
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metalloporphyrins in bio-inspired photocatalytic conversions
Asterios
Charisiadis
 a,
Vasilis
Nikolaou
b,
Emmanouil
Nikoloudakis
cd,
Kalliopi
Ladomenou
*e,
Georgios
Charalambidis
*f and
Athanassios G.
Coutsolelos
*cd
a,
Vasilis
Nikolaou
b,
Emmanouil
Nikoloudakis
cd,
Kalliopi
Ladomenou
*e,
Georgios
Charalambidis
*f and
Athanassios G.
Coutsolelos
*cd
aInstituto de Ciencia de Materiales de Madrid, Consejo Superior De Investigaciones Científicas, Sor Juana Inés de la Cruz, 3, Madrid, Spain
bChimie Et Interdisciplinarité, Synthèse, Analyse, Modélisation (CEISAM), CNRS UMR 6230, Nantes, France
cLaboratory of Bioinorganic Chemistry, Department of Chemistry, University of Crete, Heraklion, Crete, Greece. E-mail: acoutsol@uoc.gr
dInstitute of Electronic Structure and Laser (IESL), Foundation for Research and Technology – Hellas (FORTH), Heraklion, Greece
eHephaestus Laboratory, School of Chemistry, Faculty of Sciences, Democritus University of Thrace, GR-65404 Kavala, Greece. E-mail: kladomenou@chem.duth.gr
fTheoretical and Physical Chemistry Institute, National Hellenic Research Foundation, Athens, Greece. E-mail: gcharal@eie.gr
First published on 20th February 2025
Abstract
Numerous natural systems contain porphyrin derivatives that facilitate important catalytic processes; thus, developing biomimetic photocatalytic systems based on synthetic metalloporphyrins constitutes a rapidly advancing and fascinating research field. Additionally, porphyrins are widely investigated in a plethora of applications due to their highly versatile structure, presenting advantageous photoredox, photophysical and photochemical properties. Consequently, such metallated tetrapyrrolic macrocycles play a prominent role as photosensitizers and catalysts in developing artificial photosynthetic systems that can store and distribute energy through fuel forming reactions. This review highlights the advances in the field of metalloporphyrin-based biomimetic photocatalysis, particularly targeting water splitting, including both hydrogen and oxygen evolution reactions, carbon dioxide reduction and alcohol oxidation. For each photocatalytic system different approaches are discussed, concerning either structural modifications of the porphyrin derivatives or the phase in which the process takes place, i.e. homogenous or heterogenous. The most important findings for each porphyrin-based photocatalytic reaction are presented and accompanied by the analysis of mechanistic aspects when possible. Finally, the perspectives and limitations are discussed, providing future guidelines for the development of highly efficient metalloporphyrin-based biomimetic systems towards energy and environmental applications.
 Asterios Charisiadis | Dr Asterios Charisiadis is a postdoctoral researcher at the Materials Science Institute of Madrid (ICMM-CSIC), Spain. He carried out his postgraduate studies at the University of Crete, working on the preparation of porphyrin-based arrays for light-harvesting applications, including H2 production and CO2 reduction. He later held a postdoctoral position at Trinity College Dublin, focusing on the synthesis of non-planar porphyrins for 1O2 generation. His current work targets the development of artificial photosynthetic assemblies towards water splitting and their investigation through X-ray absorption spectroscopy (XAS). Asterios is the author of more than 30 publications and acts as a reviewer for scientific journals. |
 Vasilis Nikolaou | Dr Vasilis Nikolaou earned his PhD and continued as a postdoc at the University of Crete under Prof. A. G. Coutsolelos, focusing on artificial photosynthesis. He then joined Prof. A. Llobet's group at ICIQ, Spain, to study electrocatalytic CO2 reduction. Most recently, Vasilis was awarded a Marie Skłodowska-Curie Individual Fellowship to develop his own project on solar-driven catalysis, hosted by Dr F. Odobel at the CEISAM Institute (Nantes). With over 50 publications, his work has earned recognition, including the Young Talent Award at the 18th International Congress of Catalysis (2024) and the Young Researcher Award from the University of Crete (2023). |
 Emmanouil Nikoloudakis | Emmanouil Nikoloudakis finished his PhD degree at the Department of Chemistry of the University of Crete in 2022. He has made short term research visits collaborating with laboratories in Grenoble and Nantes. In 2021, he visited under the Fulbright program the University of North Texas, in Denton, USA, where he worked for 4 months. His research activities involve the synthesis, purification and characterization of porphyrin chromophores and their utilization in applications such as photocatalytic hydrogen production, photoelectrocatalytic alcohol oxidation and self-assembling systems. Currently he is enrolled as a post-doctoral fellow at IESL-FORTH and has obtained funding from Bodossaki Foundation. |
 Kalliopi Ladomenou | Dr Kalliopi Ladomenou has been an Assistant Professor in the Chemistry Department at Democritus University of Thrace since 2022. She obtained her PhD from the University of Liverpool, completed postdoctoral research at the University of Edinburgh, and later joined the University of Crete. Her research focuses on synthesizing photosensitizers and catalysts for solar cells, hydrogen production, CO2 reduction, artificial photosynthesis, and biomimetic catalysis. She has published extensively in high-impact journals, authored a book chapter, and holds a patent. Additionally, she serves as a reviewer for scientific journals and an evaluator for national research proposals. |
 Georgios Charalambidis | Dr Georgios Charalambidis received his BS degree in Chemistry from the University of Crete in 2002. He then pursued graduate studies in bio-inorganic chemistry at the University of Crete and received his PhD degree in 2007. In 2023, he started his independent career as an Associate Researcher at the Theoretical and Physical Chemistry Institute of National Hellenic Research Foundation in Athens. His group is interested in the synthesis of porphyrin-related compounds and their application in photocatalytic hydrogen production and CO2 reduction, artificial photosynthesis, self-assembling nanostructures and dye-sensitized photoelectrosynthesis cells (DSPECs). |
 Athanassios G. Coutsolelos | Professor Athanassios G. Coutsolelos received his PhD from the University of Dijon in France (1985) with Prof. R. Guilard. After postdoctoral research (1985–1986) at the University of Texas in Houston with Prof. K. M. Kadish, he joined the University of Crete where he obtained a full Professor position in 1999. He was invited as a Visiting Professor to the University of Notre Dame (USA), in Orsay and in Strasbourg (France). He is the director of the Laboratory of Bioinorganic Chemistry and his primary research interests focus on the development of new materials based on porphyrin derivatives for artificial photosynthesis. |
1. Introduction
Porphyrins are one of few cases where a similar molecular framework is used in almost any imaginable application.1–3 Porphyrin systems play a prominent role in biomimetic photocatalysis due to their well-established chemistry and highly versatile structural properties.4–7 Specifically, they are aromatic systems (18-π electrons) that present exceptional light-absorption properties, high thermal and chemical stability, and an efficient production of separated ion products upon irradiation. The porphyrin macrocycle is composed of four pyrrole units that are linked at their α-carbons through methine bridges, and can be easily modified by introducing a wide range of functional groups at its periphery.8,9 Furthermore, the inner core of these macrocyclic compounds can accommodate any transition metal ion, hence enhancing their photochemical and photoredox properties.10What is more, many natural systems, such as heme, chlorophyll, cytochromes and peroxidases, contain tetrapyrrole skeletons, with multifaceted uses in oxygen and carbon dioxide (CO2) transport, electron transfer, catalysis, and photosynthesis.11–13 These “pigments of life” play a key role in sustaining aerobic life on Earth;14 thus being widely employed in systems dealing with some of the most pressing energy challenges in an environment-friendly and sustainable way. Metalloporphyrins have been recognized as ideal photosensitizers and catalytic units, based on their excellent light-harvesting properties and photocatalytic activity, respectively.15–17 This feature article focuses on recent progress in the emerging field of porphyrin-based artificial photosynthetic systems, capable of storing and distributing solar energy through fuel forming reactions.18
One of the most urgent scientific challenges targets the production of carbon-neutral energy by exploiting renewable sources, i.e. sunlight and water, due to the extensive depletion of fossil fuels and resulting global warming.19 Artificial photosynthesis, and particularly photocatalytic water-splitting systems, has attracted significant interest.20 Such systems facilitate the conversion of photon energy into chemical energy,21 by tackling different light-induced catalytic processes. Particularly, reduction of aqueous protons to form hydrogen (H2) constitutes the reductive side of water splitting, and is one of the major strategies for solar energy conversion being widely investigated.5,22,23 Another substantial strategy is the photocatalytic transformation of carbon dioxide into useful hydrocarbons, since no CO2 emission decrease is foreseen in the following years.24,25 The products of this reductive process can act either as fuels (methane or methanol) or valuable chemical substances (carbon monoxide, formic acid or formaldehyde). Notably, the water oxidation reaction is particularly challenging since efficient water oxidation catalysts are needed to accelerate the concomitant removal of four protons and four electrons, coupled with the formation of an O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond.26,27 Hence, the oxidative side of water splitting is considered the key step in both natural and artificial photosynthetic systems. Due to the high energetic barrier of this process, alternative catalytic reactions have also been targeted, with the most appealing being the oxidation of alcoholic organic compounds.28 Importantly, such transformations offer a valuable strategy to exploit biomass, which is an alcohol-rich source, and produce high-valued carbonyl compounds.29
O bond.26,27 Hence, the oxidative side of water splitting is considered the key step in both natural and artificial photosynthetic systems. Due to the high energetic barrier of this process, alternative catalytic reactions have also been targeted, with the most appealing being the oxidation of alcoholic organic compounds.28 Importantly, such transformations offer a valuable strategy to exploit biomass, which is an alcohol-rich source, and produce high-valued carbonyl compounds.29
In this feature article we will focus on our group's contributions in developing porphyrin-based assemblies towards biomimetic photocatalytic applications.5 Additionally, some of the most important findings for each bio-inspired photocatalytic transformation will be discussed. In all cases two different approaches can be followed to achieve the catalytic processes, viz homogenous or heterogenous systems.30 For all systems discussed herein, the solar-driven catalytic conversion is achieved by employing assemblies that contain metalloporphyrins acting either as photosensitizers or as catalysts. In the homogenous systems, the photosensitizer (PS) and catalyst (CAT) units are combined with a sacrificial electron acceptor (SEA: for the oxidative processes) or donor (SED: in both reduction reactions) (Scheme 1a and b).31–34 The PS and CAT moieties can interact either through diffusion forces or via covalent bonding and have well-defined structures, making the determination of the mechanistic aspects easier.
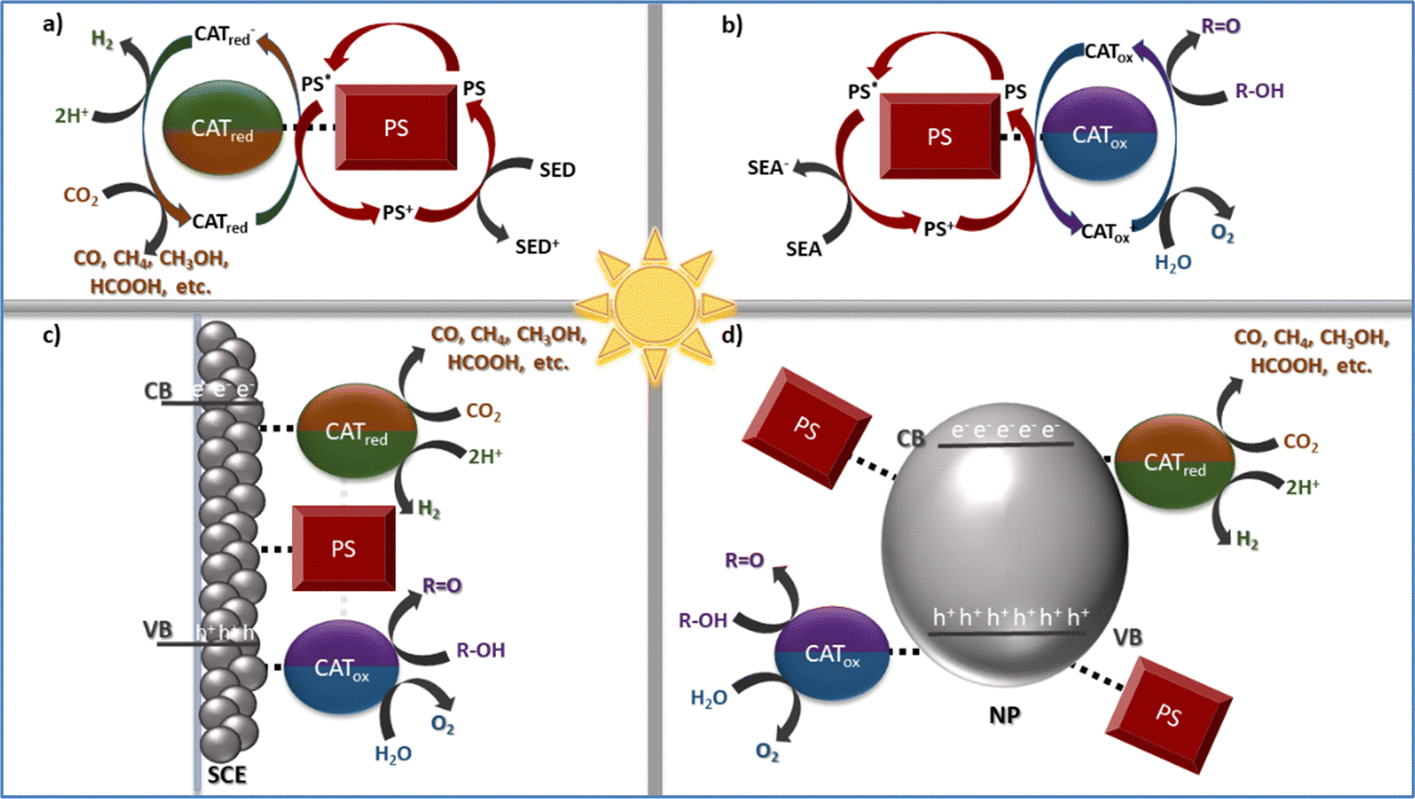 | ||
| Scheme 1 Upper part: Simplified schematic representation of homogenous photocatalytic systems toward the reduction of H+ to H2 or CO2 to CO, CH4, CH3OH, and HCOOH (a), and water or alcohol oxidation (b). PS: photosensitizer, CAT: catalyst, SEA: sacrificial electron acceptor and SED: sacrificial electron donor. Lower part: Simplified schematic representation of heterogenous photocatalytic systems by employing either a semiconducting electrode (c) or nanoparticles (d). PS: photosensitizer, CAT: catalyst, SCE: semiconductor electrode, NP: nanoparticles, CB: conduction band and VB: valence band. | ||
The presence of a sacrificial agent along with the photodegradation of the PS and/or CAT units constitutes the main drawback of such systems. An elegant approach to overcome these issues concerns the adsorption of the photosensitizer and catalyst on semiconductor electrodes or nanoparticles (Scheme 1c and d).35–38 In such heterogenous photochemical systems, the semiconducting nanoparticles facilitate the electron or hole transfer between the chromophore and catalytic entities to efficiently perform each corresponding process. What is more, the adsorption of both the PS and CAT entities on semiconducting surfaces minimizes their tendency to self-aggregate; hence increasing their stability. Heterogeneous catalytic assemblies further achieve enhanced photocatalytic efficiencies, since both structural and electronic aspects of the immobilized moieties (coordination number and environment, metal spin state, etc.) are altered through their interaction with the semiconducting substrate.
