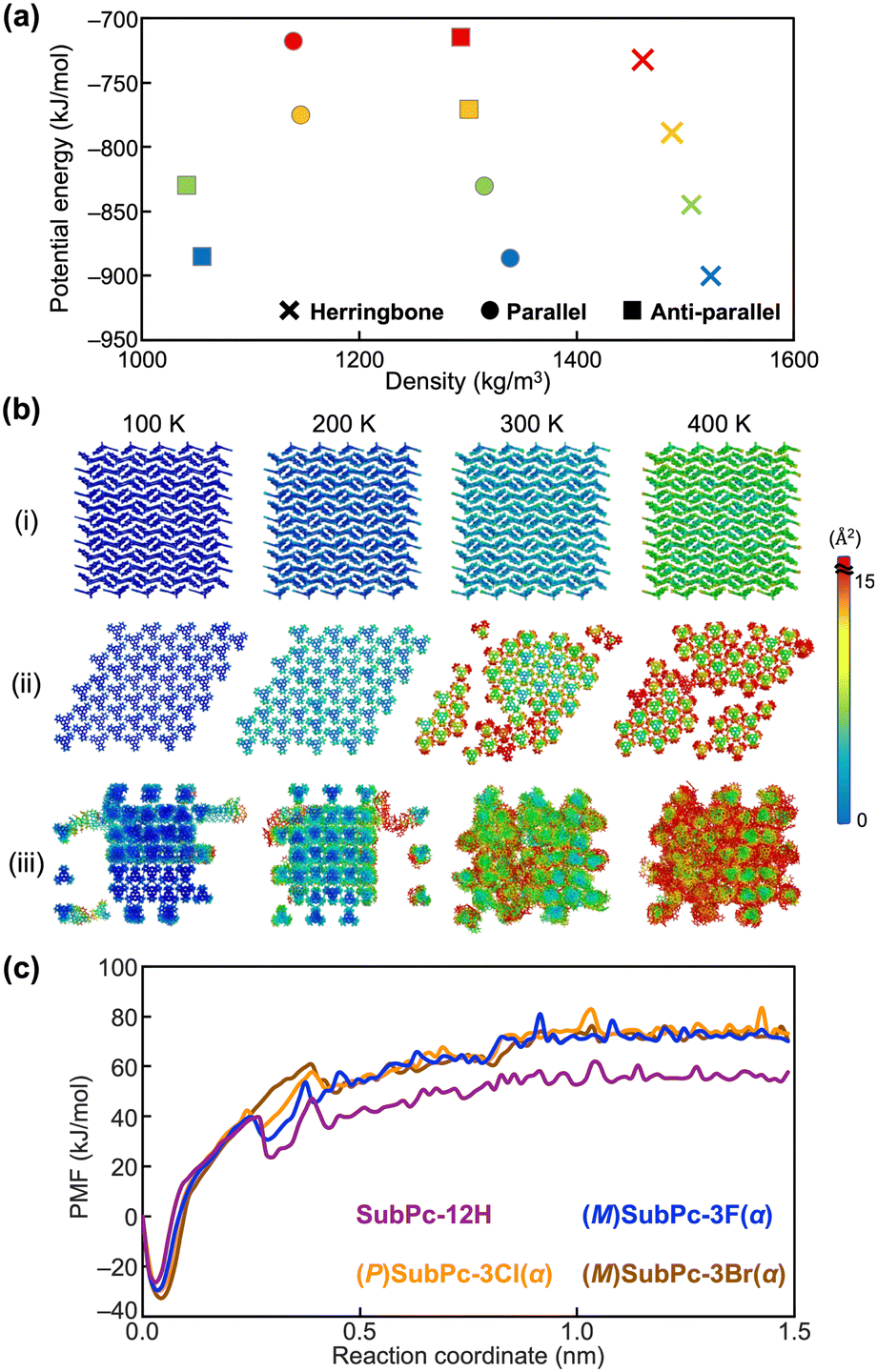 | ||
| Fig. 2 (a) Energy landscape of herringbone assembly, parallel columnar assembly, and anti-parallel columnar assembly for SubPc-12H. Plots represent 100 K (blue), 200 K (green), 300 K (orange) and 400 K (red). (b) Color-coded thermal factor distributions (Å2) obtained from MD simulations of (i) herringbone assembly, (ii) parallel columnar assembly, and (iii) anti-parallel columnar assembly for SubPc-12H. The magnitude of thermal factors is represented by the color-coded scale bar. (c) PMF profiles of SubPc-12H, (M)SubPc-3F(α), (P)SubPc-3Cl(α), and (M)SubPc-3Br(α) at 300 K. | ||
Furthermore, the stability of the molecular assemblies was also confirmed by thermal fluctuations of the molecules and conformational changes of the assemblies. We have carried out other simulations for several SubPc derivatives in a recent study20 and utilized the methods proposed in that study for the present study. According to our previous studies,15,16,20 the magnitude of thermal fluctuations was represented by the thermal factor. Fig. 2b shows the color-coded thermal factor distributions obtained from the simulations for SubPc-12H. In Fig. 2b, the colors of blue and red indicate the small fluctuation and large fluctuation, respectively. An assembly structure in which the distribution of the thermal factors was uniform, the large fluctuation of atoms and the distortion packing of molecules were not observed can be determined to be a bulk stable structure. As shown in Fig. 2b(ii) and (iii), while the structures of which the molecules forming parallel and anti-parallel columns were distorted at higher temperatures, the herringbone assembly structure was stable even with increasing the temperature. This is consistent with the fact that the column-type assembly structures of SubPc-12H have not been obtained experimentally.
