
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Design of a modular, palladium-catalyzed carbonylative synthesis of pyrido[2,1-α]isoindoles via benzyne 1,3-dipolar cycloaddition†
Karina S. Wong a,
Mohammad Ghanbari*b and
Bruce A. Arndtsen*a
a,
Mohammad Ghanbari*b and
Bruce A. Arndtsen*a
aDepartment of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, QC H3A 0B8, Canada. E-mail: bruce.arndtsen@mcgill.ca
bDepartment of Organic Chemistry, Faculty of Chemistry, University of Kashan, Kashan 8731753153, Iran. E-mail: ghanbari-m@kashanu.ac.ir
First published on 12th February 2025
Abstract
We report a palladium catalyzed, multicomponent synthesis of pyrido[2,1-α]isoindoles via carbonylative coupling of imines and 2-bromopyridines to form mesoionic dipoles, followed by cycloaddition with in situ generated arynes. This one-pot method allows efficient, modular assembly of polysubstituted products with independent control of each building block.
The development of modular and efficient approaches to assemble heterocyclic products has become an important thrust in synthetic chemistry. One common approach to this goal is via multicomponent coupling reactions.1 Unlike more classical multistep synthetic procedures, multicomponent reactions provide an avenue to couple three or more starting materials in a single reaction. This can not only create a more streamlined synthesis, but, due to their modularity, can be readily diversified to change product structures, making them attractive as well for product design. Nevertheless, a challenge in the design of multicomponent coupling reactions is accessing the reactive building blocks needed to drive the consecutive formation of multiple bonds in a single reaction. These can themselves often require a multistep synthesis and thus detracts from the efficiency benefits of employing multicomponent reactions.
A useful approach to address this challenge is to exploit metal catalysis with energetic building blocks such as carbon monoxide. Carbon monoxide is a broadly available feedstock chemical, which has helped make metal catalyzed carbonylation reactions among the most heavily employed transformations in chemical synthesis.2 In addition, an underexploited feature of carbon monoxide is its energetics. The conversion of carbon monoxide to a carboxylic acid derivative is often highly exergonic. We and others have shown that this feature can be exploited to drive the catalytic build-up of a range of reactive products,3,4 such as the modular assembly of 1,3-dipoles as a route into multicomponent heterocycle synthesis.5 An example of this chemistry is the formation of pyridine-based 1,3-dipole 1, which, following cycloaddition with electron deficient alkynes offers a multicomponent synthesis of indolizines (Fig. 1a).6
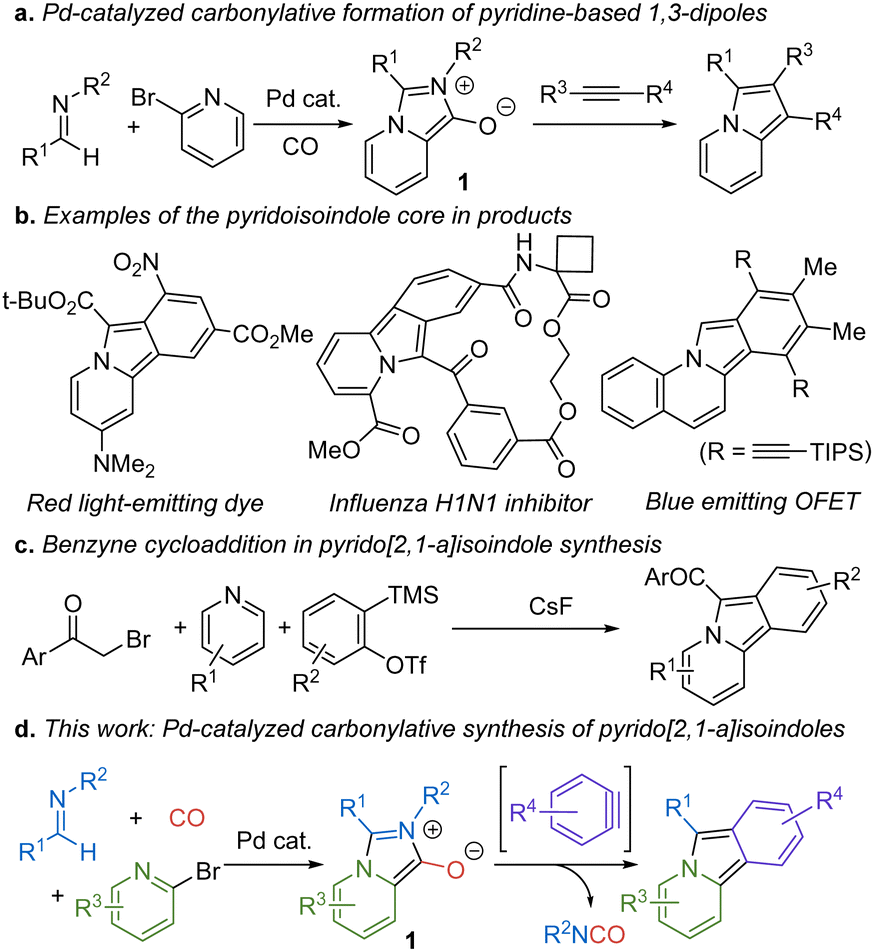 | ||
| Fig. 1 Carbonylative formation of 1,3-dipoles and benzyne cycloaddition routes to pyrido[2,1-α]isoindole synthesis. | ||