As discussed herein, metalloporphyrins have been extensively studied both as photosensitizers and catalysts for such bio-inspired catalytic processes. Water splitting is a two-part process involving both oxygen evolution and hydrogen production. While O2 evolution is often considered the more energetically demanding step, H2 evolution is equally critical, as it directly produces hydrogen gas—a clean and versatile fuel. This highlights the importance of understanding and optimizing hydrogen evolution systems, which will be the focus of the next section. Emphasis will be given to zinc-metallated porphyrins, which are among the most extensively studied photosensitizers in applications such as H2 evolution and alcohol oxidation. Zn–porphyrins have proven to be exceptional light-harvesters, often being the main components of photosynthetic reaction center mimics as they efficiently promote charge separation processes.39,40 Additionally, examples of various metallated macrocycles acting as photocatalysts towards H2 production will be presented. Regarding the transformation of CO2 to valuable carbon-based products, iron–porphyrins are amongst the most efficient catalysts that have been developed thus far.41 As far as water oxidation systems are concerned, porphyrins that contain different metal ion centres, namely Zn and Ru or Ni and Co, have been used either as photosensitizers32,38 or as catalysts,42,43 respectively. Finally, the perspectives and limitations are highlighted as they arise from our group's work, providing future guidelines for the development of highly efficient biomimetic catalytic systems based on metalloporphyrin derivatives.
2. Photocatalytic hydrogen evolution
Photocatalytic hydrogen evolution is a process bioinspired by photosynthesis.44 In natural photosynthesis, plants use chlorophyll to absorb sunlight and drive a series of reactions that convert water and carbon dioxide into glucose and oxygen. Similarly, in photocatalytic hydrogen production, artificial photocatalysts absorb sunlight to initiate the splitting of water into hydrogen and oxygen.45–48 Both processes aim to convert solar energy into a usable form of chemical energy. Conversely, artificial photocatalytic systems convert solar energy into H2, a versatile and clean fuel that can be stored and later used in fuel cells or other applications.19,49 Both natural and artificial photosynthetic assemblies employ specific light-harvesting compounds, i.e. chlorophyll and photosensitizers respectively, capable of exciting electrons. This electron excitation is the critical step that provides the energy needed to break the chemical bonds in water, leading to hydrogen production. Ideal photosensitizers should meet several key requirements, including chemical stability, an appropriate band gap energy, favorable thermodynamic redox potentials, and high activity. Among the various types explored, porphyrins rank among the best due to their unique structure and similarity to natural systems.46,50–52Porphyrins form hybrid photocatalytic systems when combined with semiconductors.5,30,53 Owing to their advantages—such as excellent light-harvesting capabilities, efficient photoinduced electron transfer, and structural versatility—they can serve multiple roles in photocatalytic hydrogen evolution systems. Particularly they can act as photosensitizers, photocatalysts, or catalysts. The photocatalytic process can be classified as either homogeneous or heterogeneous, depending on the phase in which the reaction occurs. Regarding the homogeneous systems, the well-defined structures of the individual components significantly aid in elucidating all mechanistic aspects of H2 evolution. On the other hand, heterogeneous assemblies offer several advantages, such as the presence of semiconducting surfaces, which make them suitable for practical applications, such as solar fuel cells. A photochemical system requires a photosensitizer (PS), a sacrificial electron donor (SED) and a catalyst (Scheme 2).
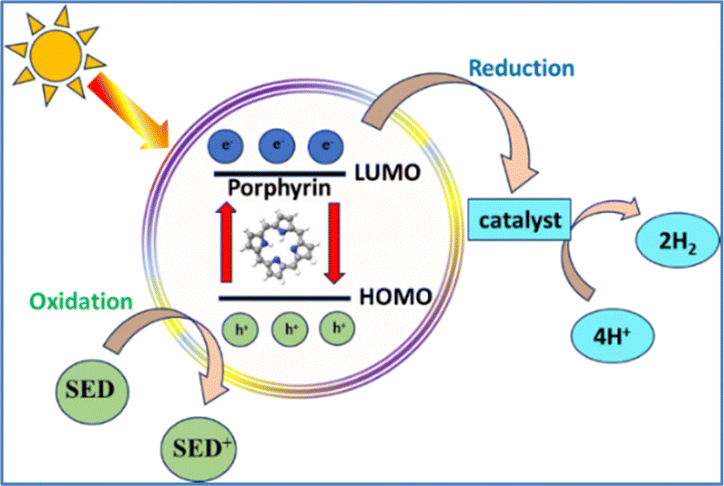 | ||
| Scheme 2 Simplified scheme of half-cycle photochemical H2 evolution. | ||
For efficient hydrogen evolution, the conduction band (CB) or LUMO energy level must be sufficiently negative relative to the hydrogen evolution potential, while the valence band (VB) level or HOMO orbital must be more positive than the oxygen generation potential. This process requires a minimum energy input of 1.23 eV. Since most dyes, including porphyrins, absorb visible light in the range of 1.5 to 3.1 eV, they possess sufficient thermodynamic power to drive the water-splitting reaction.
The primary challenges are kinetics, arising from the difference in speed between the rapid charge separation (and recombination) procedure and the slow oxidation and reduction of water. Therefore, an essential element in any photochemical water-splitting cycle is the use of multi-electron catalysts. Such units can store electrons or positive charges produced by the photosystem and transfer them to the substrate through low-energy activation pathways. Considerable research is currently directed at developing more efficient catalysts for both water oxidation and hydrogen evolution.54–57 Understanding the photophysics of photocatalytic components is crucial for designing assemblies that enhance hydrogen production and improve stability. Homogeneous catalysis can offer insights into the mechanisms behind light-driven reactions, contributing to the advancement of heterogeneous photocatalytic systems. Thus far, three major approaches were adopted by our group in the photocatalytic H2 evolution reaction (HER): (i) homogeneous photocatalytic systems, where porphyrins act as photosensitizers, (ii) self-assembled heterogeneous photocatalytic systems, and (iii) dye-sensitized heterogeneous photocatalytic systems (DSPs).
Since the late 1970s, numerous homogeneous photocatalytic hydrogen-evolution systems have been developed, revealing two distinct mechanistic behaviors: oxidative quenching and reductive quenching. These mechanisms depend on the direction of the initial electron transfer in the excited state: either from the photosensitizer to the catalyst (oxidative quenching) or from the sacrificial electron donor (SED) to the photosensitizer (reductive quenching). Coutsolelos and colleagues reported for the first time a bioinspired noble-metal free photocatalytic system based on water-soluble porphyrin PS and cobalt-based CAT. More specifically, our group synthesized water soluble porphyrins, both free-base (TMPyP) and metalated with zinc (ZnTMPyP), and studied their activity as a PS in the presence of cobalt complexes such as cobaloxime (Cat1) (Fig. 1).58
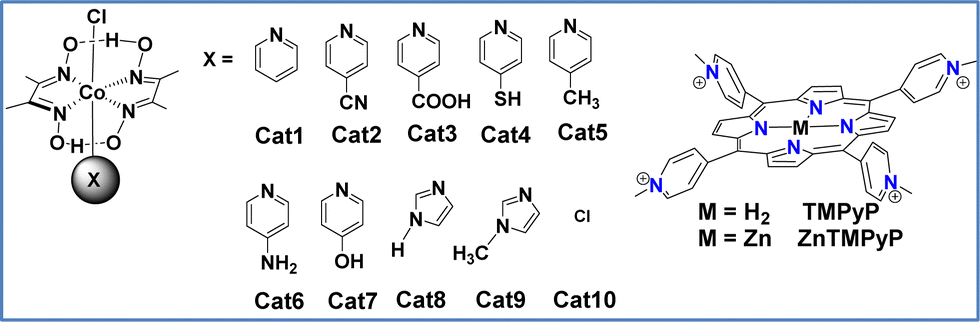 | ||
| Fig. 1 Chemical structures of porphyrin-based photosensitizers and cobaloxime-based catalysts. | ||
The system produces hydrogen under visible light irradiation (λ > 400 nm) in an acetonitrile–water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solution, using triethanolamine (TEOA) as the SED, reaching a maximum turnover number (TON) of 280 after 25 hours. In a follow-up study, our group tested the activity of the same ZnTMPyP photosensitizer with various cobaloxime catalysts featuring different axial ligands (Fig. 1).59 The catalysts were modified by incorporating diverse pyridine and imidazole axial ligands. Notably, the catalyst with N-methyl imidazole (Cat9) achieved the highest catalytic performance with a TON of 1135 after 50 hours of irradiation, while the imidazole ligand-based catalyst (Cat8) reached 565 TONs. Among the catalysts with pyridine axial ligands, those with electron-donating groups were more effective for hydrogen production. One of the main drawbacks of this type of homogenous system was its instability. To address this issue, we further improved it by employing heterogeneous photocatalysis.
:1) solution, using triethanolamine (TEOA) as the SED, reaching a maximum turnover number (TON) of 280 after 25 hours. In a follow-up study, our group tested the activity of the same ZnTMPyP photosensitizer with various cobaloxime catalysts featuring different axial ligands (Fig. 1).59 The catalysts were modified by incorporating diverse pyridine and imidazole axial ligands. Notably, the catalyst with N-methyl imidazole (Cat9) achieved the highest catalytic performance with a TON of 1135 after 50 hours of irradiation, while the imidazole ligand-based catalyst (Cat8) reached 565 TONs. Among the catalysts with pyridine axial ligands, those with electron-donating groups were more effective for hydrogen production. One of the main drawbacks of this type of homogenous system was its instability. To address this issue, we further improved it by employing heterogeneous photocatalysis.
A second approach concerns the use self-assembled porphyrin molecules designed to replicate the organized structures of chlorophyll in photosynthesis. These self-assembled chromophores exhibit distinct properties compared to their monomeric counterparts, such as broader and intensified light absorption.60–62 We synthesized a peptide nucleic acid (PNA)–porphyrin derivative, PNA-TPP63 and a peptide–(Zn)porphyrin hybrid, Fmoc-FF-(Zn)Por,64 to study their self-assembly characteristics and catalytic activity for H2 evolution (Fig. 2). Both conjugates form supramolecular architectures when exposed to a mixed solvent system using the “good-bad” solvent protocol. For PNA-TPP, spherical structures formed in a dichloromethane–ethanol (2:8) mixture (Fig. 2(a)). Under visible light irradiation in the presence of Pt nanoparticles, PNA-TPP achieved an H2 production of 74.43 nmol over 4.5 hours. Control experiments with a mixed solution of 0.1 M ascorbic acid and 5 μM K2PtCl4 resulted in minimal H2 production (12 nmol in 4.5 hours), indicating the effectiveness of the assembled nanostructure in light harvesting. The porphyrin–peptide hybrid Fmoc-FF-(Zn)Por self-assembled into nanospheres or fibrils, depending on the solvent system used. Nanospheres (Fig. 2(b)) formed when Fmoc-FF-(Zn)Por was dissolved in tetrahydrofuran (THF) and diluted in methanol (THF/MeOH, 2:8). Conversely, using a dichloromethane/heptane (1:1) mixture resulted in the formation of micrometer-long fibrils instead (Fig. 2(c)). By adjusting the solvent mixture, the same compound can self-assemble into two distinct nanostructures. When evaluated for their photocatalytic activity in the HER, each morphology demonstrated varying levels of efficiency. The fibril-like nanostructures of Fmoc-FF-(Zn)Por showed the highest catalytic performance, achieving a maximum H2 evolution rate of 1.96 mmol g−1 h−1 and remaining highly stable for over 400 hours under irradiation, with a maximum turnover number (TON) of 155. In contrast, the spherical nanostructures displayed minimal photocatalytic activity, reaching only 10 TONs after 360 hours of irradiation. Additionally, the catalytic efficiency of the amorphous solid was 17 TONs after 378 hours of irradiation, somewhat better than the spherical nanostructures but still significantly lower than the fibrillar nano-assemblies.
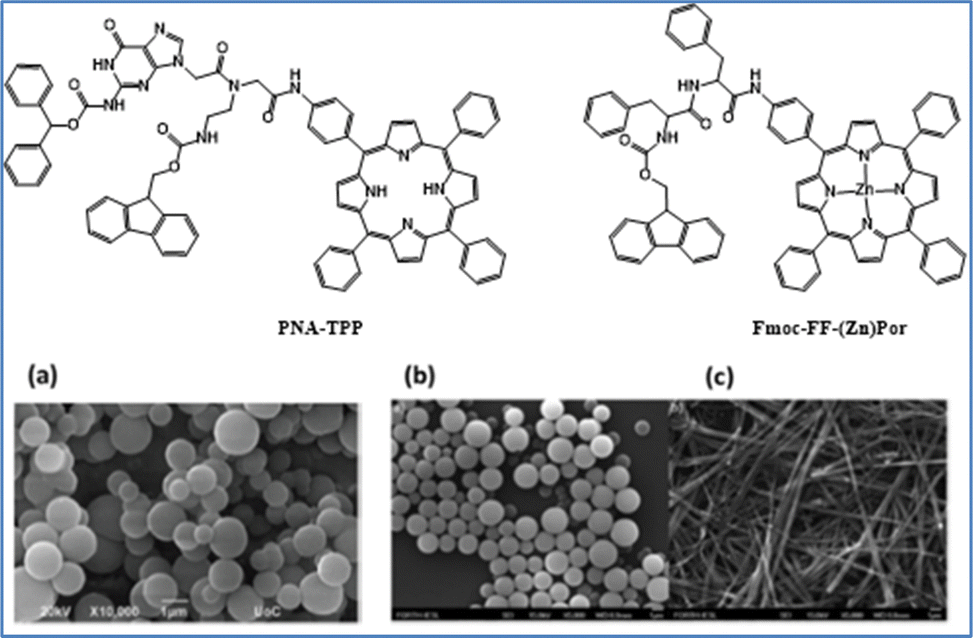 | ||
| Fig. 2 Chemical nanostructures of self-assembled porphyrins and SEM images of (a) PNA-TPP CH2Cl2–EtOH, 2:8 (7 mM), (b) Fmoc-FF-(Zn)Por THF/MeOH, 2:8 (1 mM), and (c) Fmoc-FF-(Zn)Por CH2Cl2/Hept, 1:1 (1 mM). Reproduced from ref. 63 with permission from Royal Society of Chemistry, copyright 2019. | ||
Bai, Zhu, and colleagues investigated self-assembled porphyrin nanoleaves that effectively enhanced photochemical hydrogen evolution (Fig. 3).65 A Pd(II)-tetra(4-carboxyphenyl)-porphyrin PdTCPP, was self-assembled in N,N-dimethylformamide (DMF) with the assistance of the surfactant 1-decanesulfonic acid through π–π interactions and hydrogen bonding. Both experimental data and theoretical calculations indicated the formation of a strong internal electric field, enabling the directional transport of photogenerated electrons and holes, and supporting efficient photocatalytic hydrogen production. Photocatalytic experiments were conducted under visible light irradiation (λ > 400 nm) with ascorbic acid as the sacrificial agent. After 5 hours of irradiation, the nanoleaves achieved a H2 production rate of 9.9 mmol g−1 h−1 without additional cocatalysts, a rate 40 times higher than that of commercial PdTCPP powder (0.25 mmol g−1 h−1). The nanoleaves also demonstrated excellent stability, maintaining a steady H2 production rate over 100 hours of visible light exposure. This study highlights the significance of a long-range conjugated structure in enhancing photocatalytic performance in porphyrin assemblies.
 | ||
| Fig. 3 Chemical structure of PdTCPP, (a) SEM image of nanoleaves, and (b) photocatalytic H2 production of the commercial PdTCPP powder and nanoleaves without cocatalysts. Reproduced from ref. 65 with permission from American Chemical Society, copyright 2022. | ||
In a similar work, our group reported a noble-metal free Zn(II)–porphyrin derivative (ZnTPP) that was self-assembled into three distinct structures. Subsequently, these three different self-assemblies were studied as photosensitizers in a photocatalytic hydrogen evolution system in water (Fig. 4).66 These structures were prepared with the “good-bad” solvent self-assembly protocol, maintaining the same “bad” solvent while varying the “good” solvent. This approach produced three different structural forms, each displaying unique catalytic activity for H2 production in the presence of 5% w/w Pt nanoparticles as catalysts and ascorbic acid as a sacrificial electron donor. The octahedral form achieved the highest hydrogen evolution rate of 185.5 μmol g−1 h−1. Additionally, the porphyrin could be recycled and reassembled into the optimal structure for H2 production. This work demonstrates that simple porphyrin-based chromophores can be effectively optimized for photocatalytic hydrogen evolution through controlled self-organization in mixed solvent systems.