In addition, to investigate why SubPc-12H does not form columnar assemblies, unlike other systems, the free energy analyses by the potential of mean force (PMF) during the process of pulling away one of the SubPc derivatives forming the column were performed. It can be seen that a higher PMF value for a given column would indicate greater stability. According to our previous study,23,24 the PMF for each system was obtained from a series of umbrella sampling (US) simulations, and the details of these simulations have been described in the ESI.† It should be noted that PMFs obtained from the simulations were large values because the solvent was not included in the system. A comparison of the calculated PMFs in SubPc-12H and other enantiomers at 300 K revealed that the PMF in SubPc-12H was significantly smaller than that of the other three SubPc derivatives (Fig. 2c). This indicates that SubPc-12H has only a small energy gain by forming columnar assembly. The lower PMF values observed at about 0.2 Å may be due to the fact that the SubPc molecules were densely packed in the column and the intermolecular distance along the column was shorter than that at the minimum of their interaction energy.
For the SubPc derivatives with bromine as the substituent: (M)SubPc-3Br(α) and rSubPc-3Br(α), the results of the energy landscape obtained from MD simulations of bulk assemblies was shown in Fig. 3a and Tables S4, S5, S11, and S12 (ESI†). It was confirmed that, for the enantiopure system of SubPc-3Br(α) which is (M)SubPc-3Br(α), the anti-parallel columnar assembly observed in the experiments is more stable than other molecular assemblies.
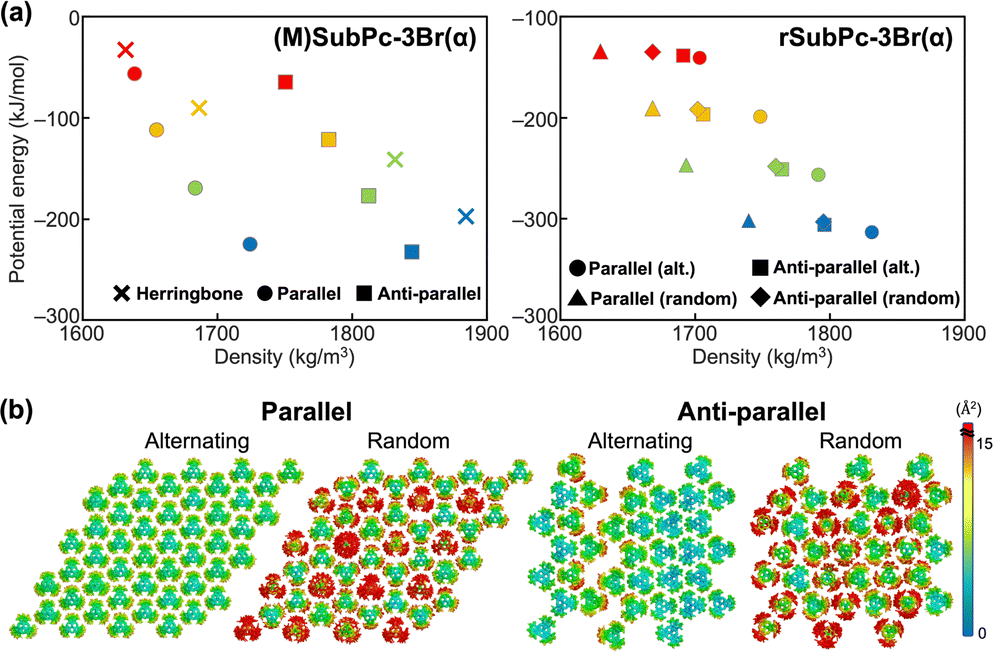 | ||
| Fig. 3 (a) Energy landscapes of herringbone packing structure, parallel columnar structure, and anti-parallel columnar structure for (M)SubPc-3Br(α) and rSubPc-3Br(α). The plots represent the following temperatures: 100 K (blue), 200 K (green), 300 K (orange), and 400 K (red). (b) Color-coded thermal factor distributions (Å2) obtained from MD simulations for the simulated systems of rSubPc-3Br(α) at 300 K. The magnitude of thermal factors is represented by the color-coded scale bar. | ||
In rSubPc-3Br(α), it has been found that M- and P-type molecules were alternately stacked in the parallel columns by the experiment (Table S1, ESI†). As shown in Fig. 3, the results of the energy landscape and thermal factor distributions of rSubPc-3Br(α) showed that the alternating molecular arrangement in the parallel columnar assembly was more stable than the other three assembly structures not obtained by the experiments. To evaluate the influence of the slight differences in the molecular arrangement in the column on the structural stability of the molecular assembly, we calculated the PMFs of a SubPc-3Br(α) molecule moving away from the single column of the following three types: a column of (M)-SubPc-3Br(α), a column of rSubPc-3Br(α) with M- and P-type molecules alternately stacked, and a column of rSubPc-3Br(α) with M- and P-type molecules randomly stacked. Fig. S22 (ESI†) shows that the molecular arrangement of the individual columns had little effect on the energetic stability of the column formation. Therefore, the main reason why M- and P-type molecules were alternately stacked in the column of rSubPc-3Br(α) would not be due to the molecular interactions along the column, but to those in two-dimensions perpendicular to the column. It was also confirmed by MD simulations at 300 K for two bulk systems of rSubPc-3Br(α), consisting of the columns with alternately stacked molecules and with randomly stacked molecules, respectively. In the system of the column with alternately stacked molecules, the molecules could be densely packed without overlapping, whereas the packing of the molecules randomly stacked in the column of the system was more sparsely (Fig. S18, ESI†). For rSubPc-3Br(α), the positional ordering of M- and P-type molecules in the intercolumnar direction was dominant in forming the parallel column structure in which these molecules were alternately stacked, with a secondary effect of the C–H⋯Br hydrogen bonding interaction in the intracolumnar direction.
On the other hand, in the case of (M)SubPc-3Br(α), the columns formed by M-type SubPc-3Br(α) were parallel to realize the dense packing and the intercolumnar C–H⋯Br hydrogen bonding network as observed in bulk MD simulations (Fig. S19, ESI†). The fact that the columns in which two different enantiomers were alternately stacked align in anti-parallel for the racemate while those formed by one type of enantiomer were parallel, was observed not only for SubPc-3Br(α) but also for SubPc-3Cl(α) (see Fig. S4, S5, S14, and S15 and Tables S6, S7, S13, and S14, ESI†).
For rSubPc-3F(α) substituted with fluorine, MD simulations predicted that the parallel columnar structure could be energetically and thermodynamically stable, as shown in Fig. S7 and S17 and Tables S9 and S16 (ESI†). It was also shown that there was no significant differences of potential energy and temperature dependence of the structural stability between random stacking and alternating stacking of molecules in the column. These predictions did not contradict with the experimental results that both (M)SubPc-3F(α) and rSubPc-3F(α) exhibit the parallel columnar alignment, and the order of the stacking of M- and P-type molecules in rSubPc-3F(α) is random. Since the van der Waals radius of F substituted with SubPc-3F(α) is about the same as that of H, the introduction of F did not much affect its molecular packing. As discussed above, the alignment of neighboring columns and the order of the molecular stacking in the column depends on the molecular packing density in the two-dimensional plane perpendicular to the column. Therefore, the small effect of the substitution of F into SubPc on the two-dimensional molecular packing would not induce the antiparallel alignment of (M)SubPc-3F(α) and the alternating stacking in the column of rSubPc-3F(α). For rSubPc-3F(α), entropy effects may result in the formation of random molecular stacking patterns in the column.
