
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence2-Aminopyridine nucleobases enable DNA invasion by peptide nucleic acid clamps under physiological conditions†
Brandon R. Tessier and
Eriks Rozners *
*
Department of Chemistry, Binghamton University, Binghamton, NY 13902, USA. E-mail: erozners@binghamton.edu
First published on 10th February 2025
Abstract
Peptide nucleic acid (PNA) clamps modified with 2-aminopyridine (M) nucleobase invaded double-stranded DNA under physiological salt conditions. In contrast, PNAs carrying common nucleobases could not fully invade DNA under these conditions. M-modified PNAs may overcome the problematic requirement for low salt concentration, a long-standing DNA invasion problem.
Peptide nucleic acid (PNA) was first reported by Nielsen and co-workers in 1991 as a DNA analog designed for triple-helical recognition of double-stranded DNA (dsDNA).1 Among the various modified oligonucleotide analogs, PNA having the entire sugar-phosphate backbone replaced by achiral pseudopeptide linkages (Fig. 1) is one of the most dramatic departures from the native structure of DNA. The advantages of PNA are: (1) the neutral backbone that eliminates electrostatic repulsion when binding to the negatively charged dsDNA; (2) high chemical and enzymatic stability of the pseudopeptide backbone; and (3) simple synthesis of the achiral amide backbone of monomers and oligomers. Because of high binding affinity and sequence specificity for single-stranded DNA and RNA, PNAs have become highly useful tools, probes, and diagnostics in basic research and biotechnology applications.2,3
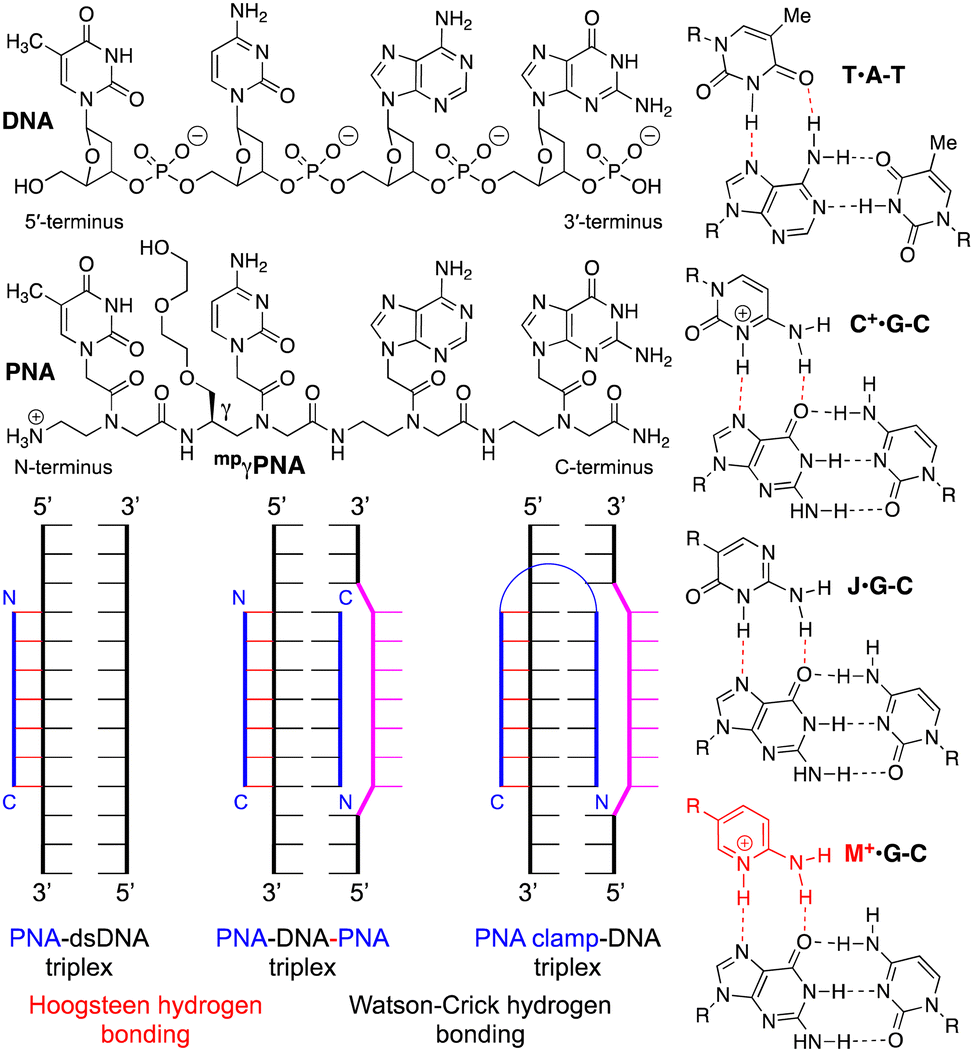 | ||
| Fig. 1 Structures of DNA, PNA, Hoogsteen hydrogen-bonded base triples, and PNA–dsDNA binding modes. | ||
The early studies discovered that PNA–dsDNA triplex formation occurred through an unexpected binding mode where polypyrimidine PNA invaded the double helix, clamping on the purine-rich strand of DNA and forming a PNA–DNA–PNA triplex (Fig. 1).1,4,5 In this novel binding mode, one PNA strand forms an antiparallel Watson–Crick duplex to the polypurine strand of DNA, while the other PNA forms a parallel Hoogsteen triplex. Because of the opposite orientation of the two PNAs, several groups recognized that a linker could easily connect the two strands to create a single molecule that was called a PNA clamp (Fig. 1).6–9 The ability of PNA to invade dsDNA, forming a PNA–DNA–PNA triplex, is a novel and unique binding mode that remains an underexplored antigene strategy.2
