
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isolating the ring-redox states of boron sub-phthalocyanines†
Wen
Zhou
a,
Declan
McKearney
a,
Yusuke
Okada
b,
Nagao
Kobayashi
 *b and
Daniel B.
Leznoff
*a
*b and
Daniel B.
Leznoff
*a
aDepartment of Chemistry, Simon Fraser University, 8888 University Drive, Burnaby, BC V5A 1S6, Canada. E-mail: dleznoff@sfu.ca
bFaculty of Textile Science and Technology, Shinshu University, Ueda 386-8567, Japan. E-mail: nagaok@shinshu-u.ac.jp
First published on 10th February 2025
Abstract
Ring-reduced and ring-oxidized boron subphthalocyanine complexes have been isolated and structurally characterized. The doubly reduced species forms a trinuclear cluster with intramolecular B–Nmeso bonds, while the mono-oxidized system shows minimal bond localization. UV-visible absorption spectra and accompanying TD-DFT calculations are indicative of the ring-oxidation state.
Containing three isoindole moieties, boron subphthalocyanines (subPcBX) were first synthesized and structurally characterized in the early 1970s.1,2 They are comprised of a 14π electron conical macrocyclic ring and, similar to their phthalocyanine cousins, they exhibit two intense electronic spectral features (at approx. 320 nm and 570 nm) which give them a strong purple colour; they are also often highly fluorescent.3subPcBX (where X is an axial ligand, commonly a halide or oxygen-donor) materials have been the subject of great interest due to their potential applications in organic photovoltaic cells, organic light emitting diodes and field effect transistors in particular, due to a combination of their absorption and ring-based redox properties.4–6 They are also used as a precursor for synthesizing A3B unsymmetric phthalocyanines by ring expansion with a diiminoisoindoline.7,8
Many of these applications in materials science harness the aforementioned redox-activity of the subphthalocyanine (subPc) ring. From the standard subPc(2-), both ring-oxidation to subPc(1-) and ring-reductions to subPc(3-) and subPc(4-) have been accessed (as is the case for phthalocyanines),9 and these key states have been extensively studied in solution.6,10,11 However, to our knowledge, no ring-oxidized or reduced subPcBX complexes have been isolated or structurally characterized, in part due to their air- and moisture-sensitivity (especially on the reduction side). Given the importance of these ring-redox states for a wide range of materials properties, we hereby report the isolation, structural characterization and analysis of ring-oxidized and ring-reduced subPcBX materials.
Ring-reduced K sub PcB trimer (1): electrochemical data (CV) has been previously reported for subPcBCl in CH2Cl2, with the first and second ring reductions identified at −0.99 V and −1.48 V, while ring oxidation occurred at 1.14 V (vs. SCE).11 These reduction couples are more negative than a range of early transition-metallophthalocyanines (PcM; M = Cr, Zr)12,13 and can be compared with reduction and oxidation potentials of −0.9, −1.2 and 0.78 V respectively for PcZn.14 This augmentation of the ring-redox potentials can be attributed to the relative stabilization of the HOMO (a2) in the subPcBCl complex when compared to PcM systems, as shown in the previously reported MO diagram for subPcBCl, where the HOMO–LUMO gap is larger than typical metallophthalocyanines, but the LUMO (e.g.) level of subPcBCl is nearly the same as in PcM.15 To access ring-reduced states of subPcB chemically, the strong reducing agent KC8 was reacted with subPcBCl in THF. However, targeting the mono-ring reduced species subPc(3-) by addition of one equiv. of KC8 instead generates a mixture of di-reduced subPc(4-), mono-reduced subPc(3-) and insoluble unreacted species due to the insolubility of both KC8 and subPcBCl; similar mixtures are obtained using one equivalent of soluble KEt3BH as a reducing agent.10,13 On the other hand, using two or more equivalents of KC8 (Scheme 1) or KEt3BH generated a purple solution, the UV-visible absorption spectrum of which featured a Q-band at λmax = 460 nm and an additional broad peak at 750 nm. Extremely air and moisture-sensitive purple crystals of 1 were isolated from this reaction with KC8 and recrystallization from DME.