To reproduce the process of the formation of the column and the assembly of the columns, we performed simulated annealing (SA). In order to simplify the system without solvent effects, the initial structure was 40 molecules were randomly dispersed in the vacuum with reference to the previous studies.25,26 The details of the simulation conditions of the SA process are described in the ESI.† Remarkably, the spontaneous formation of four anti-parallel columns was reproduced, and the obtained structure was similar to the crystal structure of (M)SubPc-3Br(α) (Fig. 4 and see also ESI,† Movie). This suggests that the formation of anti-parallel columns causes significant stabilization for enantiomers of SubPc derivatives partially substituted with large van der Waals radii halogens. Our results shows that the SA process using MD simulations can predict the structure of molecular assemblies in crystals from only the molecular structure of the target molecule. However, the parallel columnar structure observed experimentally could not be correctly reproduced with using the present simulation method. This is due to the lack of the two-dimensional molecular interaction network, which could be realized by incorporating substrates and solvents into the simulation model.
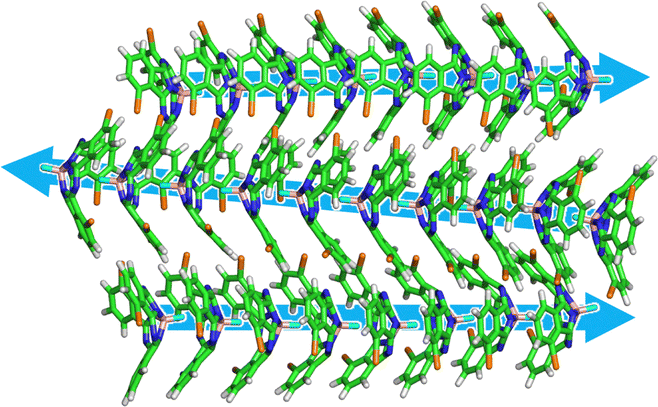 | ||
| Fig. 4 Snapshot after 20 cycles of SA simulations for (M)SubPc-3Br(α). Three columns are shown for clarity. | ||
In summary, the present study shows that MD simulation could evaluate the structural stability of bulk crystal structures for the rationally designed SubPc derivatives. The analyses of the atomic thermal fluctuation of the assembly structures and the free energy of the molecule to form the column structure quantitatively clarify the influence of the substitution of different halogen atoms on the preference of forming the columnar structure, the columnar orientations, and molecular order in the column. In addition, reproducing the anti-parallel columnar structure formed by randomly dispersed enantiomers helped to understand the mechanism of their self-assembly process. Our proposed method based on MD simulations has a high potential to predict the existing crystal structure and will play an important role in the development of CSP.
This work was financially supported by JSPS KAKENHI Grant-in-Aid for Challenging Research (Exploratory) JP22K18953 and Grant-in-Aid for JSPS Fellows JP24KJ1922 and JST CREST JPMJCR23O1 and JPMJCR24S1. The computations were partially performed at the Research Center for Computational Science, Okazaki, Japan (Project: 23-IMS-C038 and 24-IMS-C038).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- M. Kaltenbrunner, T. Sekitani, J. Reeder, T. Yokota, K. Kuribara, T. Tokuhara, M. Drack, R. Schwödiauer, I. Graz, S. B. Gogonea, S. Bauer and T. Someya, Nature, 2013, 499, 458–463 CrossRef CAS PubMed.
- Y. Noda, T. Yamada, K. Kobayashi, R. Kumai, S. Horiuchi, F. Kagawa and T. Hasegawa, Adv. Mater., 2015, 27, 6475–6481 CrossRef CAS PubMed.
- R. K. Vijayaraghavan, S. C. J. Meskers, M. A. Rahimc and S. Das, Chem. Commun., 2014, 50, 6530–6533 RSC.
- Y.-H. Kim, Y. Zhai, H. Lu, X. Pan, C. Xiao, E. A. Gaulding, S. P. Harvey, J. J. Berry, Z. V. Vardeny, J. M. Luther and M. C. Beard, Science, 2021, 371, 1129–1133 CrossRef CAS PubMed.
- S. L. Price, Phys. Chem. Chem. Phys., 2008, 10, 1996–2009 RSC.
- T. S. Thakur, R. Dubey and G. R. Desiraju, Annu. Rev. Phys. Chem., 2015, 66, 21–42 CrossRef CAS PubMed.
- D. H. Bowskill, I. J. Sugden, S. Konstantinopoulos, C. S. Adjiman and C. C. Pantelides, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 593–623 CrossRef CAS PubMed.
- R. Nikhar and K. Szalewicz, Nat. Commun., 2022, 13, 3095 CrossRef CAS PubMed.