In considering the reactivity of 1, we questioned if this 1,3-dipolar cycloaddition manifold might be directed toward other products. For example, benzynes have attracted significant research attention due to the reactivity of their strained triple bonds, including their use in nucleophilic additions, pericyclic chemistry, and related transformations.7 This synthetic utility is facilitated in part by the low lying LUMO energy of the triple bond in benzynes, which imparts similar reactivity to electron deficient alkynes.8 The latter suggests that benzyne cycloaddition might be employed in this sequence with 1 to access, in this case, fused ring pyrido[2,1-α]isoindoles. Although less extensively studied as indolizines, pyrido[2,1-α]isoindoles have found use in pharmaceutically relevant products, and their delocalized 14π-electron aromatic system has led to their application in electronic materials and dyes (Fig. 1b).9 Additionally, reduced forms of pyrido[2,1-α]isoindoles are found in various alkaloids and therapeutic agents.10 Pyrido[2,1-α]isoindoles are most commonly prepared via cyclization reactions. While effective, these typically require the multistep build-up of the appropriately substituted precursor.11 Benzyne cycloaddition to pyridinium ylides can also access these structures (Fig. 1c), although these are limited to forming products with strong electron withdrawing C-acyl units to access the dipole itself.12 As an alternative, we describe here how pyrido[2,1-α]isoindoles can be generated in a modular fashion via palladium-catalyzed carbonylation reactions. This involves the cycloaddition of benzyne derivatives to in situ generated 1 and provides a method to assemble these structures in one pot, with minimal waste, and where each of the imine, bromopyridine, and benzyne units can be systematically modified to access a range of new variants of these structures (Fig. 1d).
There are a number of methods available to generate arynes.13 One of the most versatile is the fluoride induced 1,2-elimination from o-trimethylsilylphenyl triflates developed by Kobayashi.14 Considering the mild conditions of this reaction and the stability of o-trimethylsilylphenyl triflates, our initial studies explored if this approach could allow benzyne generation to be compatible with the formation of 1,3-dipole 1. The palladium-catalyzed carbonylative coupling of the imine p-tolyl(H)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NBn, 2-bromopyridine, and o-trimethylsilylphenyl triflate in the presence of CsF as an activating agent in acetonitrile does lead to the formation of 6-(p-tolyl)pyrido[2,1-α]isoindole 2a, but does so in very low yield (9%, Table 1, entry 1). Examination of the reaction mixture by 1H NMR analysis shows significant amounts of unreacted imine and 2-bromopyridine. Increasing the amount of benzyne precursor completely inhibits the reaction (entry 2). Similar results were observed with other palladium catalysts (entries 3–7, see Table S1 for full development, ESI†).