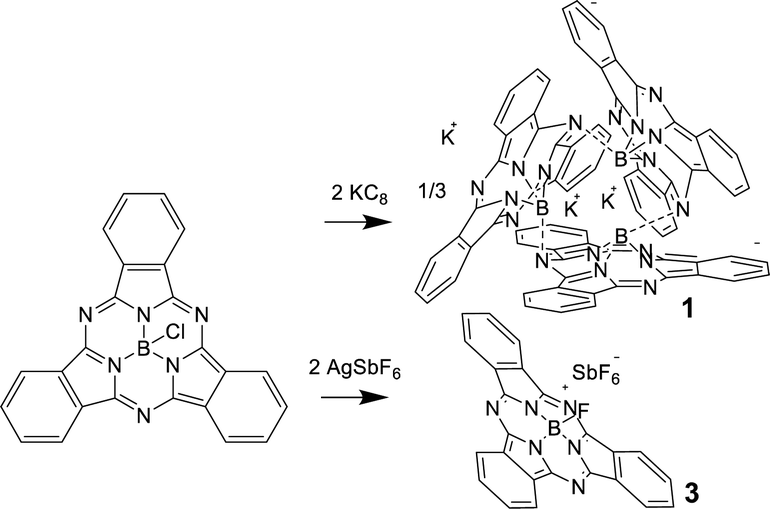 | ||
| Scheme 1 Synthetic route to (K+[subPcB−])3 (1) and [subPcBF]SbF6 (3). | ||
The single-crystal structure of 1 revealed a cyclic trimer (Fig. 1) of the form (K+[subPcB−])3 (1) where each subPc(2-) unit has been doubly ring-reduced to subPc(4-). One equivalent of KC8 is oxidized to K+, which presumably abstracts the axial chloride from the reduced subPcBCl and eliminates KCl, generating a subPc(3-) ring; the second equivalent of KC8 delivers the second electron to the ring to make [subPc(4-)B]− and the resulting K+ cation charge-balances this anion (chloride abstraction could also occur from the di-reduced species instead; the detailed order of reduction and abstraction steps was not investigated). The resulting open site on boron in [subPc(4-)B]− is filled by the intermolecular Lewis acid–base axial coordination of a meso-ring nitrogen from an adjacent molecule (Fig. 1); B–N bond distances are 1.48(3)–1.58(3) Å (within one subPcB unit) and 1.49(3)–1.54(3) Å (for the intermolecular B–N interactions). Perhaps due to this inter-unit B–N bond formation, the boron centre sits farther out of the non-planar subPc ligand, with a distance of 0.580–0.673 Å, compared to typical subPcBX structures of 0.55 Å.4,16 One of the potassium cations is solvated by DME, and the other two potassium cations are bonded to each subPcB benzo-moiety; this interaction impacts the B–N bonds immediately around the binding site.
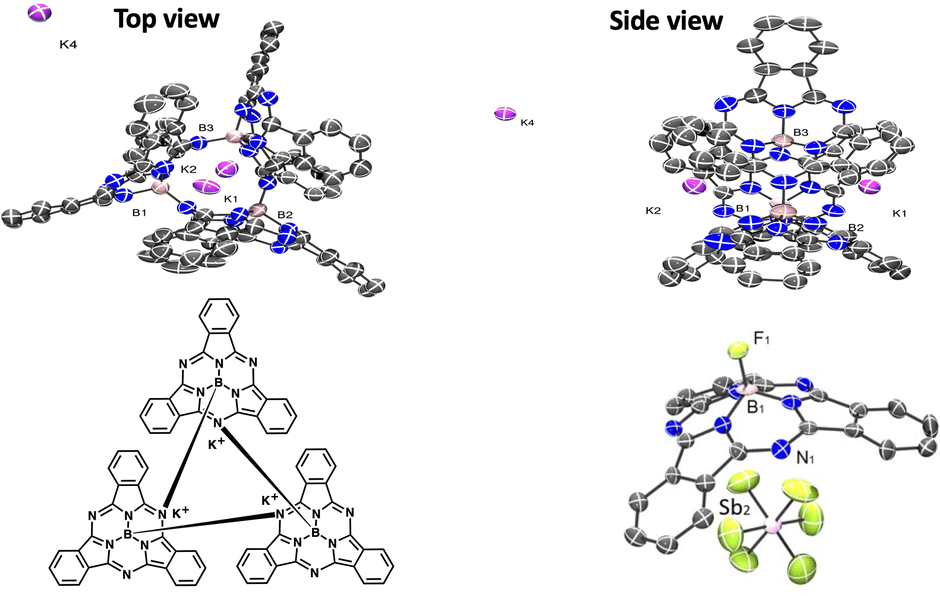 | ||
| Fig. 1 Two views of the molecular structure (50% ellipsoids) of 1 (top), Chemdraw depiction of the trinuclear ring (bottom left). Bottom right: Solid-state structure of 4 with selected bond lengths [Å]: B-F 1.399(8) and B-N 1.463(9) to 1.493(8). Colour scheme: B: light brown, K: purple, Sb: pink, F: green, N: blue, and C: gray. All H atoms, DME molecules and co-crystallized Ag(DME)3SbF6 (in 4) are omitted for clarity. | ||
Consistent with a doubly reduced 16-electron subPc(4-) ring, the C–C bond lengths in some of the benzo-moieties were localized, with a significant difference of 0.1–0.15 Å (Fig. 2, left), which is similar to that observed in related Pc(4-) structures;17 the C–N bonds with the meso-nitrogen atoms are also localized, and lengthened further by the meso-nitrogen (Nmeso) coordination to the adjacent boron centre (1.423 and 1.382 Å compared to 1.366/1.307 and 1.370/1.360 Å for the unbound C–Nmeso units; see Fig. 2, left). This represents the first example of the isolation and structural characterization of a ring-reduced subPc-based system.