Mechanistically, the invasion/clamping mode is more complicated than a simple triplex or duplex formation because it involves coupled steps of DNA duplex dissociation, PNA–DNA Watson–Crick duplex formation, and PNA–DNA Hoogsteen triplex formation. Mechanistic studies strongly support a pathway that proceeds through an initial formation of the PNA–dsDNA Hoogsteen triplex, followed by the DNA duplex's opening and the final PNA–DNA–PNA triplex formation.10 This was evidenced by a strong pH dependence for strand invasion when using cytosine-modified clamps.10 The overall invasion is a kinetically controlled process, and the dissociation of the PNA–DNA–PNA triplex (off-rate) is very slow.5,7 The early studies also demonstrated that DNA invasion was relatively fast only in low salt buffers (e.g., ∼10 mM Na+) while increasing the salt concentration to near physiological strongly inhibited invasion.5
Chemical modifications have been used to optimize the properties of PNA, including PNA clamps.2 Pseudoisocytosine (J), a neutral analog of protonated cytosine, formed J·G–C triples (Fig. 1) stable at physiological pH, while the formation of native C+·G–C triples required slightly acidic conditions (due to pKa of C ∼ 4.5).6,11 Backbone modification at the gamma position, such as the minipeg-modified γPNA (mpγPNA, Fig. 1), introduced a chiral center that preorganized PNA in a right-handed helical conformation favorable for duplex formation and DNA invasion.12 Glazer and co-workers combined J and mpγPNA modifications to enhance DNA invasion of PNA clamps for genome modification.13–15 However, slow invasion kinetics at physiological salt remains the major bottleneck for developing PNA clamps as probes and therapeutic agents.
In our research on triplex-forming PNAs for sequence-specific recognition of dsRNA,16 we introduced the more basic (pKa ∼ 6.7) 2-aminopyridine nucleobase (M, Fig. 1) as an alternative to protonated cytosine.17 M formed about 3-fold more stable M+·G–C triples with higher selectivity for the target dsRNA than J.18 The superior performance of M was due to the partial protonation at physiological pH, which enhanced the binding affinity while maintaining high sequence specificity. In the present study, we hypothesized that the partial protonation may also improve the strand invasion of dsDNA. We used electrophoretic mobility shift assays (EMSA) to demonstrate that M-modified PNA clamps invaded dsDNA faster and at lower concentrations than the J-modified counterparts. Most importantly, the M-modified PNA formed the invasion complex under physiological salt and pH, while the J-modified PNA clamp showed no invasion under these conditions. Our results suggest that the M modification has a strong potential to overcome the key bottleneck and revitalize the PNA clamp technology.