- D. Firaha, Y. M. Liu, J. van de Streek, K. Sasikumar, H. Dietrich, J. Helfferich, L. Aerts, D. E. Braun, A. Broo, A. G. DiPasquale, A. Y. Lee, S. L. Meur, S. O. N. Lill, W. J. Lunsmann, A. Mattei, P. Muglia, O. D. Putra, M. Raoui, S. M. R.-Edens, S. Rome, A. Y. Sheikh, A. Tkatchenko, G. R. Woollam and M. A. Neumann, Nature, 2023, 623, 324–328 CrossRef CAS.
- A. M. Reilly and A. Tkatchenkob, J. Chem. Phys., 2013, 139, 024705 CrossRef PubMed.
- J. Hoja, H.-Y. Ko, M. A. Neumann, R. Car, R. A. DiStasio Jr. and A. Tkatchenko, Sci. Adv., 2019, 5, eaau3338 CrossRef PubMed.
- G. J. O. Beran, Chem. Sci., 2023, 14, 13290–13312 RSC.
- A. Gavezzotti, J. Am. Chem. Soc., 2000, 122, 10724–10725 CrossRef CAS.
- P. G. Karamertzanis, P. Raiteri, M. Parrinello, M. Leslie and S. L. Price, J. Phys. Chem. B, 2008, 112, 4298–4308 CrossRef CAS PubMed.
- C. P. Yu, N. Kojima, S. Kumagai, T. Kurosawa, H. Ishii, G. Watanabe, J. Takeya and T. Okamoto, Commun. Chem., 2021, 4, 155 CrossRef CAS PubMed.
- C. P. Yu, S. Kumagai, M. Tsutsumi, T. Kurosawa, H. Ishii, G. Watanabe, D. Hashizume, H. Sugiura, Y. Tani, T. Ise, T. Watanabe, H. Sato, J. Takeya and T. Okamoto, Adv. Sci., 2023, 10, 2207440 CrossRef CAS PubMed.
- C. G. Claessens, D. G. Rodríguez and T. Torres, Chem. Rev., 2002, 102, 835–854 CrossRef CAS PubMed.
- S. Shimizu and N. Kobayashi, Chem. Commun., 2014, 50, 6949–6966 RSC.
- C. Zhang, Y. Guo, D. He, J. Komiya, G. Watanabe, T. Ogaki, C. Wang, A. Nihonyanagi, H. Inuzuka, H. Gong, Y. Yi, K. Takimiya, D. Hashizume and D. Miyajima, Angew. Chem., Int. Ed., 2021, 60, 3261–3267 CrossRef CAS PubMed.
- D. He, B. Dhara, C. Zhang, A. Nihonyanagi, H. Inuzuka, E. Hara, S. Sato, K. Adachi, G. Watanabe, D. Hashizume, K. Okuro and D. Miyajima, Angew. Chem., Int. Ed., 2025 DOI:10.1002/anie.202424162.
- M. V. Fulford, D. Jaidka, A. S. Paton, G. E. Morse, E. R. L. Brisson, A. J. Lough and T. P. Bender, J. Chem. Eng. Data, 2012, 57, 2756–2765 CrossRef CAS.
- J. Labella and T. Torres, Trends Chem., 2023, 5, 353–366 CrossRef CAS.
- Y. Kawashima, T. Hamachi, A. Yamauchi, K. Nishimura, Y. Nakashima, S. Fujiwara, N. Kimizuka, T. Ryu, T. Tamura, M. Saigo, K. Onda, S. Sato, Y. Kobori, K. Tateishi, T. Uesaka, G. Watanabe, K. Miyata and N. Yanai, Nat. Commun., 2023, 14, 1056 CrossRef CAS PubMed.
- K. Hiratsuka, T. Muramatsu, T. Seki, C. Weder, G. Watanabe and Y. Sagara, J. Mater. Chem. C, 2023, 11, 3949–3955 RSC.
- C.-T. Chen, V. Ball, J. J. A. Gracio, M. K. Singh, V. Toniazzo, D. Ruch and M. J. Buehler, ACS Nano, 2013, 7, 1524–1532 CrossRef CAS PubMed.
- N. Saikia, K. Waters, S. P. Karna and R. Pandey, ACS Omega, 2017, 2, 3457–3466 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of molecular dynamics simulations. See DOI: https://doi.org/10.1039/d4cc06482h |
| This journal is © The Royal Society of Chemistry 2025 |