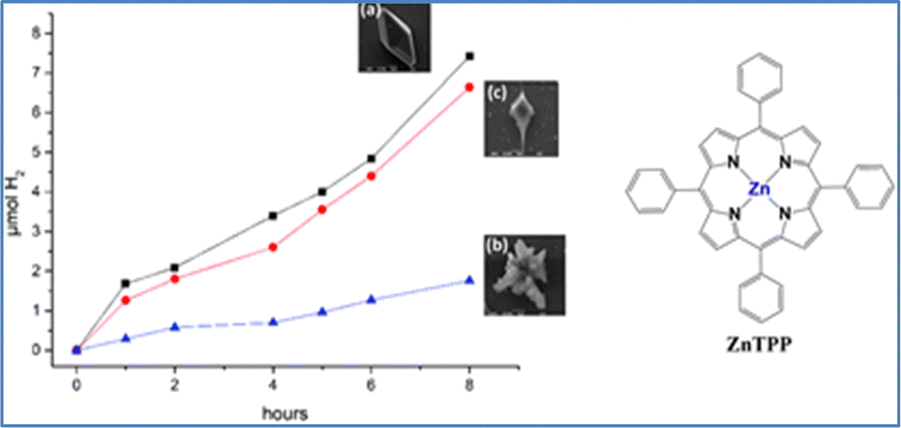 | ||
| Fig. 4 Photocatalytic hydrogen production of ZnTPP with (a) octahedral (THF/MeOH 1:4), (b) “flower” (CHCl3/MeOH 1:4), and (c) “manta-ray” (Tol/MeOH 1:4) structures, in the presence of Pt nanoparticles. Reproduced from ref. 66 with permission from Royal Society of Chemistry, copyright 2022. | ||
An alternative approach for efficient H2 production evolution is the dye-sensitized photocatalytic systems. In this kind of system TiO2 nanoparticles act as a scaffold were a photosensitizer and a catalyst are adsorbed forming a heterogeneous photocatalyst. Numerous studies have explored various molecular photosensitizers and catalysts in TiO2-based dye-sensitized systems.67,68 In addition, the deposition of Pt nanoparticles onto the TiO2 semiconductor, results in highly efficient and stable systems towards H2 evolution.69–71 Coutsolelos and coworkers further advanced this approach by using porphyrin-based photocatalysts ameliorating the H2 production of the system.71 In a recent report, our group prepared two types of metallated porphyrin derivatives bearing carboxylic acids for the attachment onto Pt–TiO2 nanoparticles.72 In one variant, the carboxylic acid was attached via a three-carbon alkyl chain at the para-position of the phenyl ring (M-Tc3CP, M = Zn, Pd, Pt) (Fig. 5).
 | ||
| Fig. 5 Chemical structures of porphyrin molecules M-TCP, M-Tc3TCP adsorbed onto TiO2 nanoparticles, (a) PS and CAT on TiO2 nanoparticles, (b) PS onto Pt–TiO2 nanoparticles, (c) PS-CAT onto TiO2 nanoparticles, and (d) PS-CAT onto Pt–TiO2 nanoparticles. Reproduced from ref. 72 with permission from Royal Society of Chemistry, copyright 2023. | ||
In the other series, the carboxylic acid group was directly attached to the para-position of the four meso-phenyl rings (M-TCP, M = Zn, Pd, Pt). The photocatalytic performance of the porphyrin-based dye-sensitized photocatalysts was notably affected by the amount of porphyrin adsorbed onto the TiO2 nanoparticles. Analysis of the optimal photocatalytic results for each system indicates that all M-Tc3TCP derivatives outperform their M-TCP counterparts in terms of both stability and efficiency. However, Zn-TCP and Zn-Tc3TCP showed nearly equivalent TONs and similar H2 evolution rates, suggesting that the photocatalytic activity of Zn–porphyrin derivatives is unaffected by the type of anchoring group, verifying their role solely as photosensitizers. In contrast, both Pd-Tc3TCP and Pt-Tc3TCP demonstrated superior TONs and H2 evolution rates compared to their respective Pd-TCP and Pt-TCP counterparts. Notably, Pt-Tc3CP exhibited outstanding performance, achieving 11607 TONs and 458 mmol g−1 h−1 of H2 evolution, far exceeding Pt-TCP, which reached 2525 TONs and 378 mmol g−1 h−1 of H2. The enhanced efficiency of the M-Tc3CP over the M-TCP derivatives may be due to their adsorption via four anchoring groups, which promotes a well-ordered self-assembly of the M-Tc3CP structures. While all reported examples rely on a sacrificial donor, a dye-sensitized system would be more sustainable if dye regeneration were achieved through an oxidation reaction, potentially transforming abundant substrates into valuable products. This approach will be discussed in a later section on photocatalytic alcohol oxidation. While hydrogen evolution is a critical component of artificial photosynthesis for generating clean energy, another equally vital strategy focuses on mitigating carbon dioxide emissions through its transformation into valuable fuels and chemicals. This approach addresses the dual challenges of renewable energy production and environmental sustainability, which leads us to the next significant process in artificial photosynthesis: photocatalytic CO2 reduction.
3. Photocatalytic CO2 reduction
The reliance on fossil fuels as primary energy sources has led to widespread concerns about greenhouse gas emissions, particularly carbon dioxide.73 Addressing CO2 emissions is essential to combat climate change and ensure sustainable development. A key strategy to tackle this issue involves converting CO2 into valuable chemicals and fuels using renewable, carbon-neutral energy sources; thus, facilitating the transition to a “low-carbon” economy.74 Solar-driven photocatalysis, which directly converts sunlight into chemical energy, offers a sustainable and scalable solution for reducing CO2 emissions.75 This approach not only provides a sustainable and carbon-neutral method for energy production but also contributes to mitigating climate change.76 Recently, artificial photosynthetic systems have emerged as advanced solutions for CO2 conversion. These systems use solar energy to drive chemical reactions, potentially offering a cost-effective means for the large-scale production of fuels and chemicals.77Although the CO2 reduction reaction (CO2RR) is still under development, it shows great potential for converting CO2 into valuable products.78 Carbon monoxide (CO), a notable product of CO2RR, is especially valuable because it can be produced at low overpotentials with high selectivity.79 However, optimizing reaction kinetics and minimizing concurrent hydrogen evolution remain critical challenges for enhancing CO production. As the field progresses, developing molecular catalysts with high activity, selectivity, and durability is crucial.80,81 Two main strategies have emerged to advance molecular CO2 reduction catalysis: (i) modifying ligands to adjust the redox properties of metal centers, and (ii) utilizing second coordination sphere effects, such as incorporating charged or hydrogen-bonding groups to improve CO2 capture and conversion.
Regarding the first approach, which focuses on ligand modifications, Savéant and colleagues, in their seminal work, demonstrated that the introduction of electron-withdrawing and electron-donating groups to the periphery of the iron–porphyrin macrocycle significantly impacts electrocatalytic performance (Fig. 6a).82 By incorporating penta-fluorinated electron-withdrawing units, two effects were observed: (i) a more positive shift in the standard potential of the iron–porphyrin, and (ii) a decrease in the catalytic rate constant, resulting in lower turnover frequency (TOF) values. Furthermore, Ott and co-workers illustrated that strategic ligand modifications can unlock unconventional catalytic pathways that operate at lower overpotentials. This was achieved by introducing a methyl group at the ortho position of a bipyridine ligand of a ruthenium-terpyridine-bipyridine catalyst (Fig. 6b).83 In 2001, a series of iron complexes were synthesized and evaluated for their electrocatalytic efficiency. These iron complexes, based on 2,2′:6′,2′′-terpyridine and methylated bis(2-pyridylmethyl)amine ligands, exhibited selective CO2 reduction to CO at remarkably low overpotentials (Fig. 6c).84 Additionally, McCrory and colleagues demonstrated that, in cobalt pyridyldiimine complexes, the effective overpotential is primarily governed by the redox potential of the ligand. By designing cobalt complexes with extended conjugation and incorporating electron-withdrawing groups, they achieved significant improvements in catalytic activity (Fig. 6d).85
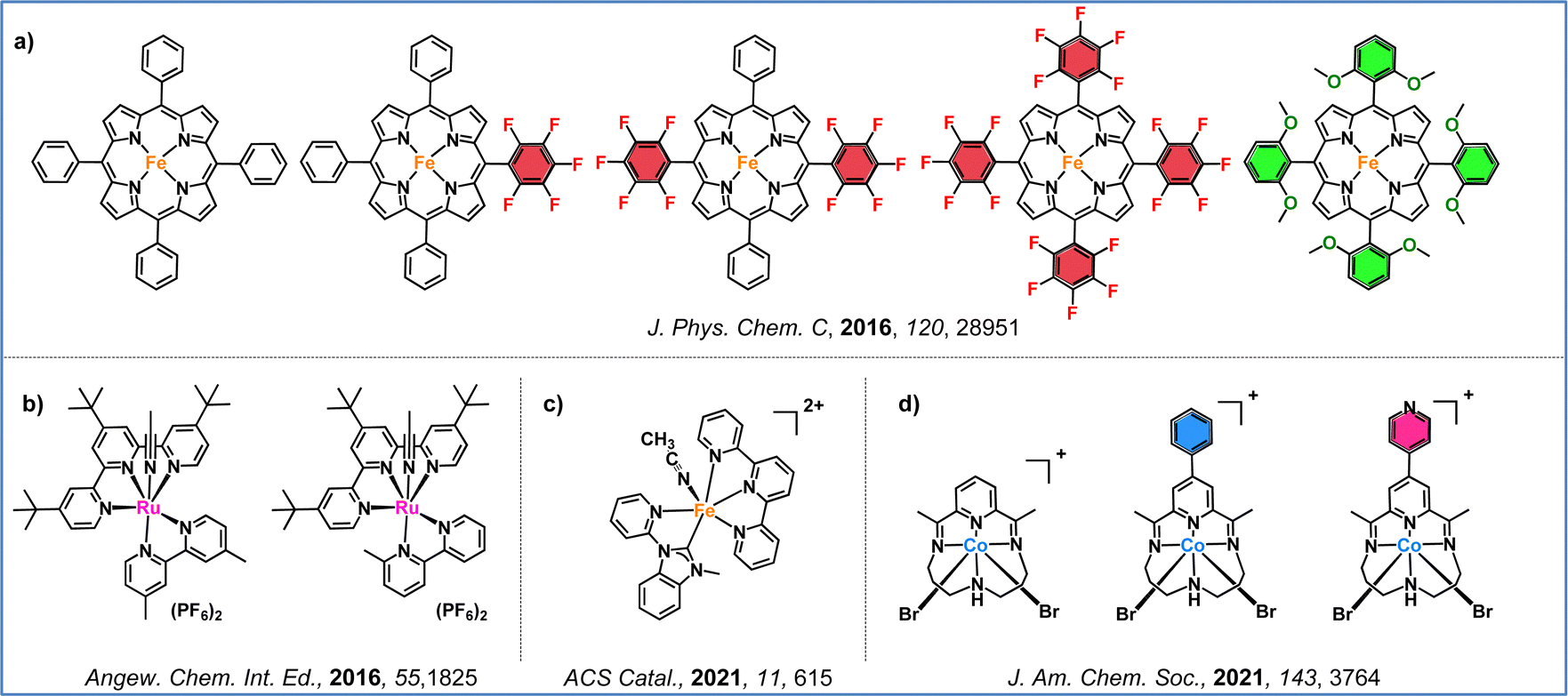 | ||
| Fig. 6 Representative examples from the literature illustrating ligand modification strategies: (a) introduction of electron-withdrawing groups to the iron–porphyrin macrocycle, (b) strategic methylation of ruthenium-complexes in the ligand framework, (c) iron-complex with modified ligands, and (d) cobalt-complexes with extended conjugation and electron-withdrawing units. | ||
Another effective strategy involves utilizing second coordination sphere effects, either through the incorporation of charged groups or the formation of hydrogen bonds. These modifications play a key role in enhancing CO2 capture and conversion. The first example was presented in 2012 by Savéant and colleagues, who demonstrated that introducing 2,6-hydroxyphenyl groups at the periphery of an iron–porphyrin complex significantly enhanced catalytic activity (Fig. 7a).86 This increased efficiency results from the high local proton concentration generated by the phenolic hydroxyl substituents, which enhance the catalytic activity. Further advances came in 2016, when the same research group introduced four positively charged trimethylanilinium groups into an iron–porphyrin, resulting in enhanced CO selectivity and stability (Fig. 7b).87 The improved catalytic performance was driven by the coulombic interactions between the negatively charged Fe(0)–CO2 adduct and the positively charged trimethylanilinium groups within the porphyrin macrocycle. Inspired by the active site of carbon monoxide dehydrogenase, Aukaullo, Halime, and coworkers functionalized iron–porphyrins with urea groups, which serve as multipoint hydrogen-bonding sites. These urea groups help stabilize CO2 and promote its efficient reduction to CO (Fig. 7c).88 They also explored the impact of atropisomerism, developing αα and αβ forms of the same urea-functionalized iron–porphyrin, demonstrating that the catalytic behavior is strongly influenced via the different hydrogen-bonding configurations.89 Moreover, they discovered that multipoint hydrogen-bonding interactions might shift the redox activation from the commonly accepted Fe(0) state to the Fe(I) state, revealing new mechanistic insights into CO2 reduction.90 Chang and colleagues developed iron–porphyrins functionalized with imidazolium pendant units, which promoted synergistic effects through both positive charge and hydrogen-bond interactions (Fig. 7d).91 This modification significantly enhanced both the catalytic activity toward CO formation and CO2 binding compared to non-functionalized iron–tetraphenyl porphyrin, which is widely used in CO2 reduction studies.