To test our hypothesis, we chose a model system of a bis-PNA clamp targeting a p262 DNA fragment previously studied by Nielsen and co-workers.19 The M20 and J6,21 monomers, and PNA clamps22 were synthesized following our previously reported procedures (for details, see ESI†). We first compared the DNA invasion ability of M- and J-modified PNA clamps (PNA1 and PNA2, Fig. 2) under low salt conditions. Fluorescently labeled (Cy5 or TYE 665) 65 base pairs long p262 DNA construct (DNA1, 100 nM) was incubated with the specified amount of PNAs for 4 hours in a 10 mM sodium phosphate buffer (no added salt). The complexes formed were resolved with 15% PAGE (Fig. 2). The EMSA results showed that under these low salt conditions, both M- and J-modified PNA clamps were able to invade DNA1. However, the invasion of M-modified PNA1 was more facile, with an apparent EC50 of 0.4 ± 0.1 μM (Fig. 2A), while the J-modified PNA2 showed an apparent EC50 of 1.1 ± 0.3 μM (Fig. 2B). The formation of multiple higher running bands in gels shown in Fig. 2 was previously observed by Nielsen and co-workers19 and was attributed to alternative conformations of the invasion triplex.
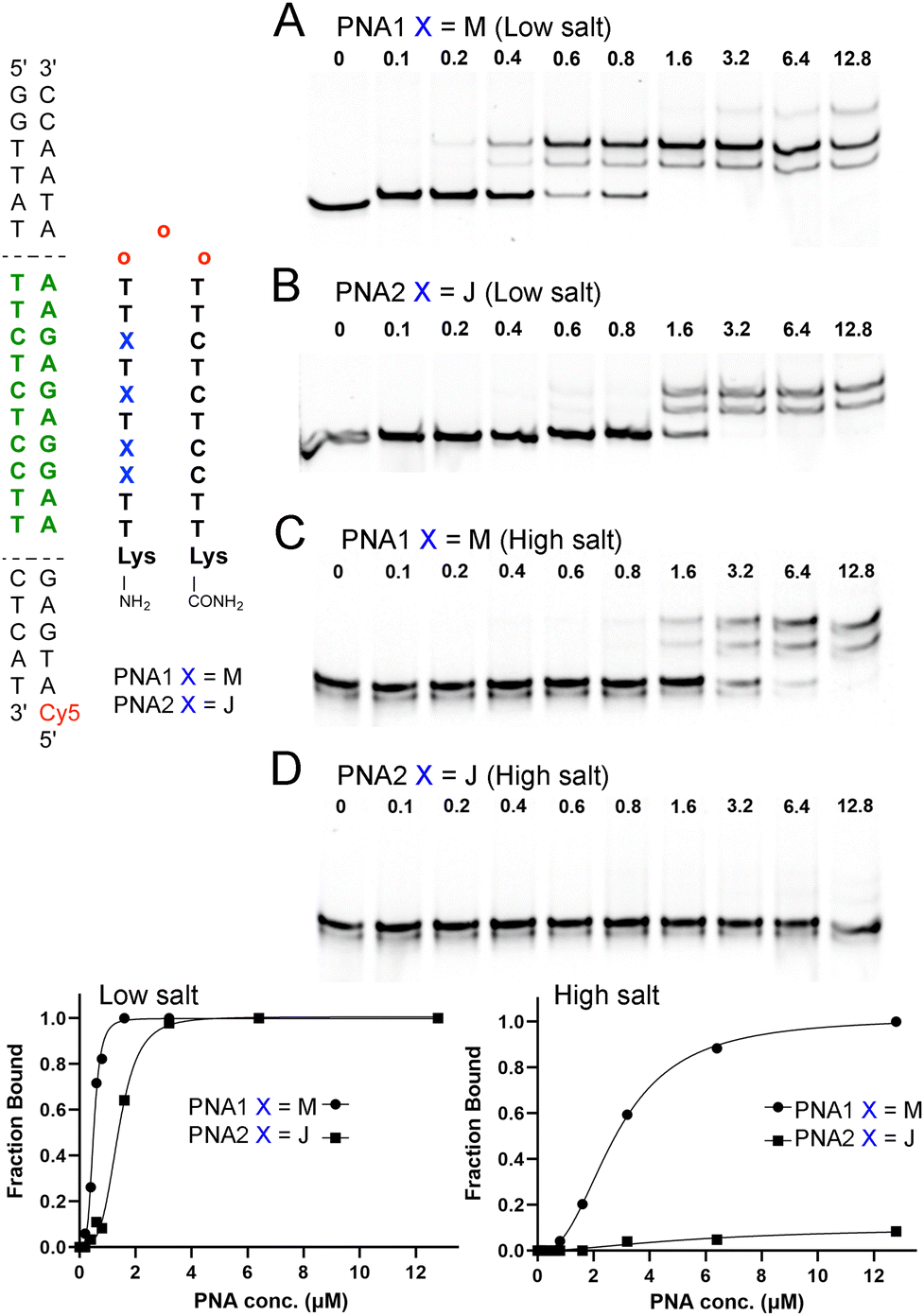 | ||
| Fig. 2 Electrophoretic mobility shift assays (EMSA) of M- and J-modified PNA clamps at low salt conditions. 100 nM of 65 bp dsDNA was incubated with a specified amount of PNA (A) and (B) in 10 mM sodium phosphate, pH 6.45, and (C) and (D) in 2 mM MgCl2, 90 mM KCl, 10 mM NaCl, 50 mM potassium phosphate, pH 7.4 for 4 h at 37 °C. Complexes were resolved with 15% PAGE in the TBE running buffer (∼12 h run time at 50 V). Red “O” denotes 2-(2-(2-aminoethoxy)ethoxy)acetic acid (AEEA). | ||
These results showed that even at the low salt concentration, known to be conducive to DNA invasion, the M-modification improved the performance of the PNA clamp. Next, we compared DNA invasion by M- and J-modified PNA1 and PNA2 in a buffer mimicking the physiological salt (Fig. 2C and D). Under these conditions (2 mM MgCl2, 90 mM KCl, 10 mM NaCl, 50 mM potassium phosphate), PNA1 was still able to invade DNA1, but the concentration required for complete complex formation increased to 12.8 μM (Fig. 2C). Moreover, the apparent EC50 of PNA1 increased nearly 7-fold to 2.8 μM, highlighting the challenge of invading under physiological salt conditions. In the buffer mimicking the physiological salt and pH, the J-modified PNA2 showed no significant formation of the DNA invasion complex (Fig. 2D), confirming that the M-modified clamp clearly outperformed the J-modified counterpart.
To further compare M- and J-modified PNA1 and PNA2, we performed a series of EMSA experiments at different PNA concentrations and monitored the DNA invasion at various time points (Fig. 3). The results showed a >90% shift of the dsDNA target at >12.8 μM of the M-modified PNA1 after 4 h. Lower concentrations of PNA1 required a longer time of ∼24 h to complete the invasion. In contrast, the J-modified PNA2 after 4 h showed relatively little invasion, ∼7% shift at 6.4 μM that increased to ∼17% at 25.6 μM.
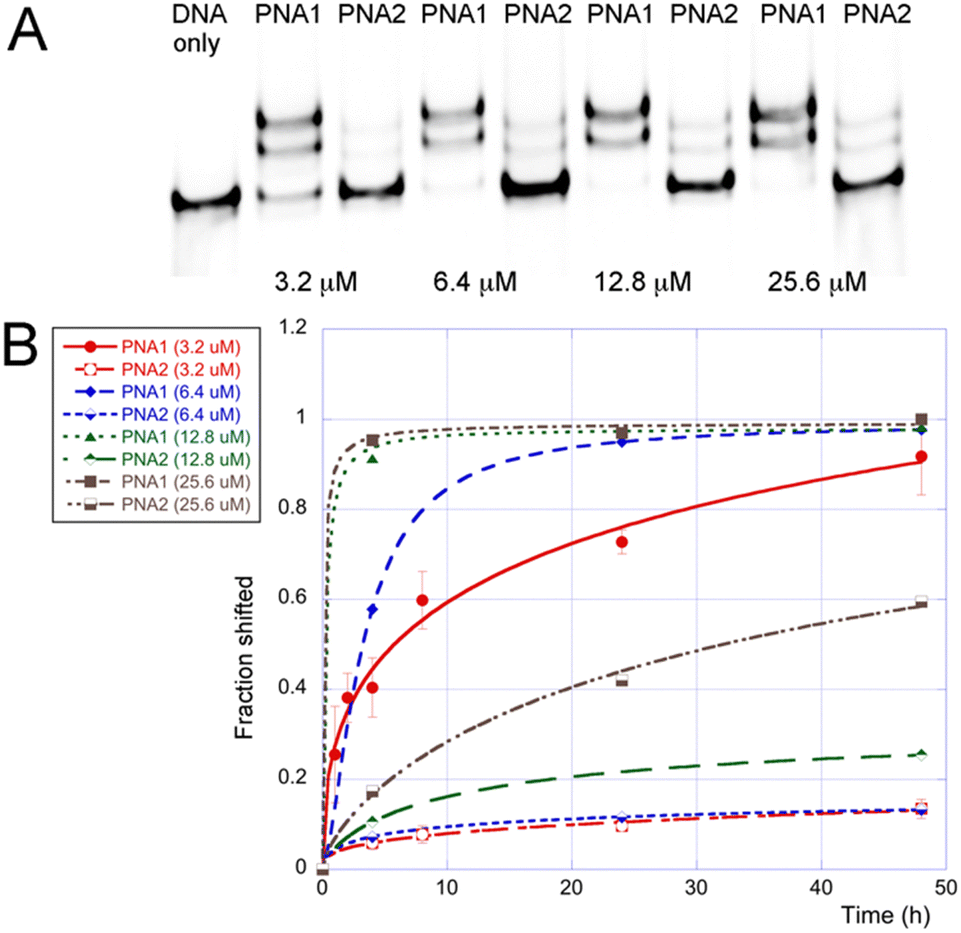 | ||
| Fig. 3 Results of EMSA of M- and J-modified PNA clamps at physiological salt conditions: (A) representative gel after 24 h incubation, and (B) graphical summary of results at various PNA concentrations and incubation times. In all experiments, 100 nM of 65 bp dsDNA was incubated with the specified amount of PNA in 2 mM MgCl2, 90 mM KCl, 10 mM NaCl, and 50 mM potassium phosphate buffer (pH 7.4) at 37 °C. Complexes were resolved with 15% PAGE in the TBE running buffer (∼12 h run time at 50 V). | ||