NBn, 2-bromopyridine, and o-trimethylsilylphenyl triflate in the presence of CsF as an activating agent in acetonitrile does lead to the formation of 6-(p-tolyl)pyrido[2,1-α]isoindole 2a, but does so in very low yield (9%, Table 1, entry 1). Examination of the reaction mixture by 1H NMR analysis shows significant amounts of unreacted imine and 2-bromopyridine. Increasing the amount of benzyne precursor completely inhibits the reaction (entry 2). Similar results were observed with other palladium catalysts (entries 3–7, see Table S1 for full development, ESI†).
|
||||
|---|---|---|---|---|
| Entry | Procedure | Ligand | Solvent | % 2ad |
| Procedure A: p-tolyl(H)CNBn (34 mg, 0.16 mmol); 2-bromopyridine (38 mg, 0.24 mmol); o-trimethylsilylphenyltriflate (48 mg, 0.16 mmol); CsF (109 mg, 0.72 mmol); i-Pr2NEt (25 mg, 0.19 mmol); Pd2(dba)3·CHCl3 (4.1 mg, 0.004 mmol); ligand (0.008 mmol bidentate, 0.016 mmol monodentate); Bu4NCl (67 mg, 0.24 mmol); 3.2 mL solvent; 5 atm CO; 80 °C. Procedure B: all reagents except benzyne precursor/CsF in C6H6 80 °C, 4 h, then o-trimethylsilylphenyl triflate (33 mg, 0.11 mmol); CsF (75 mg, 0.50 mmol), 3 mL CH3CN, RT, 24 h.a 0.35 mmol.b 0.11 mmol.c 0.16 mmol o-trimethylsilylphenyl triflate.d 1H NMR yield.e Isolated yield.f 48 h. |
||||
| 1 | A | Xantphos | CH3CN | 9% |
| 2a | A | Xantphos | CH3CN | 0% |
| 3b,f | A | DPEphos | C6H6 | 5% |
| 4b,f | A | dppe | C6H6 | 0% |
| 5b,f | A | P(t-Bu)3 | C6H6 | 6% |
| 6b,f | A | P(Ph)3 | C6H6 | 0% |
| 7b,f | A | JohnPhos | C6H6 | 2% |
| 8c | A | Xantphos | C6H6 | 53% |
| 9b | A | Xantphos | C6H6 | 65% (60%)e |
| 10b,f | A | Xantphos | C6H6 | 57% |
 |
||||
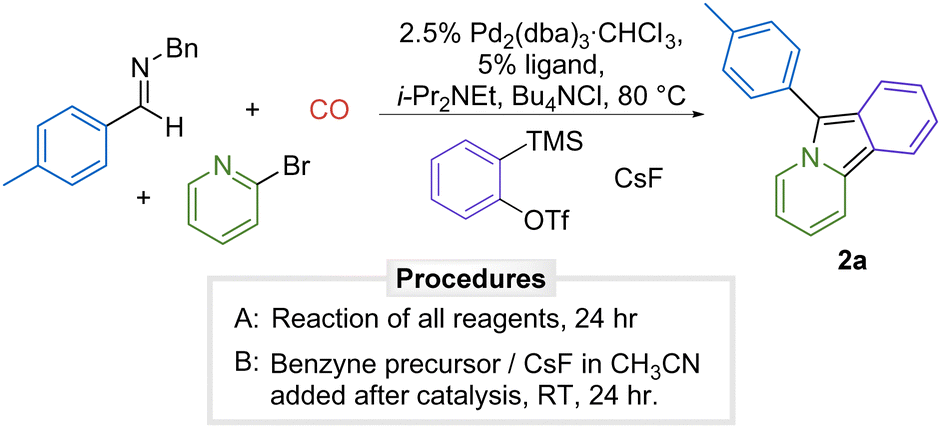
Our previous results have shown that 1,3-dipole 1a is generated in high yield with these substrates,6 suggesting the presence of benzyne inhibiting catalysis. As such, a straightforward solution is to add o-trimethylsilylphenyl triflate and CsF to the catalytically generated 1,3-dipole, which results in cycloaddition to afford 2a in 53% yield (entry 8). We observe the formation of side products in this reaction that may arise from the cycloaddition of benzyne to the product 2a.15 This can be minimized by employing benzyne as the limiting reagent and leads to the formation of 2a in slightly enhanced yield of 65% (entry 9).16 Under these conditions in benzene solvent, benzyne generation can be carried out during the palladium catalyzed reaction in somewhat lower yield (57%, entry 10).