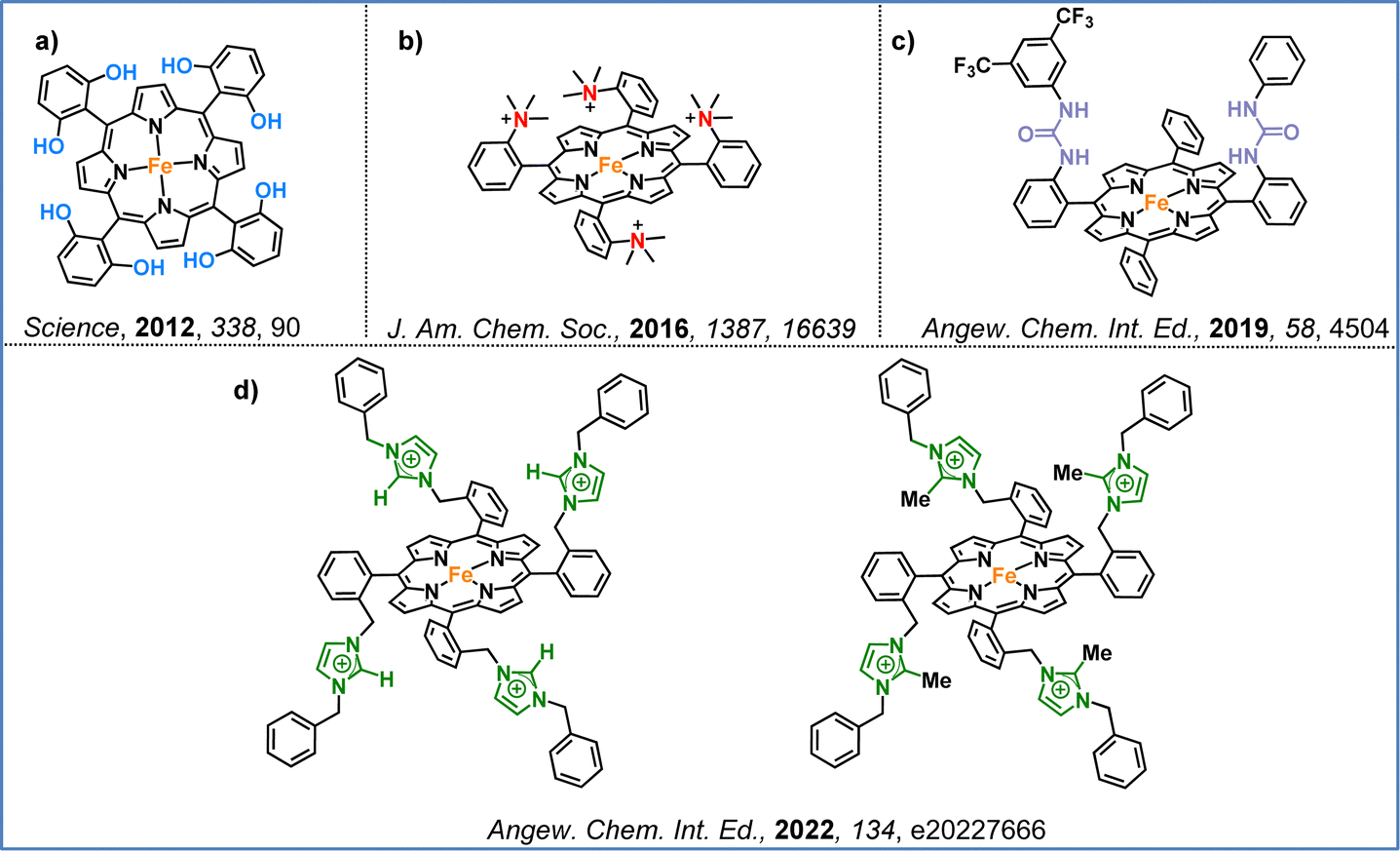 | ||
| Fig. 7 Representative examples from the literature illustrating second coordination sphere effects: (a) high local proton concentration facilitated by the phenolic hydroxyl substituents in porphyrins, (b) positively charged trimethylanilinium groups in a porphyrin framework, (c) urea groups help stabilize CO2, and (d) synergistic effects through both positive charge and hydrogen-bond interactions. | ||
All the systems already discussed in this section focused on electrocatalytic CO2 reduction, however our group has explored photocatalytic CO2 reduction. We focused on developing a series of iron porphyrins, either covalently attached to a photosensitizer or used in combination with a photosensitizer in solution.92,93 In our recent work, we synthesized four modified iron porphyrins, each featuring distinct substitution patterns around the porphyrin core.93 These structural variations had a significant impact on their catalytic performance in light-driven CO2 reduction. The most efficient catalyst, Fe-4F(TMA)-Por, featured four fluorine atoms at the ortho and meta positions of the meso-phenyl groups and trimethylammonium groups (TMA) at the para positions (Fig. 8). This design enhanced the electron-withdrawing effects of the fluorine atoms and the CO2 stabilization provided by the TMA groups, leading to an exceptionally high TON of 5500 and a TOF of 1375 h−1. In contrast, Fe-5F-Por, which includes five fluorine atoms on each phenyl ring but lacks the ammonium groups, showed significantly lower activity. Although the electron-withdrawing fluorine atoms shifted the reduction potential, the absence of stabilizing cationic groups hindered CO2 binding. This resulted in the lowest catalytic performance, with a TON of only 226. In another variation, Fe-4F(DMA)-Por, we replaced the trimethylammonium groups with dimethylamino groups (DMA). While the electron-withdrawing fluorine atoms still shifted the redox potential, the weaker CO2 stabilization due to the lack of charged groups led to moderate performance (282 TONs). Finally, we prepared Fe-TMA-Por, which lacks fluorine atoms but retains the trimethylammonium groups at the para position of the phenyl rings. Although the positively charged groups improved CO2 binding, the absence of fluorines limited electron transfer efficiency, resulting in a TON of 272. This work highlights the potential for developing more efficient molecular catalysts by tailoring their electronic and electrostatic properties. Our findings emphasize the critical balance between electron-withdrawing effects and CO2 stabilization in optimizing photocatalytic performance. By fine-tuning this balance, a molecular catalyst can achieve both efficient charge transfer and enhanced CO2 coordination.
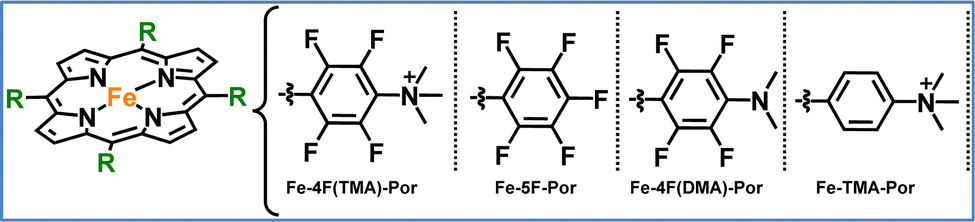 | ||
| Fig. 8 The molecular structures of the iron–porphyrins studied in this work.93 | ||
In our second study, we developed and investigated a porphyrin-catalyst dyad specifically designed for photocatalytic CO2 reduction.92 The FePor-Ru(bpy) dyad was systematically evaluated against its individual components, namely an iron porphyrin (FePor) and a ruthenium trisbipyridine (Ru(bpy)), to assess the impact of their covalent attachment (Fig. 9). This covalent connection strategy was employed to mitigate diffusion limitations commonly encountered in multi-component systems and to facilitate rapid intermolecular electron transfer from the photosensitizer to the catalyst. The results demonstrated that the dyad exhibited superior catalytic activity compared to the photocatalytic system with the individual components (FePor and Ru(bpy)). This enhancement can be primarily attributed to the dyad's ability to facilitate faster electron transfer and more effective charge separation than the standalone components. Although the excited state of the photosensitizer is initially quenched within the dyad, the covalent linkage fosters the formation of catalytically active species at lower thermodynamic costs, which is crucial for efficient CO2 reduction. This interaction allows the dyad to initiate the catalytic cycle more effectively than when FePor and Ru(bpy) function independently. In summary, the dyad not only outperformed its individual components in terms of catalytic activity, but also revealed a unique mechanism that underscores the significance of covalent coupling. This study highlights the potential for designing hybrid systems that leverage the strengths of both components, ultimately leading to more effective strategies for CO2 reduction.
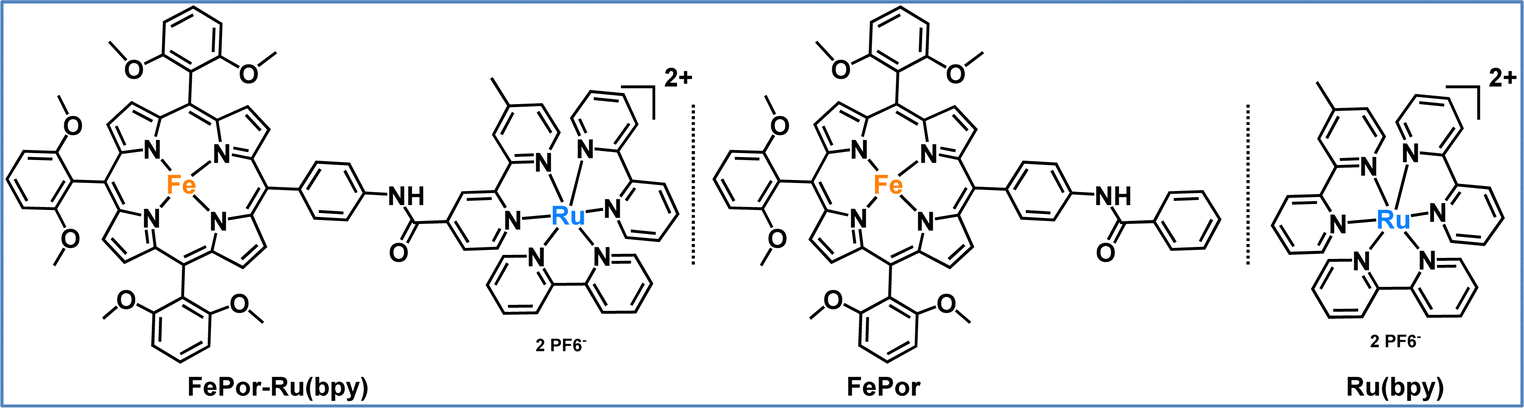 | ||
| Fig. 9 Molecular structures of the photosensitizer-catalyst dyad (FePor-Ru(bpy)) and its individual components, the catalyst (FePor) and the photosensitizer (Ru(bpy)), as studied in this work.92 | ||
Apart from the reduction processes already covered in the previous two sections, the oxidative half reaction of water splitting plays a huge part in developing efficient biomimetic artificial photosynthetic systems. More specifically, due to water oxidation being a particularly challenging process, necessitating the simultaneous removal of four protons and electrons, several arrays have been studied targeting the photocatalytic oxygen evolution reaction, as discussed in the following section.