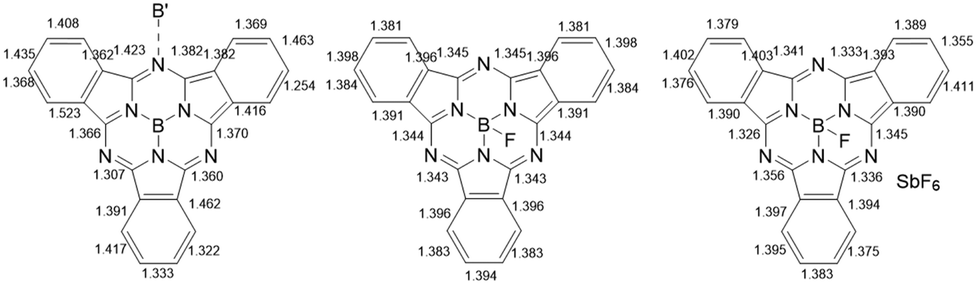 | ||
| Fig. 2 From left to right, metrical parameters for doubly ring-reduced subPc(4-) in (K+[subPcB−])3 (1), standard subPc(2-) in subPcBF (2) and ring-oxidized subPc(1-) in [subPcBF][SbF6]·Ag(DME)3SbF6 (4). | ||
Ring-oxidized [ sub PcBF] cations: for the oxidation, AgSbF6 is a convenient reagent (1.4 V vs. SCE) to access the desired ring-oxidized species.18 However, AgSbF6 can also react via a salt metathesis reaction pathway and indeed, reaction of subPcBCl with one equiv. of AgSbF6 as a slurry in toluene generated the previously reported subPcBF (2), presumably by loss of AgCl and subsequent fluoride-abstraction by the resulting [subPcB]+ species from the normally inert SbF6− anion. Reaction in THF generates polymeric THF-based species and thus is not clean. This is consistent with our reports for a ring-oxidized Pc(1-)Zn system where hydrogen-atom transfer led to the polymerization of the solvent.19 Recrystallization from benzene yielded crystals of subPcBF·benzene (Fig. 2, centre), where two concave-shaped subPcBF units encapsulate two benzene molecules via π–π interactions (Fig. S1 and S2, ESI†).
With this alternative reactivity in mind, the addition of two equiv. of AgSbF6 to subPcBCl was successful in generating, via a combination of metathesis and oxidation, the final ring-oxidized product [subPcBF][SbF6]·toluene (3) (Scheme 1 and Fig. S3, ESI†). The UV-visible absorption spectrum of this ring-oxidized product 3 exhibits B and Q bands with λmax = 310, 561 nm and a lower-energy band at 606 nm (Fig. 4), which is absent in the neutral species.