A useful feature of this transformation is its modularity, where systematic changes to the imine, bromopyridine, and benzyne reagents can be employed to readily access a range of new variants of pyrido[2,1-α]isoindoles. For example, as shown in Table 2, a number of substituted C-aryl imines can be incorporated into the reaction. The reaction conditions for the build-up of the 1,3-dipole are influenced by the imine, with more nucleophilic imines accelerating the reaction (2a–d), while those with electron-withdrawing substituents (2f, 2h) requiring more pressing conditions or longer reaction times (see ESI,† for details). Regardless of the reaction rate, imines with both electron donating and electron withdrawing C-aryl substituents form the corresponding pyrido[2,1-α] isoindole in good overall yield (2a-e, 2h). Heterocycles such as pyridine (2f) and thiophene (2g) can also be incorporated into the product from the appropriate imine. The use of polyaromatic substituted imines lead to the corresponding pyrido[2,1-α]isoindoles (2i-j), as does the more sterically hindered o-tolyl imine (2e). C-Aliphatic imines were also viable substrates and form cycloaddition products in reasonable yields. In the case of 2k-l, the formation of the 1,3-dipole must be performed in the more polar solvent, which presumably enhances the ability of this imine to trap an in situ generated acid chloride (vide infra).
a Imine (0.16 mmol); 2-bromopyridine (0.24 mmol); Pd2(dba)3·CHCl3 (4.1 mg, 0.004 mmol); xantphos (4.6 mg, 0.008 mmol); Bu4NCl (67 mg, 0.24 mmol); i-Pr2NEt (25 mg, 0.19 mmol); CO (5 atm); C6H6 (3.2 mL), 80 °C, 4 h, see ESI for reaction time variations.b o-Trimethylsilylaryltriflate (0.11 mmol); CsF (75 mg, 0.50 mmol); CH3CN (3.2 mL). Step (a) a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24 h. b120 °C, Bu4NCl (0.48 mmol), 5% Pd2(dba)3·CHCl3, 10% xantphos.c In CH3CN. 24 h. b120 °C, Bu4NCl (0.48 mmol), 5% Pd2(dba)3·CHCl3, 10% xantphos.c In CH3CN. |