4. Photocatalytic water oxidation
The growing demand for renewable energy sources has increased the interest in water oxidation catalysis, a key reaction for sustainable energy production through water splitting.94 Water oxidation to molecular oxygen constitutes the oxidative half-reaction of water splitting, which provides the necessary electrons and protons for the hydrogen generation reductive half-reaction. Solar-driven water oxidation mimics natural photosynthesis, providing a renewable and carbon-neutral approach for producing H2 fuel, a clean and sustainable energy carrier.95 Recent advancements in artificial photosynthetic systems have highlighted the potential of water oxidation catalysts (WOCs).96 Notably, Sun and coworkers developed a molecular ruthenium catalyst for water oxidation that demonstrated activity similar to the oxygen-evolving complex of photosystem II in vivo, in terms of turnover frequency and stability.97 However, achieving high efficiency and durability remains challenging due to the energetically demanding nature of the oxygen evolution reaction (OER).98 Current strategies to enhance water oxidation catalysis include: (i) morphology modulation, atomic doping, and anchoring of single-atom catalysts,99 and (ii) introduction of additional ligands and secondary coordination sphere interactions to facilitate the reaction kinetics.100Porphyrins have attracted extensive attention in this field due to their bio-inspired design, mimicking chlorophylls in photosystem II. In water oxidation catalytic systems, porphyrins serve as ligands, stabilizing the WOC metal centers. They also facilitate multi-electron transfer processes essential for converting water (H2O) into oxygen (O2), protons (H+), and electrons.101–104 The porphyrin macrocycle offers both structural rigidity and electronic tunability, allowing for precise control over the redox properties of the central metal ion.18 Metalloporphyrins with transition metals such as iron, manganese, nickel and cobalt can achieve the high oxidation states needed to form metal-oxo species, which are the active intermediates in the OER.105–107 Their macrocyclic ring also supports the stabilization of these high-valent species, preventing their decomposition and thus improving the overall catalytic efficiency.108 Additionally, porphyrins can facilitate proton-coupled electron transfer (PCET), a key mechanism in water oxidation, by modulating the electronic environment around the metal center.109,110
Inspired by the above, Bonnet and co-workers investigated hydrophilic nickel(II) porphyrins as catalysts for water oxidation in neutral to acidic conditions and in the presence of [Ru(bpy)3]2+ as the PS. They found that electron-poor substituents improved the catalytic efficiency by increasing the catalysts’ oxidation potential. Ni–porphyrins with electron-deficient groups, like Ni-3 and Ni-4, achieved 6-fold higher TOFs than electron-richer analogues Ni-1 and Ni-2 (Fig. 10a). However, the highly electron-poor Ni-5, was inactive since it presented an extremely high oxidation potential that the photosensitizer molecule [Ru(bpy)3]2+ was unable to achieve. Their electrochemical studies confirmed that electron-withdrawing substituents increased their oxidation potentials, balancing the driving force for water oxidation with electron transfer from the catalyst to the photosensitizer. Ni-3 and Ni-4 highlighted the potential of electron-tuned Ni–porphyrins to act as promising photocatalysts in the OER, as they showcased both stability and catalytic activity.102
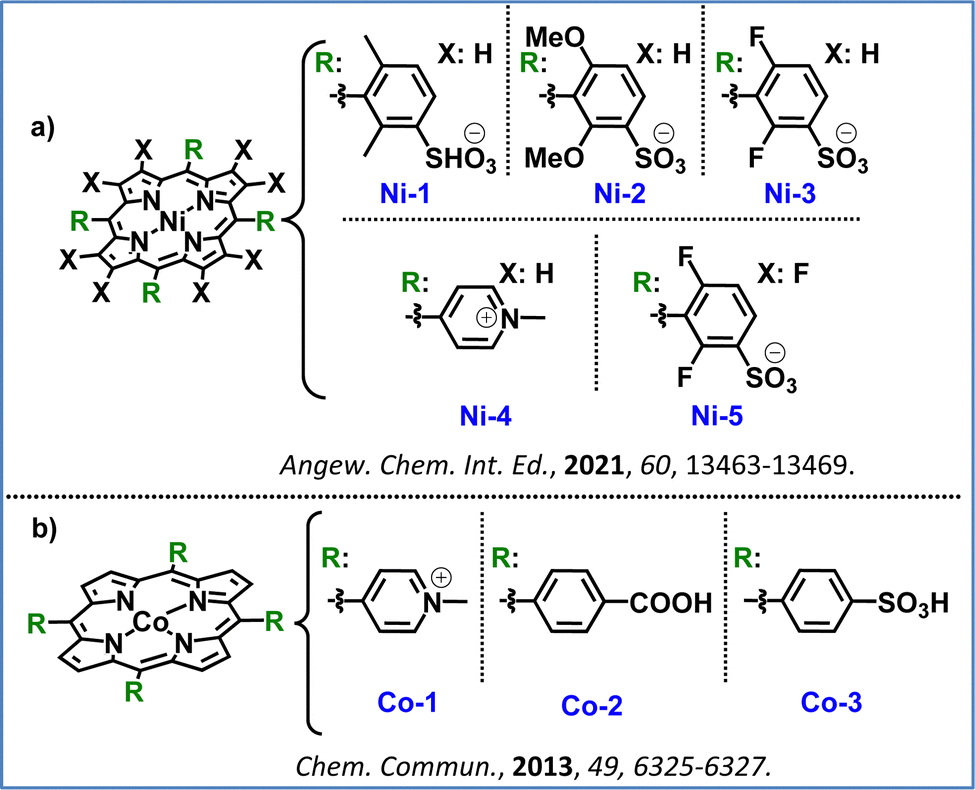 | ||
| Fig. 10 Representative examples from the literature illustrating ligand modification strategies: (a) hydrophilic Ni(II)–porphyrins with electron-withdrawing groups, and (b) water-soluble Co(II)–porphyrins with electron-withdrawing groups. | ||
Another noble metal free set of porphyrins was investigated by Sakai and collaborators who applied water-soluble Co–porphyrins (Co-1, Co-2, Co-3, Fig. 10b) as WOCs. They tested these three catalysts in the presence of the Ru(bpy)32+ photosensitizer and sodium persulfate (Na2S2O8) as the sacrificial electron acceptor at pH 11, with Co-3 presenting the highest activity. A second-order dependence on the catalyst concentration was observed in this work, suggesting that a bimolecular radical coupling step controls the reaction rate. Post-reaction analysis via ESI-MS indicated oxidative cleavage of the porphyrin ring, with oxidation products remaining bound to the cobalt center, ruling out cobalt nanoparticle formation.42
In an effort to prepare WOCs with increased stability, researchers have developed 2D and 3D materials that contain noble-metal free porphyrins. Paille et al. successfully developed a novel POM@MOF system, consisting of the porphyrinic MOF (photosensitizer) with an immobilized polyoxometalate catalyst, [(PW9O34)2Co4(H2O)2]10−, that presented high photocatalytic activity and stability (Fig. 11a). Computational studies revealed the role of TCPP porphyrin ligands in boosting oxidizing power and the MOF structure in stabilizing the catalytic site.111 Notably, Wang and coworkers introduced an ionic-type 2D covalent organic framework (COF), CoTPP-CoBpy3, that integrates cobalt porphyrin and cobalt bipyridine units to enhance photocatalytic water oxidation (Fig. 11b). This COF exhibits superior hydrophilicity and dispersibility, forming large nanosheets that facilitate local water collection at active sites. Key characteristics, including ultrafast charge transfer (1.8 ps) and prolonged charge-separated state (1.2 ns), support the efficient hole accumulation at CoTPP centers, achieving a high oxygen production rate of 7323 μmol g−1 h−1.112
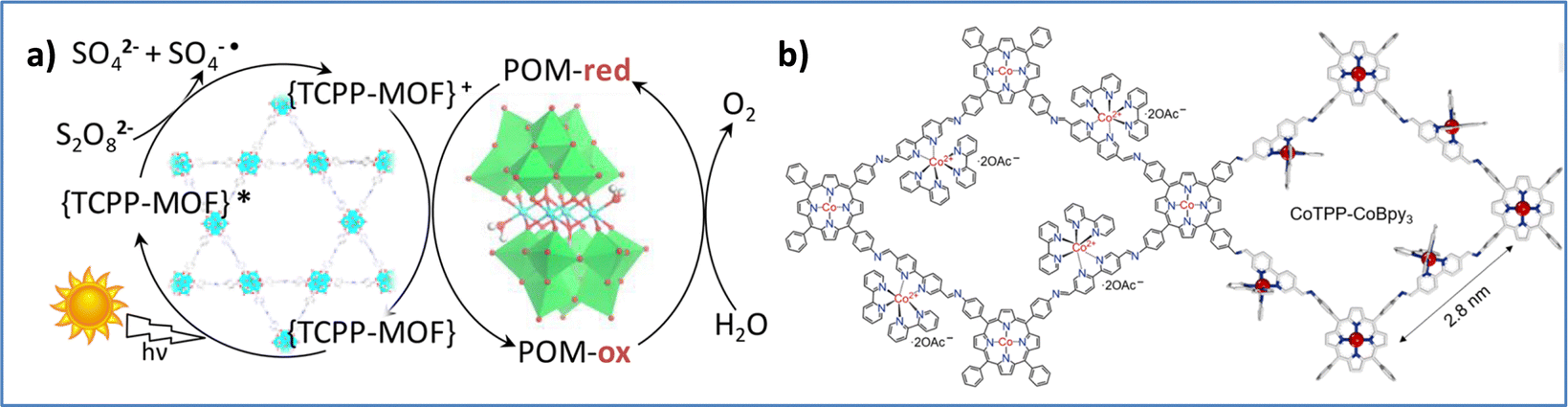 | ||
| Fig. 11 (a) Schematic illustration of the light-driven OER by P2W18Co4@MOF-545. Reproduced from ref. 111 with permission from American Chemical Society, copyright 2018. (b) Chemical structure of CoTPP-CoBpy3 COF. Reproduced from ref. 112 with permission from American Association for the Advancement of Science, copyright 2024. | ||
Having described homogenous and heterogenous photocatalytic systems for the OER in previous reports, we now turn our attention to analogous electrocatalytic systems. Han et al. applied the water-soluble cationic nickel(II) porphyrin Ni-4 (Fig. 10a) as a molecular electrocatalyst for water oxidation in neutral aqueous conditions, achieving an onset potential of ∼1.0 V (vs. NHE). Ni-4 operated as a homogeneous electrocatalyst without forming NiOx films, exhibiting consistent activity across a broad pH range (2.0–8.0) and stability over 10 hours.113 This example illustrates the potential of a single noble metal free porphyrin to act both as photo- and electro-catalyst for the OER.102,113 In another report Wang et al. investigated a series of cationic cobalt porphyrins as WOC electrocatalysts (Fig. 12a), with Co-4 demonstrating the highest efficiency in neutral aqueous conditions. This catalyst achieved an O2 production rate of 170 nmol cm−2 min−1, with a faradaic efficiency close to 90%. Buffer bases with higher pKa values were found to lower the catalytic onset potential, suggesting a critical role in proton-coupled electron transfer and O–O bond formation. The study confirmed Co-4 robustness, which showed no degradation or metal leaching over extended operation.114
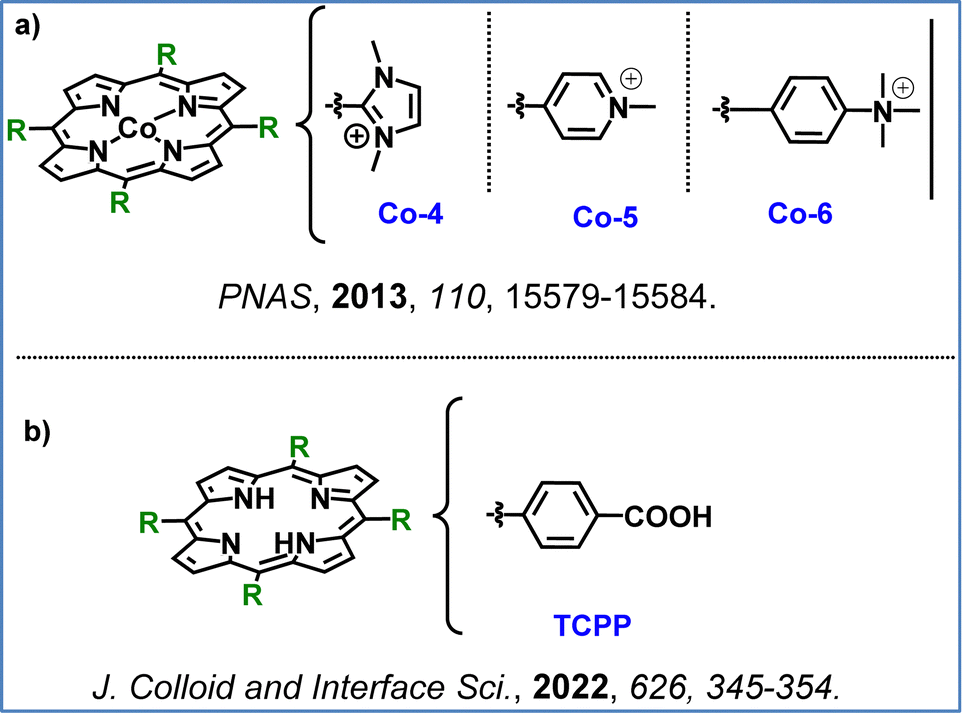 | ||
| Fig. 12 (a) Chemical structures of water-soluble Co(II)–porphyrins used as electrocatalysts for the OER by Wang et al.,114 and (b) the chemical structure of TCPP. | ||
Recently, a promising approach to water oxidation catalysis has emerged, which integrates the beneficial aspects of both photocatalytic and electrocatalytic systems: the development of photoanodes for photo-electrocatalytic (PEC) oxygen evolution reaction. Towards this direction, Xie and coworkers developed a hybrid photoanode, CoPi/TCPP/Ti–Fe2O3, integrating Ti-doped hematite, a simple free base porphyrin (TCPP, Fig. 12b), and cobalt phosphate (CoPi) cocatalyst to enhance photocatalytic water oxidation. The strategic band alignment of Ti–Fe2O3, TCPP, and CoPi allows directional transfer of photogenerated holes from Ti–Fe2O3 to CoPi through TCPP; hence improving charge separation at the interface and facilitating efficient hole injection into the catalytic site. This design achieved a photocurrent density of 1.84 mA cm−2 at 1.23 V vs. RHE, significantly outperforming CoPi/Ti–Fe2O3 alone. This study highlights the critical role of porphyrins in promoting charge transfer in photoanodes, offering a promising approach for designing efficient PEC systems for water oxidation.115
Inspired by the above reports and in alignment with the new PEC approach for water oxidation systems, our research team synthesized a molecular dyad comprising of a ruthenium tris(bipyridyl) photosensitizer coupled with a cost-effective nickel porphyrin catalyst (Fig. 13). The NiP-Ru dyad effectively combines light absorption and catalytic water oxidation functionalities, utilizing the Ru-complex as a light-harvester and Ni–porphyrin as the active water oxidation site. In initial experiments conducted in organic media, the covalently linked dyad demonstrated superior photocatalytic efficiency compared to a non-covalent, two-component system, as confirmed by the direct detection of O2via an optical oxygen sensor. Moving one step forward, we employed the NiP-Ru dyad in a photoelectrochemical cell, where it was immobilized on a TiO2 photoanode via carboxylate anchoring groups. This work demonstrated a proof of concept, where the dyad-based PEC device performs visible light induced water oxidation in neutral aqueous media.43
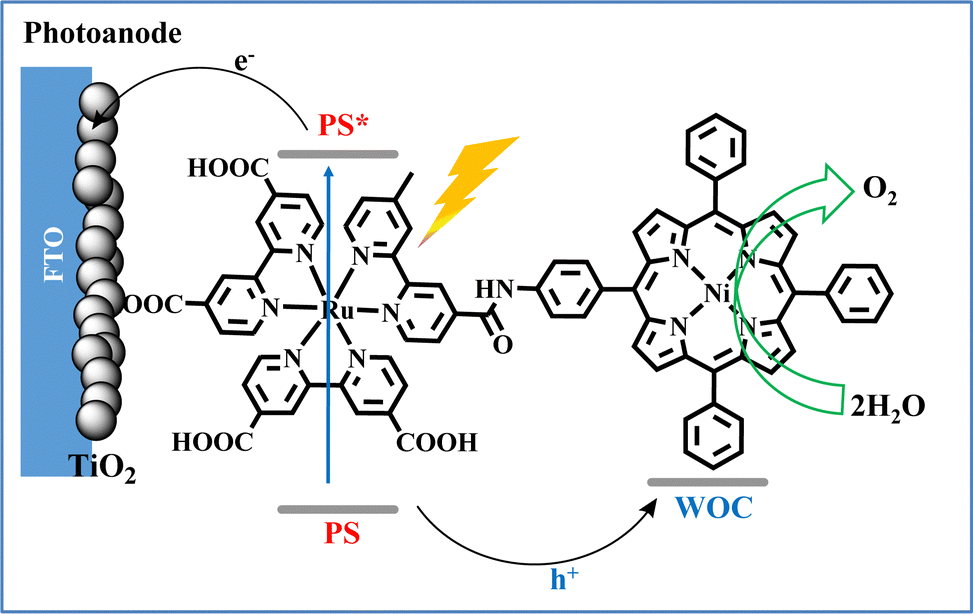 | ||
| Fig. 13 Schematic illustration of the photo-electrocatalytic water oxidation by the NiP–Ru dyad photoanode. Reproduced from ref. 43 from Royal Society of Chemistry, copyright 2022. | ||
5. Photocatalytic alcohol oxidation
The rapid expansion of global energy demand, coupled with escalating environmental concerns, has driven the search for sustainable and carbon-neutral energy solutions. Photocatalytic processes for H2 generation and CO2 reduction have garnered significant attention, offering the potential to both harness solar energy and contribute to a reduced carbon footprint.5,116 A central challenge in solar fuel photocatalytic systems, however, lies in efficiently managing the oxidative half-reactions occurring at the anode. Traditionally, the oxygen evolution reaction has been employed as the anodic counterpart to H2 production or CO2 reduction.117 Nonetheless, the OER presents several critical disadvantages that hinder its efficacy in photocatalytic and electrocatalytic systems. Primarily, the OER suffers from sluggish kinetics, as it requires a four-electron transfer process to produce oxygen from water and additionally it requires high potential (1.23 V vs. NHE at pH = 0).118 Furthermore, O2 has low economic value and the OER demands catalysts that can withstand harsh oxidative conditions, often necessitating the use of costly noble metals like iridium or ruthenium. Additionally, the OER's production of oxygen introduces safety concerns due to the potential formation of explosive H2/O2 mixtures. These limitations make the OER an energetically inefficient and economically challenging process.119 Alternatively, to enhance H2 evolution and CO2 reduction rates, researchers often introduce co-catalysts and various SEDs (e.g., ascorbic acid, formic acid, triethylamine, triethanolamine, and lactic acid) as hole scavengers into the reaction system.50 This approach leads to significant waste of resources but also results in the generation of by-products causing environmental pollution.120Recognizing the challenges posed by the OER or the presence of SEDs, researchers have begun to explore alternative strategies for the anode reaction in photo-driven solar fuel production systems. The motivation behind this effort is not only to find a way to provide the necessary electrons and protons for the reductive half-reactions with lower anodic overpotential and more favorable kinetics, but also to find new means of generating value-added chemical products via the oxidation reaction. In this regard, photocatalytic oxidation half-reactions such as biomass conversion, alcohol oxidation,121 amine oxidation,122 degradation of organic pollutants in wastewater,123 photo-reforming of waste plastics,124 and H2O2 production120 have attracted great interest.