Using a small excess (2.2 equiv.) of AgSbF6 with subPcBCl in toluene generates isolable but poor-quality crystals of [subPcBF][SbF6]·toluene (3); washing with DME also yields a minor product with co-crystallized AgSbF6, namely [subPcBF][SbF6]·Ag(DME)3SbF6 (4) (Fig. 1 and Fig. S4, ESI†). The isolation of the additional salt adduct (in low yield) indicates that excess Ag+ does not access a further oxidation product; this adduct 4 is thus not observed when AgSbF6 is used stoichiometrically. The structures of the [subPcBF]+ cation in 3 and 4 are geometrically similar to each other and also to that of the neutral subPcBF (2), with tetrahedral B(III) centres protruding from the concave subPc ligand 3 (0.593 Å) and 2 (0.614 Å). In ring-oxidized 3, the B–N bond lengths are 1.44(3)–1.47(3) Å, and the axial B–F bond length is 1.43(3) Å, all of which are slightly shorter than in subPcBF·benzene, 2. These data are consistent with previously reported structures containing the subPcBF unit.20 The oxidized ring shows only a low level of localization in the phenyl C–C bond lengths of 1.376(8)–1.411(9) Å as compared with those in neutral subPcBF of a tighter 1.381(3)–1.398(3) Å (Fig. 2); this was similarly observed in a related ring-oxidized zinc phthalocyanine structure, where the localization is much less pronounced than in the di-reduced species.19 To our knowledge, these represent the first structurally characterized ring-oxidized subPcB complexes.
As has been observed and indeed targeted in related subPcB-containing systems,21 the conical subPcB frameworks in 2–4 are also adept at encapsulating and/or recognizing other small-molecule units, which co-crystallize in the concave π-surface of the subPcB molecule or cation: specifically benzene co-crystallizes with a π-interaction distance of 5.427 Å in the structure of subPcBF (2), toluene in [subPcBF][SbF6] (3); (interaction distance = 4.424 Å) and SbF6− in [subPcBF][SbF6]·AgSbF6 (4; interaction distance = 4.945 Å).
The 1H NMR spectrum of di-reduced 1 shows two peaks at δ = 5.24 and 4.27 ppm, significantly upfield from the equivalent protons in subPcBCl at δ = 8.81 and 7.91 ppm. On the other hand, the 1H NMR peaks for ring-oxidized 3 are at a similar δ = 8.67 and 7.40 ppm but are significantly broadened, as expected for this paramagnetic system. An ESR spectrum of [subPcBF][SbF6] (3) in toluene at room temperature showed a single signal at g = 2.001 (Fig. S5, ESI†), also consistent with a ring-based unpaired electron, similar to ring-oxidized [Pc(1-)Zn]+ radical cations.19