|---|
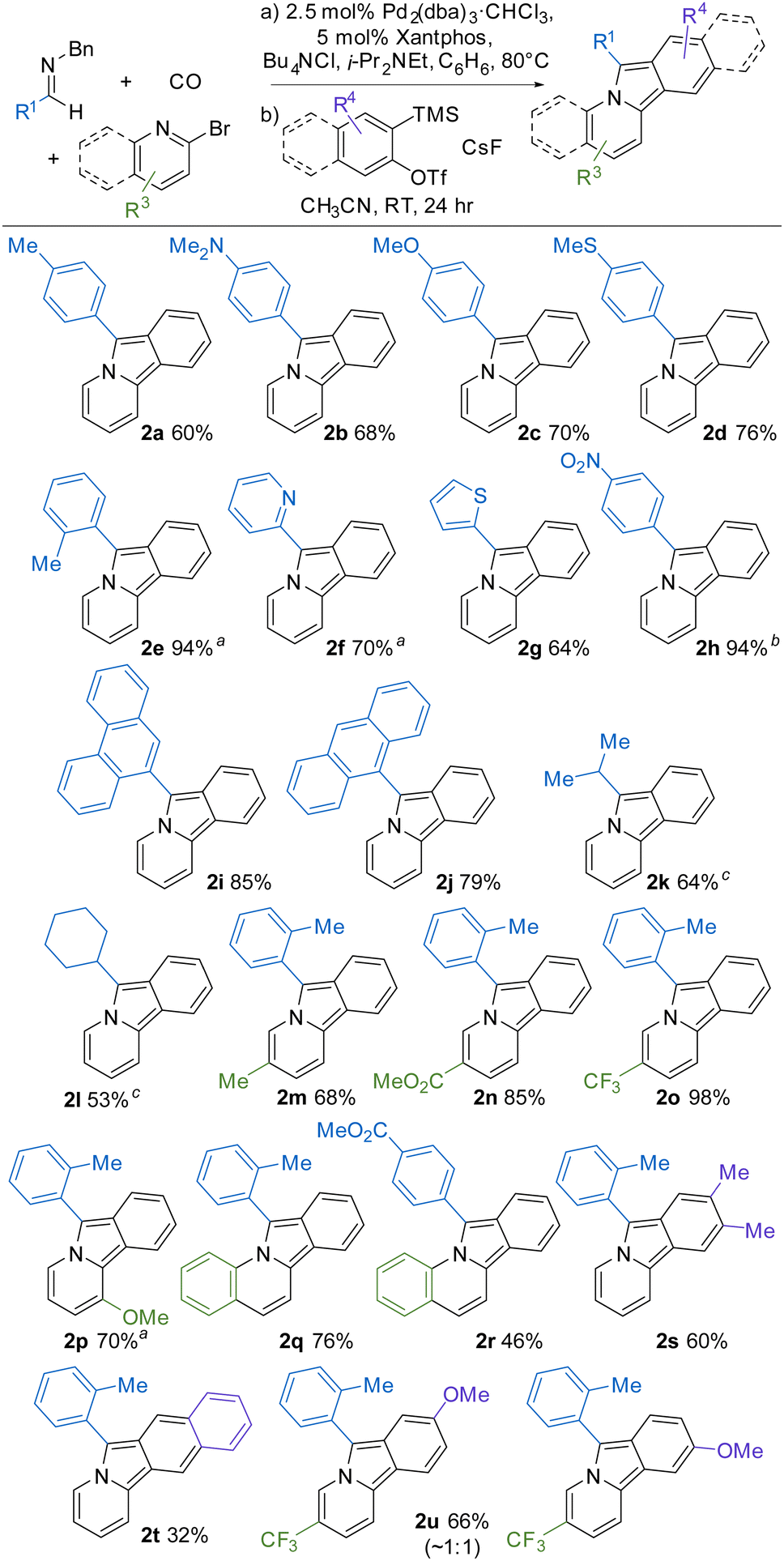 |
The 2-bromopyridine unit can also be modulated. As examples, electron donating and withdrawing groups on the pyridine component are well tolerated (2m-o), as are substituents closer to the pyrrole unit (e.g. 2p). The 2-bromopyridine unit can be replaced with 2-bromoquinoline to form more extended conjugated products (2q-r). Finally, the benzyne precursor can also be tuned. In the case of 4,5-dimethyl substituted trimethylsilyl aryl triflate, the product 2s is formed in good overall yield, as is the fused ring product 2t. However, unsymmetrical arynes form a mixture of cycloaddition products (2u). The latter contrasts with results using substituted alkynes,6 and presumably arises from the high aryne reactivity and the moderate electronic influence of the remote 4-methoxy substituent on selectivity.17
Pyrido[2,1-α]isoindoles are often sensitive to oxidation.18 Nevertheless, we have found that the products here are indefinitely stable when stored under an inert atmosphere. Under these conditions, crystals of 2c can be obtained from diethyl ether/acetonitrile, and its structure confirmed by X-ray crystallography. As shown in Fig. 2, 2c has a relatively planar π-conjugated core (dihedral angle: C5–C6–C7–C12: 0.3°). The C–C bonds in the pyridine ring are shortened relative to those in the pyrrole (e.g. C2–C3: 1.354(4) Å vs. C8–C13: 1.395(3) Å), suggesting a higher degree of aromaticity in the pyridine. The aromatic substituent on C13 is twisted out of the plane of the pyrido[2,1-α]isoindole presumably due to steric clashes between the ortho-arene hydrogens and the heterocyclic core (N1–C13–C14–C19: 49.8°). The crystal does not exhibit a π-stacking system between pyrido[2,1-α]isoindoles; however, there are C–H π-interactions between C12–H and a second pyrido[2,1-α]isoindole (see ESI,† for details).