Among them, the selective oxidation of alcohols to carbonyl compounds is one promising approach since it generates higher-value products, requires a significantly lower potential and only two holes/photons instead of four.125 Selective oxidation of alcohols is a crucial organic transformation with significant laboratory and industrial importance. The resulting carbonyl compounds (i.e., aldehydes and ketones) are valuable structural building blocks for the synthesis of pharmaceutical intermediates and fine chemicals.126 For example, formaldehyde, benzyl aldehyde, and 4-methyl benzyl aldehyde are widely used in the fragrance, confectionery, beverage, and pharmaceutical industries. However, traditional chemical oxidation methods involve toxic and corrosive heavy-metal oxidants. Moreover, strong oxidants and harsh operating conditions are required that generate undesired and overoxidized products, and significant waste. Thus, light-driven alcohol oxidation offers a potentially appealing alternative that is more cost-effective, safer, and environmentally friendly. Moreover, alcohols are abundant feedstocks since they can be extracted from biomass.29 Therefore, the efficient conversion of alcohols to aldehydes or ketones via oxidant-free photocatalysis has attracted significant research attention.125
Various solar-driven H2 evolving systems coupled with alcohol oxidation derivatives have been reported in the literature.127–129 Li et al. developed a hybrid photocatalytic system that employs graphitic carbon nitride (g-C3N4) as a semiconductor, alongside a ruthenium complex and platinum as the oxidation and reduction catalysts, respectively. This system performed selective oxidation of benzyl alcohols coupled with H2 evolution in pure water under visible light irradiation (Fig. 14a).130 The selectivity of benzyl alcohol oxidation towards the corresponding benzaldehydes is significantly enhanced due to the formation of the active Ru(IV)O intermediate species of the molecular catalyst, through efficient hole transfer from g-C3N4 to the Ru catalyst. Similarly, Reisner and co-workers reported a hybrid photocatalytic system composed of carbon nitride functionalized with cyanamide groups on its surface, paired with a molecular nickel(II) bis(diphosphine) catalyst.131 This system effectively integrates light-driven hydrogen evolution reaction and benzylic alcohol oxidation (Fig. 14b). Under optimized conditions, the hybrid system exhibited high activity for both the HER and aldehyde production, achieving a rate of 763 mmol g−1 h−1 and a quantum efficiency of 15%. The turnover frequency was calculated to be 76 h−1 based on the amount of nickel, with a conversion rate of 4-methylbenzyl alcohol reaching 83.0%. Transient absorption spectroscopy revealed that the cyanamide groups on the surface of carbon nitride suppressed electron–hole recombination, thereby enhancing catalytic efficiency. These groups helped suppress the recombination of excited electrons and holes, thereby increasing the overall yield. However, the system's operational lifetime was limited, likely due to the instability of the nickel catalyst under photocatalytic conditions.
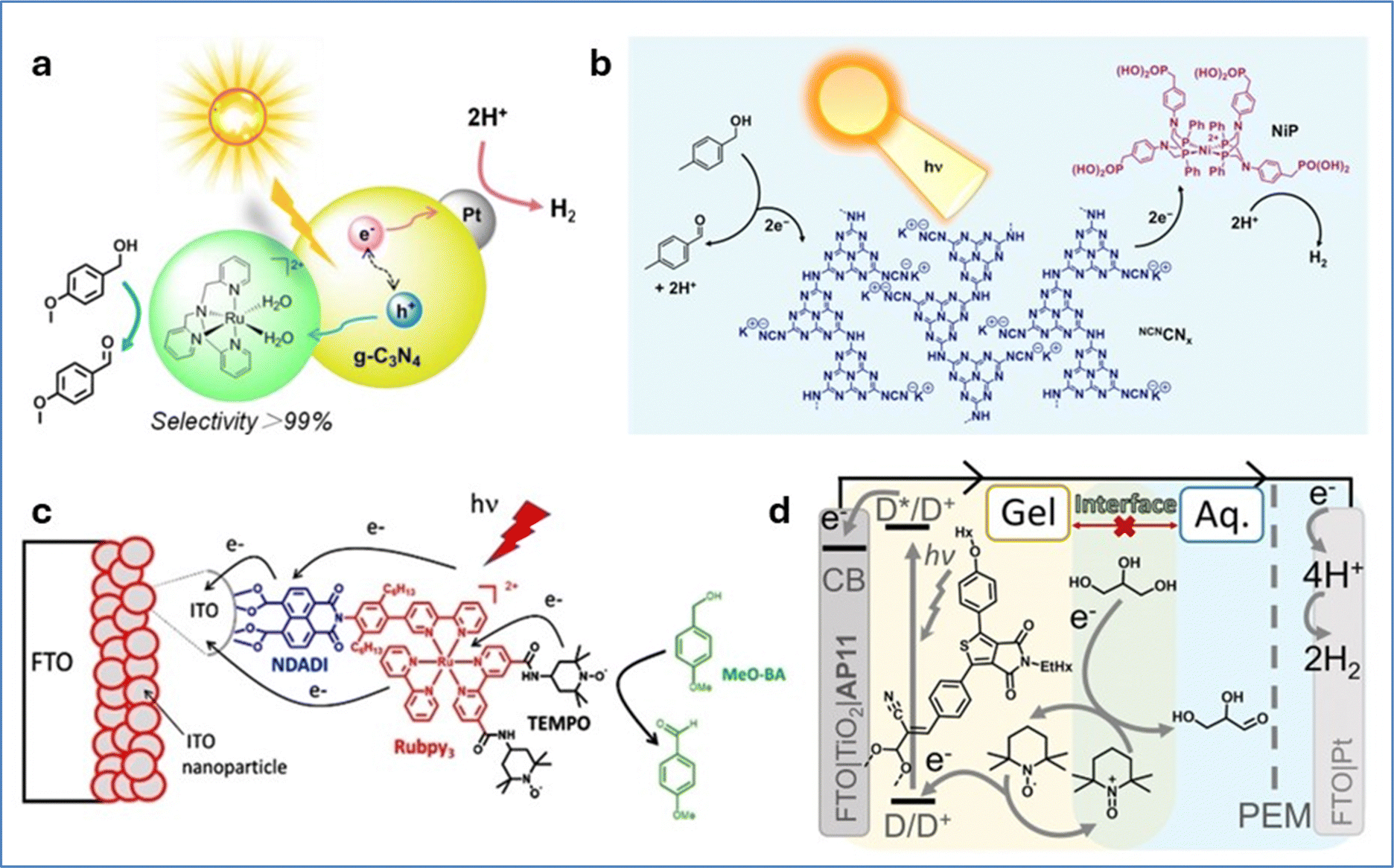 | ||
| Fig. 14 (a) Schematic representation of a redox system containing a molecular Ru catalyst and Pt modified g-C3N4 composite oxidizing benzyl alcohols to aldehydes with over 99% selectivity. Reproduced from ref. 130 with permission from Elsevier, copyright 2018. (b) Schematic representation of a closed redox system for solar-driven H2 evolution coupled with alcohol oxidation. Reproduced from ref. 131 with permission from American Chemical Society, copyright 2016. (c) Schematic representation of the photoanodic compartment for the NDADI-Ru-TEMPO triad supported on ITO nanoparticles. Reproduced from ref. 132 with permission from John Wiley and Sons, copyright 2021. (d) Schematic representation of an aqueous biphasic DSPEC with a TEMPO containing redox-gel layer for glycerol oxidation. Reproduced from ref. 133 with permission from John Wiley and Sons, copyright 2022. | ||
Odobel and co-workers developed a hybrid photocatalytic system by immobilizing a molecular triad (NDADI-Ru-TEMPO) on a nano-ITO electrode.132 The triad includes a Ru-tris-bipyridine complex as a photosensitizer, linked to 2,2,6,6-tetramethyl-1-piperidine N-oxyl (TEMPO) as an alcohol oxidation catalyst, and naphthalenedicarboxyanhydride dicarboximide (NDADI) as an electron acceptor (Fig. 14c). Linear sweep voltammograms under chopped light irradiation in the presence of 50 mM para-methoxybenzyl alcohol, demonstrated an increased photocurrent response, reaching a net photocurrent of about 150–180 μA cm−2. Under light irradiation and in pH 10 carbonate buffer, the triad achieved selective oxidation of para-methoxybenzyl alcohol to para-methoxybenzaldehyde with a TON of ∼150 in 1 hour at a bias of 0.4 V vs. SCE. Under optimal conditions, the average faradaic efficiency was on the order of 80%. Nitroxides in general and TEMPO in particular are very active, cheap, green catalysts for alcohol oxidation,134,135 that are even applied to the chemical industry.136 Reek and co-workers developed an aqueous dye-sensitized photoelectrochemical cell (DSPEC) capable of oxidizing glycerol and at the same time producing H2.133 In this work a mesoporous TiO2 electrode was sensitized with a thienopyrroledione-based organic dye and a TEMPO catalyst for glycerol oxidation (Fig. 14d). Both components were embedded in a 3 mm thick acetonitrile-based redox gel on the TiO2 surface, creating a biphasic system with the aqueous solution. Under one-sun irradiation in a 0.1 M glycerol solution (pH 8.3), the cell achieved a photocurrent density near 200 μA cm−2 with nearly quantitative faradaic efficiency for glyceraldehyde production, remaining stable for up to 48 hours. The introduction of the gel layer presented a 10-fold increase in the oxidation of glycerol compared to the aqueous system. This was attributed to the gel's ability of stabilizing the photoanode components from detachment under the alkaline conditions required for glycerol oxidation.
Our group explored the development of a TiO2-based DSPEC for alcohol oxidation, using a TEMPO organo-catalyst covalently linked, for the first time, to a zinc porphyrin (ZnP) sensitizer.137 Two different dyads (Fig. 15), each distinguished by the nature of the anchoring group (carboxylic acid vs. hydroxamic acid) attached to the porphyrin, were synthesized and their absorption, emission, and electrochemical characteristics were recorded. The photovoltaic properties of the dyads were initially tested in dye-sensitized solar cells (DSSCs) to optimize the dyeing conditions and assess their relative efficiencies. The TEMPO-substituted dyads outperformed the reference Zn–porphyrin without TEMPO, showing significantly higher short-circuit current (Jsc) and open-circuit voltage (Voc). The incident photon-to-current conversion efficiency (IPCE) reached 2.6% at 430 nm, corresponding to the Soret absorption band. Then the DSPEC systems were effectively applied to the photoelectrochemical oxidation of benzyl alcohol derivatives. The optimal performance was observed in an aqueous borate buffer at pH 8, using the hydroxamic acid-anchored hyd-ZnP-TEMPO dyad. Under these conditions, para-methoxy benzaldehyde was selectively produced, achieving an average photocurrent density of 200 μA cm−2, a faradaic efficiency of 82%, a TON of 26, and a TOF of 47 h−1. In acetonitrile, with N-methyl-imidazole as a base, the system is also active but with lower efficiency (photocurrent density ∼100 μA cm−2, faradaic efficiency 76%, TON 13, TOF 24 h−1). The reduced efficiency in acetonitrile was primarily due to photocatalyst leaching from the TiO2 electrode, whereas in aqueous conditions, porphyrin degradation was the main factor limiting the catalytic performance. Transient absorption measurements revealed rapid electron injection from the excited Zn–porphyrin into the TiO2 conduction band, forming a porphyrin radical cation. A slower electron transfer followed this from TEMPO to ZnPor+, which partially competed with charge recombination. This observation was consistent with the low driving force (0.1 eV) and the weak electronic coupling between the two components. A comparison between the hyd-ZnP-TEMPO dyad and the bi-component system, “hyd-ZnP + ac-TEMPO”, in acetonitrile showed that both systems offered similar performance. However, the covalent approach displayed easier catalyst recovery and required a smaller amount of catalyst. This photocatalytic system represents one of the most efficient systems ever reported for alcohol oxidation in DSPECs.