Fig. 3 and 4 show the absorption spectra of 1–3 in THF. 1 showed a broad band with intermediate intensity at ca. 650–850 nm and broad peaks at 460 and 304 nm, which resemble those of hitherto reported doubly ring-reduced Pc(4-) complexes.9,13,17 For example, both the isolated and spectrochemically obtained [Pc(4-)Zn]2− species have a broad band of intermediate intensity in THF at ca. 575–725 nm and two intense bands at 522 and 335 nm.22 The absorption bands of 1 are broader than that of [Pc(4-)Zn]2−, but this is acceptable, taking into account that 1 is a cyclic trimer of subPcB, and indeed, its broad peak at ca. 650–850 nm and two peaks at 304 and 460 nm are at longer wavelengths, respectively of the Q (560–570 nm) and B (Soret) bands (ca. 300 nm) of neutral subPc(2-).23,24
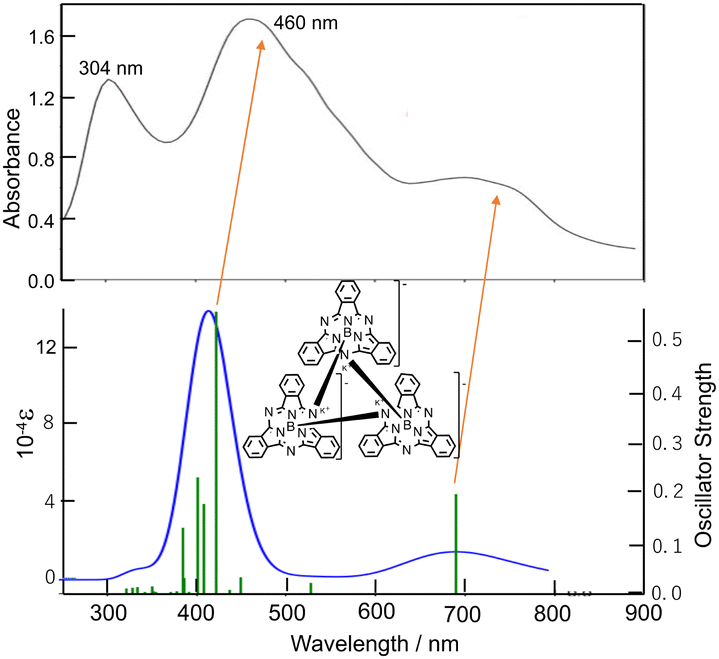 | ||
| Fig. 3 (top) Absorption spectrum of 1 in THF, and (bottom) that of calculated spectrum. Due to the large molecular size, energies in the region above 310 nm were not calculated. DFT and TDDFT calculations were carried out using the CAM-B3LYP functional with 6-31+G(d,p) basis sets. | ||
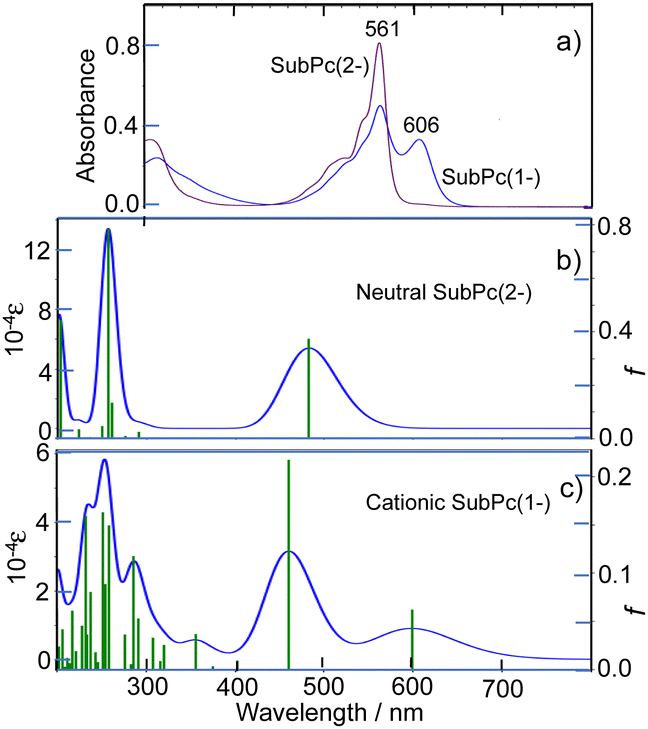 | ||
| Fig. 4 (a) UV-visible absorption spectra of subPcBF (2) (purple line) and [subPcBF][SbF6] (3) (blue line) in THF. Calculated absorption spectra of (b) neutral and (c) mono-cationic subPcB. Note scale difference of f and ε between (b) and (c). DFT and TDDFT calculations were carried out using the CAM-B3LYP functional with 6-31+G(d,p) basis sets. | ||
This spectrum is related to that reported for the mono-reduced [subPcBCl]− species generated spectro-electrochemically (containing a subPc(3-) ring), which has broad, ill-defined bands around 500 and 657 nm;10 no doubly-reduced subPcB species has been reported spectro-electrochemically to our knowledge.
The absorption spectrum of 2 is quite typical of neutral subPc(2-) species,25 while the spectrum of 3, with two intense peaks in the Q-band region, is different than the spectrum of π-cation radicals of PcM complexes, which have a single Q peak.24 Although the process from neutral to cation radical species of subPcs was previously pursued through spectro-electrochemistry, the π-cation radicals obtained in that process all appear to be unstable, since an isosbestic point emerges only at an early stage and after that the absorbance in the whole visible region decreased to almost zero, suggesting decomposition of the products.10 Indeed, it is known that almost all Pcs and tetraazaporphyrins are susceptible to oxidative decomposition and produce phthalimide derivatives as decomposition products, although they are not necessarily decomposed in strong acid.26 The two-peak Q band of 3 in this study is almost identical to the last stage spectrum that appeared maintaining an isosbestic point in the electrochemical oxidation of subPcs.10 Electrolysis in a cell under standard conditions may not be conducive to generating a stable, isolable oxidized subPcB species. However, since the spectrum herein was obtained from a solution prepared from crystals of π-cation radicals, it may be one of the most reliable spectra of π-cation radicals of subPcs.