The comparison of PNA1 and PNA2 showed (Fig. 3B) that at all concentrations tested, the M-modified PNA clamp invaded the dsDNA target faster and to a greater extent than the J-modified counterpart. It appeared that the J-modified PNA2 was not able to completely invade the dsDNA even after 48 h, reaching only ∼60% at the highest concentration of 25.6 μM. The better performance of the M-modified PNA1 was due to a faster invasion because the thermodynamic stabilities of PNA1 and PNA2 complexes with the single-stranded DNA (ssDNA1, the purine-rich strand of DNA1) were comparable as measured by isothermal titration calorimetry (Tables S2, S3 and Fig. S5, S6, ESI†). The respective association constants (Ka × 106) were 39 for PNA1–ssDNA and 29 for PNA2–ssDNA. Binding of PNA5 and PNA6, the M- and J-modified triplex-forming parts of clamps (Fig. S1, ESI†) to double-stranded DNA1 was also comparable and relatively weak with Ka 0.6 and 0.4 × 106, for PNA5 and PNA6 respectively (Tables S4 and S5, ESI†). This was consistent with our previous results that short triplex-forming PNAs bind weakly to dsDNA with Ka 0.2–0.9 × 106 for PNA 9-mers.18 The duplex-forming part of PNA clamps, PNA7 (Fig. S1, ESI†), formed a stable duplex with ssDNA1 with Ka 22 × 106 (Table S6, ESI†). The triplex and duplex parts, PNA5 and PNA7 together but without the linker were not able to invade DNA1 (Fig. S27, ESI†). Our results suggest that the Watson–Crick duplex contributed most to the thermodynamic stability of PNA1–DNA1 invasion complexes, while the Hoogsteen triplex drove the kinetics of invasion. It is conceivable that the partial cationic nature of M enhanced the on-rate that drives the triplex-first invasion process, creating a unique synergy of duplex and triplex formation in the clamps.
To evaluate the sequence specificity of dsDNA invasion by the M-modified PNAs, we swapped two nucleobases in either the DNA target or the PNA sequence (Fig. 4). The goal was to create minimal mismatches (two) while maintaining the same nucleobase composition. Changing a C–G base pair in DNA1 to a G–C in DNA2 created a mismatched M+·C–C triple in the middle of the sequence (Fig. 4A), effectively eliminating the DNA invasion by PNA1. At the highest PNA concentrations, we observed some reduction of DNA band intensity, down to 65% at 500 μM (5000-fold excess), most likely due to sequence non-specific PNA interactions with DNA2. Next, we evaluated the impact of two mismatches in either the triplex or the duplex parts of the PNA–dsDNA complex. Swapping T and M in PNA3 created two consecutive Hoogsteen mismatches in the triplex part (Fig. 4B), which strongly suppressed the DNA invasion. In contrast, swapping of T and C in PNA4 created two consecutive Watson–Crick mismatches in the duplex part (Fig. 4C), which suppressed DNA invasion but to a notably lesser extent than the two mismatches in the triplex part. The gel showed a ∼50% shift of DNA1 at 12.8 μM of PNA4 (Fig. 4C), conditions under which PNA1 achieved complete invasion of DNA1 (Fig. 2C). These results were consistent with the sequence-specific formation of the expected PNA–DNA–PNA triplexes. The results also suggested that mismatches were less tolerated in the Hoogsteen triplex part than in the Watson–Crick duplex part of the PNA–DNA–PNA triplex, consistent with the proposed DNA invasion mechanism that starts with triplex formation.10
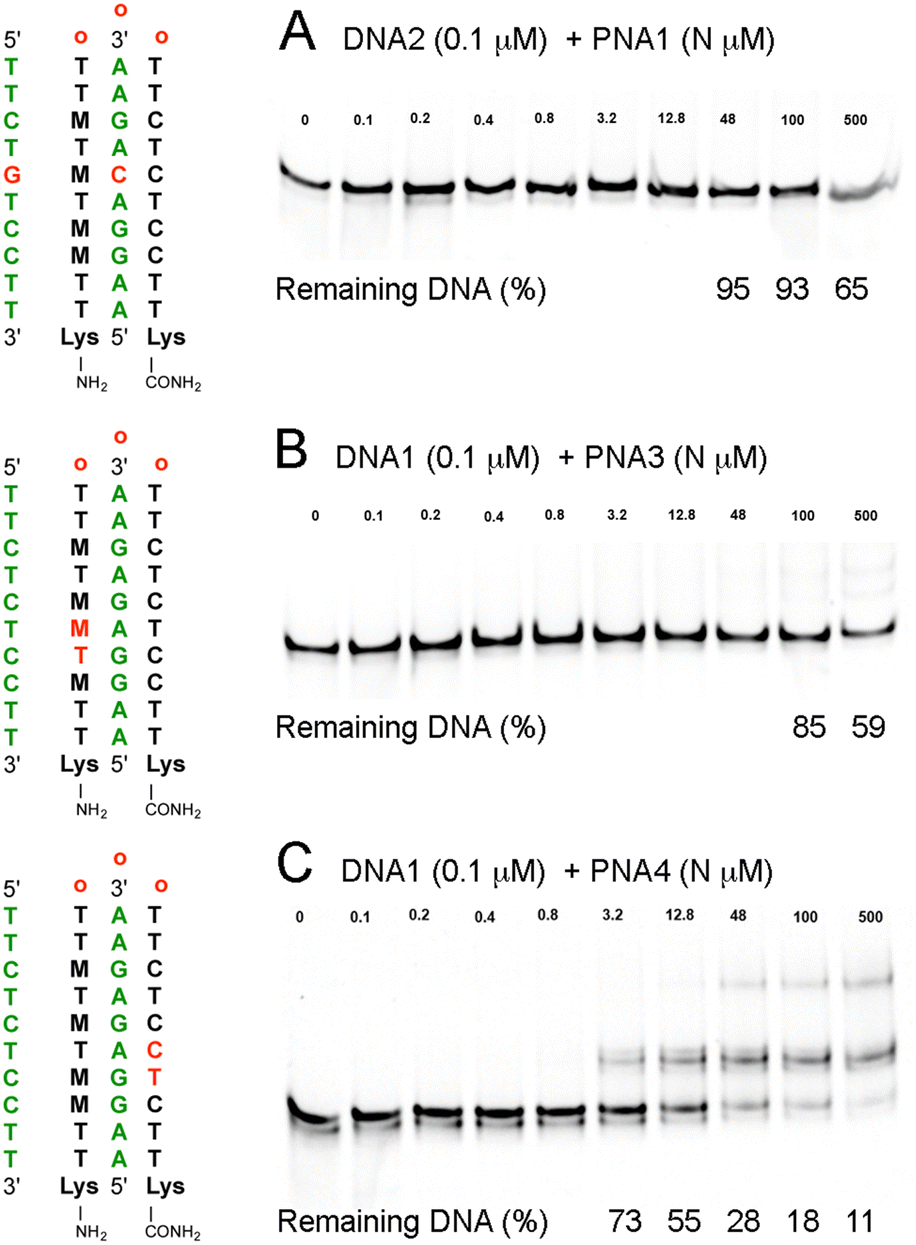 | ||
| Fig. 4 EMSA studies of sequence specificity of M-modified PNA clamps at physiological salt conditions: (A) PNA1–DNA2 triplex having a mismatched M+·C–C triple, (B) PNA3–DNA1 triplex having two mismatches in the Hoogsteen strand of PNA, and (C) PNA4–DNA1 triplex having two mismatches in the Watson–Crick strand of PNA. In all experiments, 0.1 μM of 65 bp dsDNA was incubated with the specified amount (N μM) of PNA for 4 h under conditions as in Fig. 3. | ||
The ability of PNA to invade dsDNA has been a fascinating but underutilized mode of biomedical DNA targeting technologies. The key bottleneck has been the slow and incomplete DNA invasion at physiological salt concentrations. Chemical modifications have been used to overcome this problem but with limited success.2 J nucleobase11 has been used in modified PNA clamps to form J·G–C triples stable at physiological pH.23,24 Glazer and co-workers combined J and mpγPNA modifications in PNA clamps designed to induce modification of genomic DNA.13–15,24 Lu and co-workers25 used J-modified PNA clamps to assist DNAzymes in cleaving dsDNA, followed by subsequent recombination. The above studies are all notable examples where replacing J with M may provide significant advances and bring PNA technology closer to in vivo applications.
In the present study, we demonstrated that 2-aminopyridine (M) nucleobase modifications strongly enhanced the ability of a PNA clamp to invade dsDNA under physiological salt concentration. The M-modified clamp formed a PNA–DNA–PNA triplex under conditions when J-modified PNA could not invade the dsDNA target. The positive impact was most likely due to the partial positive charge of M (pKa ∼ 6.7) that enabled highly stable yet sequence-specific M+·G–C triples under physiological salt and pH. The PNA–DNA–PNA triplex formation was sequence-specific as mismatches in either Hoogsteen or Watson–Crick parts strongly inhibited DNA invasion. These results are significant because the M-modified PNAs have the potential to overcome the problems of using PNA technologies to invade DNA under physiological conditions.
This work was supported by the US National Science Foundation (CHE-2107900 and CHE-2427911 to E. R.). We thank I. Kumpina for help with ITC and gel data analysis, and J. Harasgama for help with gel data analysis and EMSA experimental design. We thank Bapeks (www.bapeks.com) for a generous gift of a Fmoc-(AEEA)3-COOH starting material for PNA synthesis.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497 CrossRef CAS PubMed.
- N. Brodyagin, M. Katkevics, V. Kotikam, C. A. Ryan and E. Rozners, Beilstein J. Org. Chem., 2021, 17, 1641 CrossRef CAS PubMed.
- J. Saarbach, P. M. Sabale and N. Winssinger, Curr. Opin. Chem. Biol., 2019, 52, 112 CrossRef CAS PubMed.
- D. Y. Cherny, B. P. Belotserkovskii, M. D. Frank-Kamenetskii, M. Egholm, O. Buchardt, R. H. Berg and P. E. Nielsen, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1667 CrossRef CAS PubMed.
- N. J. Peffer, J. C. Hanvey, J. E. Bisi, S. A. Thomson, C. F. Hassman, S. A. Noble and L. E. Babiss, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 10648 CrossRef CAS.
- M. Egholm, L. Christensen, K. L. Dueholm, O. Buchardt, J. Coull and P. E. Nielsen, Nucleic Acids Res., 1995, 23, 217 CrossRef CAS PubMed.
- M. C. Griffith, L. M. Risen, M. J. Greig, E. A. Lesnik, K. G. Sprankle, R. H. Griffey, J. S. Kiely and S. M. Freier, J. Am. Chem. Soc., 1995, 117, 831 CrossRef CAS.
- L. Betts, J. A. Josey, J. M. Veal and S. R. Jordan, Science, 1995, 270, 1838 CrossRef CAS PubMed.
- V. V. Demidov, M. V. Yavnilovich, B. P. Belotserkovskii, M. D. Frank-Kamenetskii and P. E. Nielsen, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2637 CrossRef CAS PubMed.
- H. Kuhn, V. V. Demidov, P. E. Nielsen and M. D. Frank-Kamenetskii, J. Mol. Biol., 1999, 286, 1337 CrossRef CAS PubMed.
- A. G. Veselkov, V. V. Demidov, P. E. Nielsen and M. D. Frank-Kamenetskii, Nucleic Acids Res., 1996, 24, 2483–2487 CrossRef CAS PubMed.
- S. Rapireddy, G. He, S. Roy, B. A. Armitage and D. H. Ly, J. Am. Chem. Soc., 2007, 129, 15596 CrossRef CAS.
- R. Bahal, N. Ali McNeer, E. Quijano, Y. Liu, P. Sulkowski, A. Turchick, Y.-C. Lu, D. C. Bhunia, A. Manna, D. L. Greiner, M. A. Brehm, C. J. Cheng, F. Lopez-Giraldez, A. Ricciardi, J. Beloor, D. S. Krause, P. Kumar, P. G. Gallagher, D. T. Braddock, W. Mark Saltzman, D. H. Ly and P. M. Glazer, Nat. Commun., 2016, 7, 13304 CrossRef CAS PubMed.
- J. Y. Chin, J. Y. Kuan, P. S. Lonkar, D. S. Krause, M. M. Seidman, K. R. Peterson, P. E. Nielsen, R. Kole and P. M. Glazer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13514 CrossRef CAS PubMed.
- S. N. Oyaghire, E. Quijano, J. D. R. Perera, H. K. Mandl, W. M. Saltzman, R. Bahal and P. M. Glazer, Cell Rep. Phys. Sci., 2023, 4, 101635 CrossRef CAS PubMed.
- M. Katkevics, J. A. MacKay and E. Rozners, Chem. Commun., 2024, 60, 1999 RSC.
- T. Zengeya, P. Gupta and E. Rozners, Angew. Chem., Int. Ed., 2012, 51, 12593 CrossRef CAS PubMed.
- C. A. Ryan, N. Brodyagin, J. Lok and E. Rozners, Biochemistry, 2021, 60, 1919 CrossRef CAS PubMed.
- G. I. Hansen, T. Bentin, H. J. Larsen and P. E. Nielsen, J. Mol. Biol., 2001, 307, 67 CrossRef CAS PubMed.
- I. Kumpina, V. Baskevics, G. D. Walby, B. R. Tessier, S. F. Saei, C. A. Ryan, J. A. MacKay, M. Katkevics and E. Rozners, Synlett, 2024, 649 CAS.
- R. H. E. Hudson and F. Wojciechowski, Can. J. Chem., 2008, 86, 1026 CrossRef CAS.
- M. M. Rahman, C. A. Ryan, B. R. Tessier and E. Rozners, Heliyon, 2024, 10, e339142 Search PubMed.
- K.-H. Kim, P. E. Nielsen and P. M. Glazer, Biochemistry, 2006, 45, 314 CrossRef CAS.
- J. Y. Chin, F. Reza and P. M. Glazer, Mol. Ther., 2013, 21, 580–587 CrossRef CAS PubMed.
- M. Lyu, L. Kong, Z. Yang, Y. Wu, C. E. McGhee and Y. Lu, J. Am. Chem. Soc., 2021, 143, 9724 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, HPLC purification, and LC-MS characterization of PNA oligomers; ITC results and representative titration images; raw EMSA data and images (PDF). See DOI: https://doi.org/10.1039/d4cc06748g |
| This journal is © The Royal Society of Chemistry 2025 |