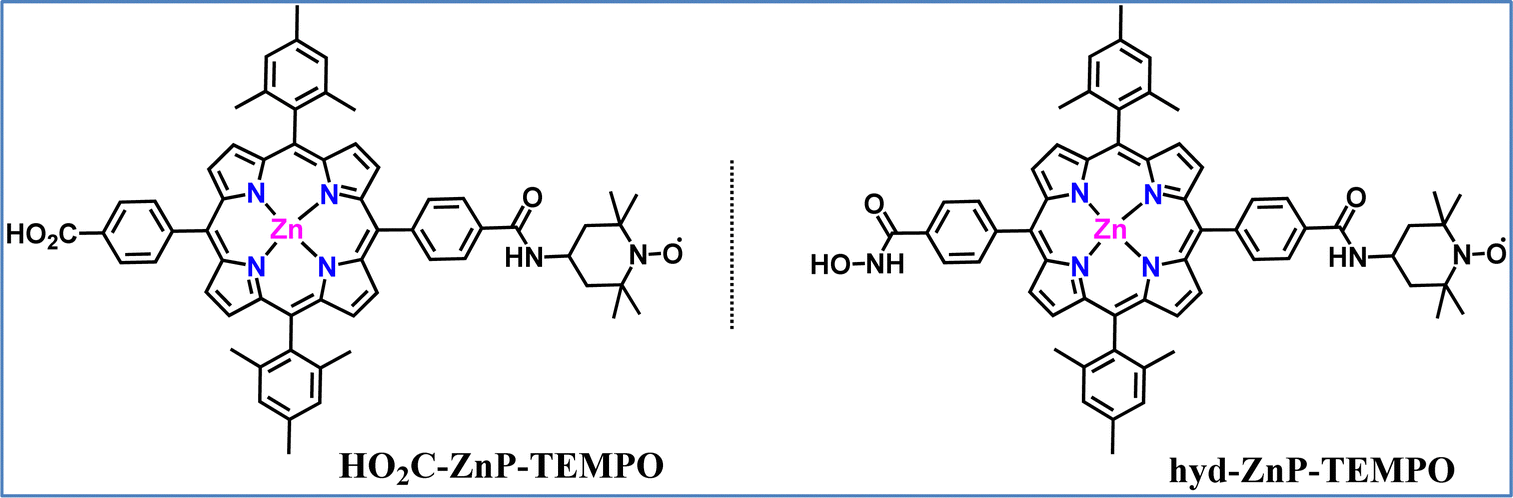 | ||
| Fig. 15 Molecular structures of the two ZnP-TEMPO dyads that were used for the development of a TiO2-based DSPEC. | ||
In our team's most recent work, a dye-sensitized photocatalytic system was reported that enables simultaneous H2 production and selective alcohol oxidation to aldehydes without using a SED.138 DSP is an appealing photocatalytic system due to its simpler preparation process compared to DSPEC, which involves the additional steps of preparing electrodes, setting up wiring, and installing a membrane to separate the anolyte and catholyte. Traditionally, DSP systems are composed of a dye and a hydrogen evolution catalyst (HEC) co-grafted onto n-type semiconductor (n-SC) nanoparticles (NPs), such as TiO2.67 In most cases their activity relies on SEDs like ascorbic acid or triethanolamine to regenerate the oxidized dye, a process that is costly and environmentally unsustainable.70 Here, a diketopyrrolopyrrole (DPP) dye covalently linked to a TEMPO-based catalyst replaces the SED, enabling the conversion of alcohols, such as benzyl alcohol and methoxybenzyl alcohol, to aldehydes while producing H2 (Fig. 16). Under visible-light irradiation in the presence of platinum NPs as the HEC on TiO2 nanoparticles, a TON of up to 50 for H2 evolution was achieved along with the simultaneous conversion of alcohols into high-value aldehyde products. Reaction conditions such as pH, dye loading, and Pt concentration were optimized to maximize both H2 and aldehyde production. The highest rate of H2 evolution (200 μmol h−1 g−1 of TiO2) was observed at pH = 8, with 24 nmol of DPP-TEMPO per mg of TiO2 and with 3.2 nmol of Pt0 per mg of TiO2. Furthermore, testing under sunlight simulation confirmed that the DSP system produces H2 with a slightly lower TON of 27, verifying its functionality under real-world light conditions. Stability studies demonstrated that H2 evolution ceases after approximately 4 hours of irradiation, primarily due to the photodecomposition of the DPP photosensitizer. In this setup, H2 gas accumulates in the reaction headspace, while aldehydes remain in solution, facilitating product separation and enhancing the system's practicality for scalable applications. This approach has promising implications for sustainable photocatalytic systems, potentially extending to CO2 or N2 reduction, broadening the scope for DSPs without SEDs, and emphasizing renewable fuel and value-added product generation.
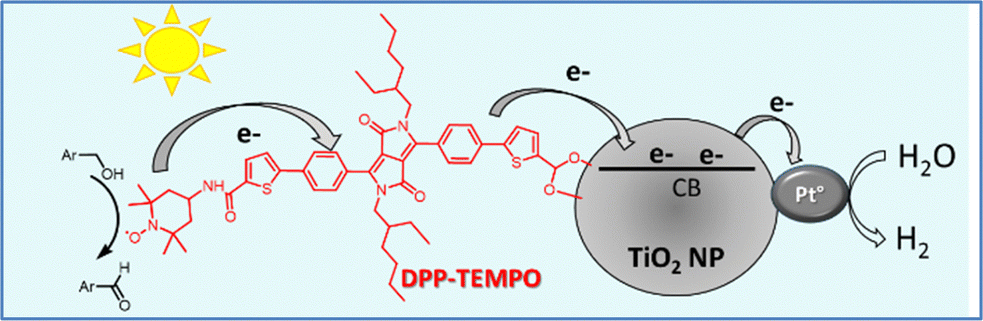 | ||
| Fig. 16 Schematic representation of the dual-functional photocatalytic system based on a DPP dye covalently linked to the TEMPO catalyst. Reproduced from ref. 138 with permission from John Wiley and Sons, copyright 2024. | ||
6. Outlook and perspectives
Light-driven biomimetic catalysis is one of the most widely investigated concepts of artificial photosynthesis, due to its importance and resemblance to natural photosynthetic systems in generating and storing solar fuels. One of the main advantages of such systems, particularly water splitting and carbon dioxide reduction, relies on their versatility towards storing solar energy through fuel forming reactions with high value. A sustainable strategy that facilitates the transition to a “low-carbon” economy is the solar-driven photocatalytic transformation of carbon dioxide into useful and valuable chemicals and fuels. Additionally, though the reductive half reaction of artificial water splitting, H2 is produced in a clean way and then gets stored for further use; hence facilitating both the exploitation and generation of green energy.On the other hand, the oxidative half-reaction of water splitting, although an appealing process, is characterized by high energetic demands. Also, its main product (O2) has very low commercial value and a high risk of generating harmful reactive species. Therefore, alternative oxidation reactions are commonly targeted for the anodic part of such artificial assemblies that can assist the reductive half-reaction while simultaneously generating value-added products. The most appealing approach is the photocatalytic oxidation of alcohols as they produce high-valued carbonyl compounds and serve as a sustainable environment remediation strategy.
In this feature article we have highlighted the unique advantages of employing metalloporphyrins, both as photosensitizers and as catalysts, in developing sustainable and efficient biomimetic photocatalytic systems capable of producing solar fuels as well as offering solutions to environmental issues. By analyzing recent reports, we were able to display important advantages of metalloporphyrins, such as structural design, light-harvesting capability and charge separation efficiency, while also displaying limitations of porphyrin-based artificial photosynthetic assemblies. Notably, for each targeted catalytic process a few distinct strategies are discussed, emphasising some of the most important advancements. Our conceptual approach along with the results of our experimental work allowed us to understand important energetic requirements of each process. Moreover, optimal structural modifications of metalloporphyrins as well as the components and substrates, needed to construct highly efficient artificial photosynthetic systems, are identified. Specifically, our work on H2 production systems reveals that the most promising and efficient approach relies on heterogeneous photocatalysts. Such systems are formed by the adsorption of metalloporphyrins and/or co-catalysts onto semiconducting nanoparticles. Additionally, our targeted carbon dioxide reduction arrays demonstrated that structural fine-tuning leads to highly efficient Fe–porphyrin photocatalysts with favorable redox potentials and enhanced CO2 coordination. However, the need for a sacrificial agent in the artificial photosynthetic assemblies described herein constitutes one of their main drawbacks. As a result, the most auspicious approach concerns the development of systems that combine efficient reductive photocatalysts that produce solar fuels, with oxidative co-catalysts that facilitate the simultaneous production of valuable chemicals and eliminate the use of detrimental sacrificial agents.
The first choice for such systems for the oxidative half-reaction would be O2 evolving catalysts. Even so, as shown in this review, metalloporphyrins can act as water oxidation catalysts, their low stability in the harsh conditions of OER systems, as well as their use in combination with noble metal-based complexes are important limitations. What is more, the product of this catalytic process has very low commercial value. For this reason, we highlight an alternative oxidative process – the solar-driven oxidation of alcohols. This reaction yields high-valued chemicals while providing the necessary electrons and protons for the reduction reaction. Therefore, we anticipate that the present article will aid in designing metalloporphyrin-based hybrid systems that combine components for the two half-reactions. Despite the promising efficiencies demonstrated in laboratory settings, translating metalloporphyrin-based photocatalytic systems into scalable and industrially viable technologies requires addressing a few challenges: material cost, catalyst regeneration, and integration into existing energy infrastructure. Emerging trends such as machine learning for catalyst optimization, tandem photocatalytic systems combining multiple reaction pathways, and hybrid organic–inorganic assemblies offer exciting avenues to enhance the performance and applicability of metalloporphyrin-based systems, paving the way for next-generation artificial photosynthesis technologies. Such biomimetic systems offer opportunities to serve as a more effective strategy for generating solar fuels and value-added chemicals in an environmentally friendly and sustainable manner.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Emmanouil Nikoloudakis gratefully acknowledges the Bodossaki Foundation for financial support. Dr. Vasilis Nikolaou acknowledges the support of the HORIZON TMA MSCA Postdoctoral Fellowships – European Fellowships (HORIZON-TMA-MSCA-PF-EF) action program under the MSCA Postdoctoral Fellowships 2021 call (HORIZON-MSCA-2021-PF-01), through grant agreement no. 101064765 (SOLAR-CAT).Notes and references
- J. Min Park, J. H. Lee and W.-D. Jang, Coord. Chem. Rev., 2020, 407, 213157 CrossRef
.
- J. M. Park, K.-I. Hong, H. Lee and W.-D. Jang, Acc. Chem. Res., 2021, 54, 2249–2260 CrossRef CAS PubMed
- Y. Shi, F. Zhang and R. J. Linhardt, Dyes Pigm., 2021, 188, 109136 CrossRef CAS
- S. Chen, J. Wei, X. Ren, K. Song, J. Sun, F. Bai and S. Tian, Molecules, 2023, 28, 4283 CrossRef CAS PubMed
- E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC
- M. K. Panda, K. Ladomenou and A. G. Coutsolelos, Coord. Chem. Rev., 2012, 256, 2601–2627 CrossRef CAS
- Y. Kuramochi, Y. Fujisawa and A. Satake, J. Am. Chem. Soc., 2020, 142, 705–709 CrossRef CAS PubMed
- S. Hiroto, Y. Miyake and H. Shinokubo, Chem. Rev., 2017, 117, 2910–3043 CrossRef CAS PubMed
- M. O. Senge, Chem. Commun., 2011, 47, 1943–1960 RSC
- R. J. P. Williams, Chem. Rev., 1956, 56, 299–328 CrossRef CAS
- M. O. Senge, S. A. MacGowan and J. M. O'Brien, Chem. Commun., 2015, 51, 17031–17063 RSC
- M. O. Senge, N. N. Sergeeva and K. J. Hale, Chem. Soc. Rev., 2021, 50, 4730–4789 RSC
- M. Tahoun, C. T. Gee, V. E. McCoy, P. M. Sander and C. E. Müller, RSC Adv., 2021, 11, 7552–7563 RSC
- V. Nikolaou, E. Nikoloudakis, K. Ladomenou, G. Charalambidis and A. G. Coutsolelos, Front. Chem. Biol., 2024, 2, 1346465 CrossRef
- A. Khaledian and S. Zakavi, Inorg. Chem. Commun., 2024, 167, 112727 CrossRef CAS
- A. G. Mojarrad and S. Zakavi, Catal. Sci. Technol., 2018, 8, 768–781 RSC
-
K. Rybicka-Jasińska and D. Gryko, in Photochemistry, ed. S. Crespi and S. Protti, The Royal Society of Chemistry, 2021, vol. 49, pp. 457–478 Search PubMed
- R. Das, P. Kumar Verma and C. M. Nagaraja, Coord. Chem. Rev., 2024, 514, 215944 CrossRef CAS
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed
- M. Đokić and H. S. Soo, Chem. Commun., 2018, 54, 6554–6572 RSC
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC
- K. Ladomenou, M. Natali, E. Iengo, G. Charalampidis, F. Scandola and A. G. Coutsolelos, Coord. Chem. Rev., 2015, 304, 38–54 CrossRef
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed
- M. D. Kärkäs, E. V. Johnston, O. Verho and B. Åkermark, Acc. Chem. Res., 2014, 47, 100–111 CrossRef PubMed
- C. R. Lhermitte and K. Sivula, ACS Catal., 2019, 9, 2007–2017 CrossRef CAS
- L. I. Granone, F. Sieland, N. Zheng, R. Dillert and D. W. Bahnemann, Green Chem., 2018, 20, 1169–1192 RSC
- J. S. O'Neill, L. Kearney, M. P. Brandon and M. T. Pryce, Coord. Chem. Rev., 2022, 467, 214599 CrossRef
- N. Chanda, D. Koteshwar, S. Gonuguntla, S. Bojja, U. Pal and L. Giribabu, Mater. Adv., 2021, 2, 4762–4771 RSC
- A. Vidal, F. Adamo, E. Iengo and E. Alessio, Inorg. Chim. Acta, 2021, 516, 120143 CrossRef CAS
- H. Yuan, Y. Yu, S. Yang, Q. Lei, Z. Yang, B. Lan and Z. Han, Chem. Commun., 2024, 60, 6292–6295 RSC
- T. Nagasawa, S. I. Allakhverdiev, Y. Kimura and T. Nagata, Photochem. Photobiol. Sci., 2009, 8, 174–180 CrossRef CAS PubMed
- A. Charisiadis, E. Giannoudis, Z. Pournara, A. Kosma, V. Nikolaou, G. Charalambidis, V. Artero, M. Chavarot-Kerlidou and A. G. Coutsolelos, Eur. J. Inorg. Chem., 2021, 1122–1129 CrossRef CAS
- G. Di Carlo, C. Albanese, A. Molinari, S. Carli, R. Argazzi, A. Minguzzi, F. Tessore, E. Marchini and S. Caramori, ACS Appl. Mater. Interfaces, 2024, 16, 14864–14882 CrossRef CAS PubMed
- Y. Dong, R. Nie, J. Wang, X. Yu, P. Tu, J. Chen and H. Jing, Chin. J. Catal., 2019, 40, 1222–1230 CrossRef CAS
- M. Yamamoto, L. Wang, F. Li, T. Fukushima, K. Tanaka, L. Sun and H. Imahori, Chem. Sci., 2016, 7, 1430–1439 RSC
- F. D’Souza and O. Ito, Chem. Commun., 2009, 4913–4928 RSC
- V. Nikolaou, A. Charisiadis, C. Stangel, G. Charalambidis and A. G. Coutsolelos, C, 2019, 5(3), 57 CAS
- P. Gotico, Z. Halime and A. Aukauloo, Dalton Trans., 2020, 49, 2381–2396 RSC
- T. Nakazono, A. R. Parent and K. Sakai, Chem. Commun., 2013, 49, 6325–6327 RSC
- E. Nikoloudakis, A. Z. Alsaleh, G. Charalambidis, A. G. Coutsolelos and F. D'Souza, Chem. Commun., 2022, 58, 12078–12081 RSC
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed
- T. Keijer, T. Bouwens, J. Hessels and J. N. H. Reek, Chem. Sci., 2021, 12, 50–70 RSC
- R. J. Cogdell, T. H. P. Brotosudarmo, A. T. Gardiner, P. M. Sanchez and L. Cronin, Biofuels, 2010, 1, 861–876 CrossRef CAS
- J. Barber and P. D. Tran, J. R.