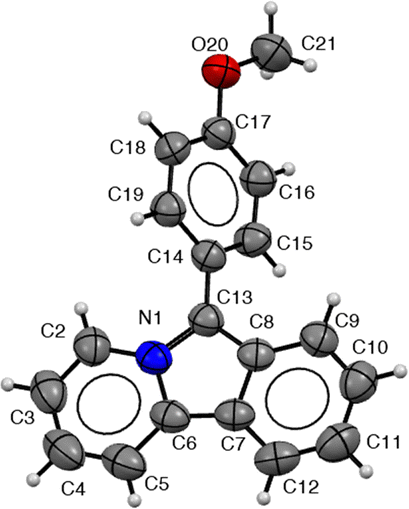 | ||
| Fig. 2 Crystal structure of 2c. Select bond lengths [Å]: N1–C13 1.385(3), N1–C2 1.396(3), N1–C6 1.415(3), C6–C7 1.403(3), C7–C8 1.418(3), C8–C13 1.395(3), C2–C3 1.354(4). Bond angles (°): C13–N1–C6 110.08(18), N1–C13–C8 106.47(19), C13–C8–C7 109.65(18), C6–C7–C8 106.87(19) dihedral angles (°): C5–C6–C7–C12 0.3, C2–N1–C13–C8 176.4, N1–C13–C8–C9 177.6. | ||
Our preliminary postulate for the mechanism of this multicomponent reaction is shown in Fig. 3. The oxidative addition of aryl bromides to Pd(0) is well-established and would form here a Pd(II)-(2-pyridyl) complex 3 that can undergo CO insertion to form the Pd-acyl complex 4. In the presence of chloride, anionic exchange can occur to form the chloride complex 5. As we have previously noted,6 chloride significantly enhances the rate of 1,3-dipole 1 formation by allowing the reductive elimination of an electrophilic acid chloride, which offers a lower barrier pathway to incorporate the weakly nucleophilic imine than direct reaction with complex 4 for subsequent cyclization to 1,3-dipole 1. The rigid, large bite-angle Xantphos ligand presumably also facilitates this step by creating a sterically encumbered palladium to drive the disfavored reductive elimination of acid chloride.6 In analogy to reports of benzyne addition to mesoionic 1,3-dipoles such as Münchnones,19 the in situ generation of aryne after 1,3-dipole formation leads to its rapid cycloaddition followed by cycloreversion to release isocyanate to form pyrido[2,1-α]isoindole 2. The isocyanate can be clearly seen by in situ 1H NMR analysis of the reaction mixture (see Fig. S1, ESI†). The inhibitory influence of benzyne generation during catalysis could be tied to the presence of fluoride, which may slow the generation of the 1,3-dipole 1, or the formation of benzyne before the 1,3-dipole is present, leading to its decomposition.
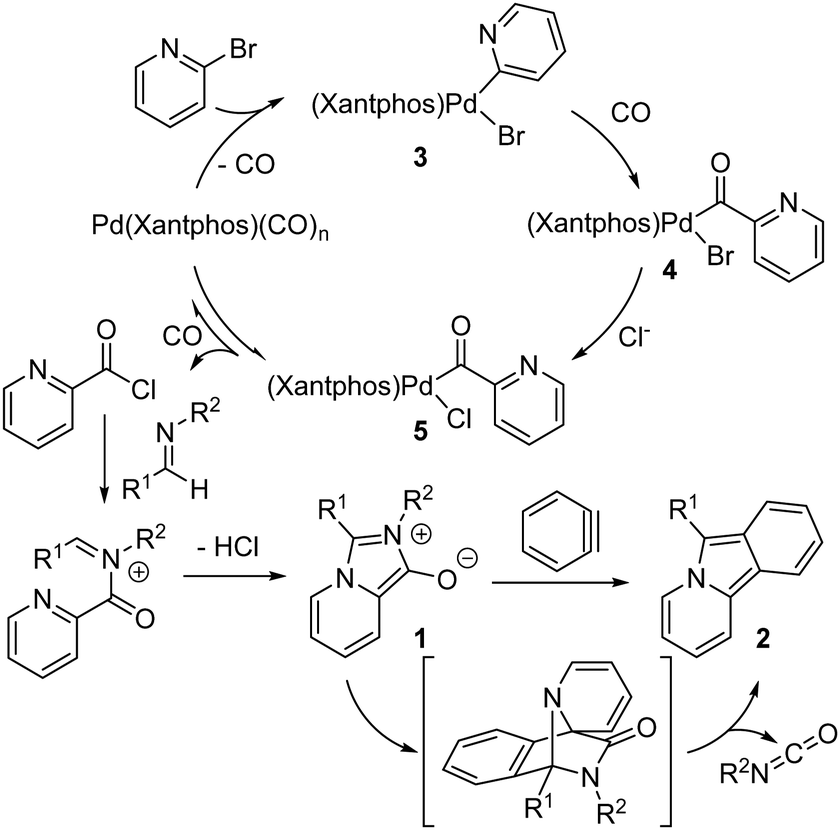 | ||
| Fig. 3 Proposed reaction mechanism of pyrido[2,1-α]isoindole formation. | ||
In conclusion, we have described a multicomponent route to form substituted pyrido[2,1-α]isoindoles. This couples the palladium-catalyzed carbonylative formation of pyridine-based 1,3-dipoles with their ability to undergo cycloaddition with in situ generated arynes. The systematic variation of any of the three substrates can allow a range of new variants of these structures to be generated in one pot, and from either commercially available or easily generated building blocks.
We would like to thank the Natural Science and Engineering Research Council of Canada (NSERC), McGill University and the FRQNT supported by the Centre for Green Chemistry and Catalysis for funding this research.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. M. D’Souza and T. J. J. Müller, Chem. Soc. Rev., 2007, 36, 1095–1108 RSC; (b) R. V. A. Orru and E. Ruijter, Synthesis of Heterocycles via Multicomponent Reactions. I, Springer-Verlag, Berlin, 2010 Search PubMed; (c) B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323–8359 CrossRef CAS PubMed.
- B. Cornils, W. A. Herrmann, M. Beller and R. Paciello, Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Four Volumes, Wiley, 2017, vol. 4 Search PubMed.
- (a) J. S. Quesnel and B. A. Arndtsen, J. Am. Chem. Soc., 2013, 135, 16841–16844 CrossRef CAS PubMed; (b) G. Kinney, J. Tjutrins, G. M. Torres, N. J. Liu, O. Kulkarni and B. A. Arndtsen, Nat. Chem., 2018, 10, 193–199 CrossRef PubMed; (c) R. G. Kinney and B. A. Arndtsen, Angew. Chem., Int. Ed., 2019, 58, 5085–5089 CrossRef CAS PubMed; (d) T. M. Levesque, R. G. Kinney and B. A. Arndtsen, Chem. Sci., 2020, 11, 3104–3109 RSC; (e) Y. Liu, A. M. Kaiser and B. A. Arndtsen, Chem. Sci., 2020, 11, 8610–8616 RSC.
- (a) R. Munday, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 2754–2755 CrossRef CAS PubMed; (b) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed; (c) M. N. Burhardt, R. H. Taaning and T. Skrydstrup, Org. Lett., 2013, 15, 948–951 CrossRef CAS PubMed.
- (a) B. A. Arndtsen, Chem. – Eur. J., 2009, 15, 302–313 CrossRef CAS PubMed; (b) S. Bontemps, J. S. Quesnel, K. Worrall and B. A. Arndtsen, Angew. Chem., 2011, 123, 9110–9113 CrossRef; (c) D. C. Leitch, L. V. Kayser, Z.-Y. Han, A. R. Siamaki, E. N. Keyzer, A. Gefen and B. A. Arndtsen, Nat. Commun., 2015, 6, 7411 CrossRef CAS PubMed; (d) J. Tjutrins and B. A. Arndtsen, Chem. Sci., 2017, 8, 1002–1007 RSC.
- S. A. Roy, J. Zgheib, C. Zhou and B. A. Arndtsen, Chem. Sci., 2021, 12, 2251–2256 RSC.
- (a) A. Biju, Modern Aryne Chemistry, Wiley-VCH, Weinheim, 2021 Search PubMed; (b) H. Ne Pellissier and M. Santelli, Tetrahedron, 2003, 59, 701–730 CrossRef.
- (a) N. G. Rondan, L. N. Domelsmith, K. N. Houk, A. T. Bownelb and R. H. Levin, Tetrahedron Lett., 1979, 3237–5240 CrossRef CAS; (b) W. F. Maier, G. C. Lau and A. B. McEwen, J. Am. Chem. Soc., 1985, 107, 4724 CrossRef CAS.
- (a) J. S. A. Badaro, B. Koszarna, M. H. E. Bousquet, E. T. Ouellette, D. Jacquemin and D. T. Gryko, Org. Chem. Front., 2022, 9, 1861–1874 RSC; (b) T. Mitsumori, M. Bendikov, O. Dautel, F. Wudl, T. Shioya, H. Sato and Y. Sato, J. Am. Chem. Soc., 2004, 126, 16793–16803 CrossRef CAS PubMed; (c) A. Pareek, M. Mehboob, M. Cieplac, M. Majdecki, H. Szabat, K. Noworyta, P. Polcynski, M. Morawiak, P. Sharma, C. Foroutan-Nejad and P. Gawel, J. Am. Chem. Soc., 2025 DOI:10.1021/jacs.4c16189.
- (a) B. Song, X. Guo, L. Yang, H. Yu, X. Zong, X. Liu, H. Wang, Z. Xu, Z. Lin and W. Yang, Angew. Chem., Int. Ed., 2023, 62, e202218886 CrossRef CAS PubMed; (b) K. Speck and T. Magauer, Beilstein J. Org. Chem., 2013, 9, 2048–2078 CrossRef PubMed; (c) A. M. Hamlin, J. K. Kisunzu and R. Sarpong, Org. Biomol. Chem., 2014, 12, 1846 RSC; (d) S. W. Pelletier, Pure Appl. Chem., 1997, 69, 119–124 CrossRef CAS.
- (a) A. A. Pokholenko, Z. V. Vojtenko and V. A. Kovtunenko, Russ. Chem. Rev., 2004, 73, 833–849 CrossRef; (b) B. Zhao, M. Yu, H. Liu, Y. Chen, Y. Yuan and X. Xie, Adv. Synth. Catal., 2014, 356, 3295–3301 CrossRef CAS; (c) W. Li, S. Zhang, X. Feng, X. Yu, Y. Yamamoto and M. Bao, Org. Lett., 2021, 23, 2521–2526 CrossRef CAS PubMed; (d) D. Shi, T. Zeng, X. Lei, X. Wu, M. Li and Y. Zhang, Synthesis, 2022, 5434–5444 CAS.
- (a) C. Xie, Y. Zhang and P. Xu, Synlett, 2008, 3115–3120 CAS; (b) X. Huang and T. Zhang, Tetrahedron Lett., 2009, 50, 208–211 CrossRef CAS.
- (a) F. I. M. Idiris and C. R. Jones, Org. Biomol. Chem., 2017, 15, 9044–9056 RSC; (b) H. Yoshida, Comprehensive Aryne Synthetic Chemistry, Elsevier Science, 2022 Search PubMed.
- (a) Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 1211–1214 CrossRef CAS; (b) J. Shi, L. Li and Y. Li, Chem. Rev., 2021, 121, 3892–4044 CrossRef CAS PubMed.
- H. Hennige, R. Kreher and J. Uhrig, Synthesis, 1982, 842–844 CrossRef CAS.
- All benzyne is consumed under these conditions, and small amounts of products from its addition to 2a are formed, suggesting this is the highest efficiency accessible in the reaction.
- T. Ikawa, S. Masuda, A. Takagi and S. Akai, Chem. Sci., 2016, 7, 5206 RSC.
- S. Asako, T. Kobashi and K. Takai, J. Am. Chem. Soc., 2018, 140, 15425 CrossRef CAS PubMed.
- (a) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191–218 RSC; (b) J. M. Lopchuk and G. W. Gribble, Tetrahedron Lett., 2015, 56, 3208–3211 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, NMR spectra, and X-ray crystal data. CCDC 2386038. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06483f |
| This journal is © The Royal Society of Chemistry 2025 |