To further interpret the observed spectra, simulated absorption spectra of 1–3 were generated via TD-DFT calculations. The calculated electronic absorption spectrum for 1 is shown in Fig. 3 (bottom), with calculated data summarized in Table S2 (ESI†), and with energies and shapes of some frontier MOs shown in Fig. S6 and S7 (ESI†). The bands at 460 and ca. 650–850 nm are nicely reproduced. As can be judged from composition and the shape of MOs, the six orbitals from H−2–L+2 participate in the Q transition. Since 1 is composed of three subPcB units, this corresponds to the HOMO–LUMO transition in a putative monomeric subPc(4-)B unit.27 In 1, the higher energy region, unoccupied MOs such as L+5, L+9–11, L+24, L+25, L; 28, L+29, and L+34 are participating in some transitions, some of which (L+5, L+9–11) do not have large coefficients in the core region of each subPc(4-)B unit.
The calculated electronic absorption spectra for 2 and 3 are shown in Fig. 4b and c, with calculated data summarized in Table S4 (ESI†). In addition, the energies and shapes of some key frontier MOs for 2 and 3 are shown in Fig. S11 and S13 (ESI†) and some additional MOs are collected in Fig. S12 and S14 (ESI†) respectively; this confirms that, as observed, only a low level of bond localization should be expected in the oxidized species, since one electron is removed from a symmetric HOMO in 2. Although the calculated energies are slightly overestimated, the calculations are consistent with the experimental spectra. In particular, the split Q band of subPcB(1-) is reproduced, including the relative intensity difference of the split Q peaks. According to the data in Table S4 (ESI†), the Q absorption peaks at longer and shorter wavelengths appear to be associated with alpha and beta spins, respectively.
In conclusion, we have reduced and oxidized subPcBCl and characterized their products structurally, spectroscopically and using MO calculations (Fig. S15, ESI†). X-ray analysis revealed that the two-electron reduced product was a cyclic trimer of subPcB connected via B–Nmeso bonding. Substantial bond localization in the di-reduced species but not significantly in the oxidized species correlate with the MO calculations. The calculated absorption spectra of the three species reproduced the experimental spectra, with a slight overestimation of the energies. To our knowledge, these are the first structurally characterized ring-reduced and ring-oxidized subPcB complexes.
We are grateful to NSERC of Canada for generous research support via a Discovery Grant (DBL) and a PGS-D Scholarship (DM). We also thank Prof. Charles Walsby and Dr Greg MacNeil (SFU) for assistance with collecting the ESR spectrum.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Meller and A. Ossko, Monatsh. Chem., 1972, 103, 150–155 CrossRef CAS.
- H. Kietaibl, Monatsh. Chem., 1974, 105, 405–418 CrossRef CAS.
- C. G. Claessens, et al. , Chem. Rev., 2002, 102, 835–854 CrossRef CAS PubMed; C. G. Claessens, et al. , Chem. Rev., 2014, 114, 2192–2277 CrossRef PubMed; Z. X. Li, et al. , Coord. Chem. Rev., 2023, 493, 215325 CrossRef; T. Fukuda and N. Kobayashi, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific Press, 2010, vol. 9, pp. 1 Search PubMed.
- G. Lavarda, et al. , Chem. Soc. Rev., 2022, 51, 9482–9619 RSC; J. Labella and T. Torres, Trends Chem., 2023, 5, 353–366 CrossRef CAS; R. Zigelstein and T. P. Bender, Mol. Syst. Des. Eng., 2024, 9, 856–874 RSC.
- W. Y. Zhou, et al. , Mater. Adv., 2021, 2, 165–185 RSC.
- G. E. Morse and T. P. Bender, ACS Appl. Mater. Interfaces, 2012, 4, 5055–5068 CrossRef CAS PubMed; D. Gonzalez-Rodrigues, et al. , J. Am. Chem. Soc., 2004, 126, 6301–6313 CrossRef PubMed.
- N. Kobayashi, et al. , J. Am. Chem. Soc., 1990, 112, 9640–9641 CrossRef CAS.
- N. Kobayashi, et al. , J. Am. Chem. Soc., 1999, 121, 9096–9110 CrossRef CAS.
- A. B. P. Lever, J. Porphyrins Phthalocyanines, 1999, 3, 488–499 CrossRef CAS; J. Mack and M. J. Stillman, Coord. Chem. Rev., 2001, 219, 993–1032 CrossRef; T. Nyokong and H. Isago, J. Porphyrins Phthalocyanines, 2004, 8, 1083–1090 CrossRef; J. Mack and N. Kobayashi, Chem. Rev., 2011, 111, 281–321 CrossRef PubMed; M. J. Stillman, in Phthalocyanines: Properties and Applications, ed. C. C. Leznoff and A. B. P. Lever, VCH, New York, 1993, vol. 3, pp. 227 Search PubMed.
- K. Sampson, et al. , J. Phys. Chem. A, 2018, 122, 4414–4424 CrossRef CAS PubMed.
- G. E. Morse, et al. , ACS Appl. Mater. Interfaces, 2011, 3, 3538–3544 CrossRef CAS PubMed; G. E. Morse, et al. , J. Phys. Chem. C, 2011, 115, 11709–11718 CrossRef.
- J. Obirai and T. Nyokong, J. Electroanal. Chem., 2004, 573, 77–85 CAS.
- W. Zhou, et al. , Chem. – Eur. J., 2017, 23, 2323–2331 CrossRef CAS PubMed; W. Zhou, et al. , Dalton Trans., 2015, 44, 13955–13961 RSC.
- A. Giraudeau, et al. , J. Am. Chem. Soc., 1980, 102, 5137–5142 CrossRef CAS; M. L’Her and A. Pondaven, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, 2003, vol. 16, pp. 117 Search PubMed.
- N. Kobayashi, et al. , J. Am. Chem. Soc., 1999, 121, 9096–9110 CrossRef CAS.
- J. D. Virdo, et al. , Acta Crystallogr., Sect. C: Struct. Chem., 2016, 72, 297 CrossRef CAS PubMed.
- J. A. Cissell, et al. , Inorg. Chem., 2007, 46, 7713 CrossRef CAS PubMed; D. V. Konarev, et al. , Chem. Asian J., 2018, 13, 1552–1560 CrossRef PubMed; M. A. Faronov, et al. , Eur. J. Inorg. Chem., 2023, 202300407 CrossRef.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed; K. Sudoh, et al. , Heteroat. Chem., 2018, 29, e21456 CrossRef.
- D. McKearney, et al. , Inorg. Chem., 2018, 57, 9644–9655 CrossRef CAS PubMed.
- M. S. Rodriguez-Morgade, et al. , Chem. – Eur. J., 2008, 14, 1342–1350 CrossRef CAS PubMed; M. V. Fulford, et al. , J. Chem. Eng. Data, 2012, 57, 2756–2765 CrossRef; N. F. Farac, et al. , Cryst. Growth Des., 2024, 24, 9447–9464 CrossRef; C. Zhang, et al. , Angew. Chem., Int. Ed., 2021, 60, 3261–3267 CrossRef PubMed.
- G. Zango, et al. , Chem. Sci., 2020, 11, 3448–3459 RSC; D. V. Konarev, et al. , CrystEngComm, 2015, 17, 3923–3926 RSC; L. A. Burt, et al. , J. Porphyrins Phthalocyanines, 2016, 20, 1034–1040 CrossRef CAS.
- D. W. Clack and J. R. Yandle, Inorg. Chem., 1972, 11, 1738–1742 CrossRef CAS; J. Mack and M. J. Stillman, Inorg. Chem., 1997, 36, 413–425 CrossRef; E. W. Y. Wong, et al. , Inorg. Chem., 2010, 49, 3343–3350 CrossRef PubMed.
- N. Kobayashi, J. Porphyrins Phthalocyanines, 1999, 3, 453–467 CrossRef CAS.
- J. Mack and M. J. Stillman, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, 2003, vol. 16, pp. 43–116 Search PubMed.
- N. Kobayashi, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, 2003, vol. 16, pp. 161–262 Search PubMed.
- A. K. Sobbi, et al. , J. Chem. Soc., Perkin Trans. 2, 1993, 481–488 RSC.
- We have also calculated the optimized structure, MOs and electronic absorption spectrum for such a putative mononuclear [subPcB]−, where the ring-oxidation state is subPc(4-) (Fig. S8–S10 and Table S3, ESI†); this shows a similar absorption spectrum to 1 and similar bond-localization, albeit less pronounced than in the trimer, likely due to the absence of the B–Nmeso bonds.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and experimental details, crystallographic information tables, crystal structure figures for 2 and 3; ESR spectrum of 3; MO energy level diagrams, graphical depictions of selected MOs, details for TD-DFT calculations, including tables of calculated transition energies and oscillator strengths for the components of the Q- and B-absorption bands. CCDC 2368551–2368554 for 1–4 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc06727d |
| This journal is © The Royal Society of Chemistry 2025 |