Soc., Interface, 2013, 10, 20120984 CrossRef PubMed
- I. Holmes-Gentle, S. Tembhurne, C. Suter and S. Haussener, Nat. Energy, 2023, 8, 586–596 CrossRef CAS
- K. Ladomenou, M. Natali, E. Iengo, G. Charalampidis, F. Scandola and A. G. Coutsolelos, Coord. Chem. Rev., 2015, 304–305, 38–54 CrossRef CAS
- L. Wang, H. Fan and F. Bai, MRS Bull., 2020, 45, 49–56 CrossRef
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Coord. Chem. Rev., 2015, 304–305, 3–19 CrossRef CAS PubMed
- E. Nikoloudakis, A. G. Coutsolelos and E. Stratakis, Energy Fuels, 2024, 38, 19222–19235 CrossRef CAS PubMed
- A. Sartorel, M. Bonchio, S. Campagna and F. Scandola, Chem. Soc. Rev., 2013, 42, 2262–2280 RSC
- P. D. Tran, V. Artero and M. Fontecave, Energy Environ. Sci., 2010, 3, 727–747 RSC
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC
- N. Coutard, N. Kaeffer and V. Artero, Chem. Commun., 2016, 52, 13728–13748 RSC
- T. Lazarides, M. Delor, I. V. Sazanovich, T. M. McCormick, I. Georgakaki, G. Charalambidis, J. A. Weinstein and A. G. Coutsolelos, Chem. Commun., 2014, 50, 521–523 RSC
- A. Panagiotopoulos, K. Ladomenou, D. Sun, V. Artero and A. G. Coutsolelos, Dalton Trans., 2016, 45, 6732–6738 RSC
- M.-Y. Yeh, T.-Y. Tseng, H.-C. Hsieh, B.-X. Wu, Y.-S. Liao, Y.-C. Yeh and J.-C. Liu, ChemPhotoChem, 2020, 4, 481–486 CrossRef CAS
- Y. Liu, H. Wang, S. Li, C. Chen, L. Xu, P. Huang, F. Liu, Y. Su, M. Qi, C. Yu and Y. Zhou, Nat. Commun., 2020, 11, 1724 CrossRef CAS PubMed
- M. Kownacki, S. M. Langenegger, S.-X. Liu and R. Häner, Angew. Chem., Int. Ed., 2019, 58, 751–755 CrossRef CAS PubMed
- E. Nikoloudakis, K. Karikis, J. Han, C. Kokotidou, A. Charisiadis, F. Folias, A. M. Douvas, A. Mitraki, G. Charalambidis, X. Yan and A. G. Coutsolelos, Nanoscale, 2019, 11, 3557–3566 RSC
- V. Nikolaou, G. Charalambidis and A. G. Coutsolelos, Chem. Commun., 2021, 57, 4055–4058 RSC
- R. Cao, G. Wang, X. Ren, P.-C. Duan, L. Wang, Y. Li, X. Chen, R. Zhu, Y. Jia and F. Bai, Nano Lett., 2022, 22, 157–163 CrossRef CAS PubMed
- E. Orfanos, K. Ladomenou, P. Angaridis and A. G. Coutsolelos, Dalton Trans., 2022, 51, 8009–8014 RSC
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2016, 45, 9–23 RSC
- S. Rajak, N.-N. Vu, P. Kaur, A. Duong and P. Nguyen-Tri, Coord. Chem. Rev., 2022, 456, 214375 CrossRef CAS
- L. Zani, M. Melchionna, T. Montini and P. Fornasiero, JPhys Energy, 2021, 3, 031001 CrossRef CAS
- J.-F. Huang, Y. Lei, T. Luo and J.-M. Liu, ChemSusChem, 2020, 13, 5863–5895 CrossRef CAS PubMed
- V. Nikolaou, G. Charalambidis, K. Ladomenou, E. Nikoloudakis, C. Drivas, I. Vamvasakis, S. Panagiotakis, G. Landrou, E. Agapaki, C. Stangel, C. Henkel, J. Joseph, G. Armatas, M. Vasilopoulou, S. Kennou, D. M. Guldi and A. G. Coutsolelos, ChemSusChem, 2021, 14, 961–970 CrossRef CAS PubMed
- V. Nikolaou, E. Agapaki, E. Nikoloudakis, K. Achilleos, K. Ladomenou, G. Charalambidis, E. Triantafyllou and A. G. Coutsolelos, Chem. Commun., 2023, 59, 11256–11259 RSC
- J. Du, D. Xiang, K. Zhou, L. Wang, J. Yu, H. Xia, L. Zhao, H. Liu and W. Zhou, Nano Energy, 2022, 104, 107875 CrossRef CAS
- T. R. Knutson, R. Zhang and L. W. Horowitz, Nat. Commun., 2016, 7, 13676 CrossRef CAS PubMed
- M. Gao, T. Zhang and G. W. Ho, Nano Res., 2022, 15, 9985–10005 CrossRef CAS
- M. Robert, ACS Energy Lett., 2016, 1, 281–282 CrossRef CAS
- C. Fu, Z. Wan, X. Yang, J. Zhang and Z. Zhang, J. Mater. Chem. A, 2024, 12, 28618–28657 RSC
- S. R. Lingampalli, M. M. Ayyub and C. N. R. Rao, ACS Omega, 2017, 2, 2740–2748 CrossRef CAS PubMed
- E. O. Eren and S. Özkar, J. Power Sources, 2021, 506, 230215 CrossRef CAS
- E. Boutin, L. Merakeb, B. Ma, B. Boudy, M. Wang, J. Bonin, E. Anxolabéhère-Mallart and M. Robert, Chem. Soc. Rev., 2020, 49, 5772–5809 RSC
- A. R. Groves, Nat. Synth., 2024, 3, 556 CrossRef CAS
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Phys. Chem. C, 2016, 120, 28951–28960 CrossRef CAS
- B. A. Johnson, S. Maji, H. Agarwala, T. A. White, E. Mijangos and S. Ott, Angew. Chem., Int. Ed., 2016, 55, 1825–1829 CrossRef CAS PubMed
- S. Gonell, J. Lloret-Fillol and A. J. M. Miller, ACS Catal., 2021, 11, 615–626 CrossRef CAS
- W. Nie, D. E. Tarnopol and C. C. L. McCrory, J. Am. Chem. Soc., 2021, 143, 3764–3778 CrossRef CAS PubMed
- C. Costentin, S. Drouet, M. Robert and J. M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed
- P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2019, 58, 4504–4509 CrossRef CAS PubMed
- P. Gotico, L. Roupnel, R. Guillot, M. Sircoglou, W. Leibl, Z. Halime and A. Aukauloo, Angew. Chem., Int. Ed., 2020, 59, 22451–22455 CrossRef CAS PubMed
- S. Amanullah, P. Gotico, M. Sircoglou, W. Leibl, M. J. Llansola-Portoles, T. Tibiletti, A. Quaranta, Z. Halime and A. Aukauloo, Angew. Chem., Int. Ed., 2024, 63, e202314439 CrossRef CAS PubMed
- M. R. Narouz, P. De La Torre, L. An and C. J. Chang, Angew. Chem., Int. Ed., 2022, 61, e202207666 CrossRef CAS PubMed
- T. Adelais, G. Philipp, H. Christian, H.-T. Minh-Huong, P. Thomas, L. Winfried, C. Georgios, C. Athanassios, H. Zakaria and A. Ally, C. R. Chim, 2021, 24, 101–114 Search PubMed
- A. Stoumpidi, A. Trapali, M. Poisson, A. Barrozo, S. Bertaina, M. Orio, G. Charalambidis and A. G. Coutsolelos, ChemCatChem, 2023, 15, e202200856 CrossRef CAS
- S. Dash, K. Arjun Singh, S. Jose, D. Vincent Herald Wilson, D. Elangovan, S. K. Surapraraju and S. K. Natarajan, Int. J. Hydrogen Energy, 2024, 83, 614–629 CrossRef CAS
- Z. A. K. Khattak, H. A. Younus, N. Ahmad, M. Alomar, H. Ullah, M. Al-Abri, R. Al-Hajri, C.-M. Kao and F. Verpoort, Int. J. Hydrogen Energy, 2024, 76, 141–151 CrossRef CAS
-
S. Ye, C. Ding and C. Li, in Advances in Inorganic Chemistry, ed. R. van Eldik and C. D. Hubbard, Academic Press, 2019, vol. 74, pp. 3–59 Search PubMed
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418–423 CrossRef CAS PubMed
- F. Creazzo and S. Luber, Chem. – Eur. J., 2021, 27, 17024–17037 CrossRef CAS PubMed
- Y. Xie, F. Luo and Z. Yang, Energy Rev., 2024, 3, 100103 CrossRef
- T. Liu, L. Fan, H. Yang, L. Wang, X. Fan and L. Sun, Artif. Photosynth., 2024, 1, 14–26 CrossRef
- A. K. Singh and L. Roy, ACS Omega, 2024, 9, 9886–9920 CrossRef CAS PubMed
- C. Liu, D. van den Bos, B. den Hartog, D. van der Meij, A. Ramakrishnan and S. Bonnet, Angew. Chem., Int. Ed., 2021, 60, 13463–13469 CrossRef CAS PubMed
- X. Wu, F. Li, B. Zhang and L. Sun, J. Photochem. Photobiol., C, 2015, 25, 71–89 CrossRef CAS
- J. Meng, P. Bi, J. Jia, X. Sun and R. Chen, ChemistrySelect, 2017, 2, 4882–4888 CrossRef CAS
- S. Fukuzumi, T. Kojima, Y.-M. Lee and W. Nam, Coord. Chem. Rev., 2017, 333, 44–56 CrossRef CAS
- W. Lai, R. Cao, G. Dong, S. Shaik, J. Yao and H. Chen, J. Phys. Chem. Lett., 2012, 3, 2315–2319 CrossRef CAS PubMed
- Y. Naruta, M.-A. Sasayama and T. Sasaki, Angew. Chem., Int. Ed. Engl., 1994, 33, 1839–1841 CrossRef
- B. Yao, Y. He, S. Wang, H. Sun and X. Liu, Int. J. Mol. Sci., 2022, 23, 6036 CrossRef CAS PubMed
- J. Rosenthal and D. G. Nocera, Acc. Chem. Res., 2007, 40, 543–553 CrossRef CAS PubMed
- C. J. Gagliardi, A. K. Vannucci, J. J. Concepcion, Z. Chen and T. J. Meyer, Energy Environ. Sci., 2012, 5, 7704–7717 RSC
- G. Paille, M. Gomez-Mingot, C. Roch-Marchal, B. Lassalle-Kaiser, P. Mialane, M. Fontecave, C. Mellot-Draznieks and A. Dolbecq, J. Am. Chem. Soc., 2018, 140, 3613–3618 CrossRef CAS PubMed
- E. Zhou, X. Zhang, L. Zhu, E. Chai, J. Chen, J. Li, D. Yuan, L. Kang, Q. Sun and Y. Wang, Sci. Adv., 2024, 10, eadk8564 CrossRef CAS PubMed
- Y. Han, Y. Wu, W. Lai and R. Cao, Inorg. Chem., 2015, 54, 5604–5613 CrossRef CAS PubMed
- D. Wang and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15579–15584 CrossRef CAS PubMed
- Q. Bu, X. Liu, Q. Zhao, G. Lu, X. Zhu, Q. Liu and T. Xie, J. Colloid Interface Sci., 2022, 626, 345–354 CrossRef CAS PubMed
- Z. Wang, J. Fan, B. Cheng, J. Yu and J. Xu, Mater. Today Phys., 2020, 15, 100279 CrossRef
- B. Zhang and L. Sun, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS PubMed
- X. Liang, X. Cao, W. Sun and Y. Ding, ChemCatChem, 2019, 11, 6190–6202 CrossRef CAS
- H. Liu, S. He, J. Qu, Y. Cai, X. Yang, C. M. Li and J. Hu, Chin. J. Catal., 2024, 65, 1–39 CrossRef CAS
- B. You, G. Han and Y. Sun, Chem. Commun., 2018, 54, 5943–5955 RSC
- X. Bao, M. Liu, Z. Wang, D. Dai, P. Wang, H. Cheng, Y. Liu, Z. Zheng, Y. Dai and B. Huang, ACS Catal., 2022, 12, 1919–1929 CrossRef CAS
- Y. Wu, X. Chen, J. Cao, Y. Zhu, W. Yuan, Z. Hu, Z. Ao, G. W. Brudvig, F. Tian, J. C. Yu and C. Li, Appl. Catal., B, 2022, 303, 120878 CrossRef CAS
- T. Uekert, H. Kasap and E. Reisner, J. Am. Chem. Soc., 2019, 141, 15201–15210 CrossRef CAS PubMed
- D. Tang, G. Lu, Z. Shen, Y. Hu, L. Yao, B. Li, G. Zhao, B. Peng and X. Huang, J. Energy Chem., 2023, 77, 80–118 CrossRef CAS
- M.-Y. Qi, M. Conte, M. Anpo, Z.-R. Tang and Y.-J. Xu, Chem. Rev., 2021, 121, 13051–13085 CrossRef CAS PubMed
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed
- S. Kampouri and K. C. Stylianou, ACS Catal., 2019, 9, 4247–4270 CrossRef CAS
- Q. Chen, Y. Liu, B. Mao, Z. Wu, W. Yan, D. Zhang, Q. Li, H. Huang, Z. Kang and W. Shi, Adv. Funct. Mater., 2023, 33, 2305318 CrossRef CAS
- F. Li, Y. Wang, J. Du, Y. Zhu, C. Xu and L. Sun, Appl. Catal., B, 2018, 225, 258–263 CrossRef CAS
- H. Kasap, C. A. Caputo, B. C. M. Martindale, R. Godin, V. W.-H. Lau, B. V. Lotsch, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2016, 138, 9183–9192 CrossRef CAS PubMed
- P. B. Pati, M. Abdellah, S. Diring, L. Hammarström and F. Odobel, ChemSusChem, 2021, 14, 2902–2913 CrossRef CAS PubMed
- D. F. Bruggeman, A. A. H. Laporte, R. J. Detz, S. Mathew and J. N. H. Reek, Angew. Chem., Int. Ed., 2022, 61, e202200175 CrossRef CAS PubMed
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed
- F. Huang, F. Zhang, Y. Wang and X. Lang, Trends Chem., 2024, 6, 115–127 CrossRef CAS
- R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2010, 14, 245–251 CrossRef CAS
- E. Nikoloudakis, P. B. Pati, G. Charalambidis, D. S. Budkina, S. Diring, A. Planchat, D. Jacquemin, E. Vauthey, A. G. Coutsolelos and F. Odobel, ACS Catal., 2021, 11, 12075–12086 CrossRef CAS
- D. Romito, C. Govind, V. Nikolaou, R. J. Fernández-Terán, A. Stoumpidi, E. Agapaki, G. Charalambidis, S. Diring, E. Vauthey, A. G. Coutsolelos and F. Odobel, Angew. Chem., Int. Ed., 2024, 63, e202318868 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |