
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhoto-ejected ligands hyperpolarized by parahydrogen in reversible exchange†
Emily E. Brown ,
Rielly J. Harrison,
Dmytro Havrylyuk,
Stephen J. McBride,
Edith C. Glazer,
Felix N. Castellano* and
Thomas Theis*
,
Rielly J. Harrison,
Dmytro Havrylyuk,
Stephen J. McBride,
Edith C. Glazer,
Felix N. Castellano* and
Thomas Theis*
Department of Chemistry, North Carolina State University, Raleigh, North Carolina 27965-8204, USA. E-mail: fncastel@ncsu.edu; ttheis@ncsu.edu
First published on 20th February 2025
Abstract
Hyperpolarization derived from signal amplification by reversible exchange (SABRE) is valued for its relative simplicity among hyperpolarization approaches. Here, we demonstrate that photoejection of the ligands pyridazine and pyrazine from prodrug Ru(II) polypyridyl complexes offers a new modality for activating SABRE hyperpolarization, achieving enhancements of over 3200-fold.
Nuclear magnetic resonance (NMR), an essential spectroscopic technique for identifying and characterizing molecular structure, is inherently sensitivity-limited by low nuclear spin polarization. Hyperpolarization methods markedly increase this polarization, generating strongly enhanced NMR signals that overcome sensitivity limitations.1–5 Among hyperpolarization methods, signal amplification by reversible exchange (SABRE) is particularly simple to implement, which makes it pertinent for biomedical applications.6–8 Traditional advantages of SABRE also include the preservation of chemical identity and replicability as opposed to hydrogenative methods, yet expanding the SABRE-substrate scope is more tedious than for other methods, such as dynamic nuclear polarization (DNP).1,3,9,10 This work demonstrates SABRE of photo-ejected ligands from prodrugs without extensive activation, providing a new modality for SABRE-based NMR hyperpolarization. Room temperature solutions containing Ru(II) complexes and the Ir-based SABRE catalyst are irradiated with visible light to photo-eject the ligands from the prodrug Ru complexes.7 These free ligands are subsequently hyperpolarized by parahydrogen (p-H2) in reversible exchange on the SABRE catalyst. After photo-ejection, p-H2 and the ejected substrate reversibly bind to the iridium hydride catalyst ([Ir(H)2(IMes)(sub)3]Cl, IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene, sub = substrate) shown in Scheme 1.8,11–14 Once bound, spin order is transferred from the p-H2-derived hydrides to the photo-ejected target molecule through J-coupling interactions.
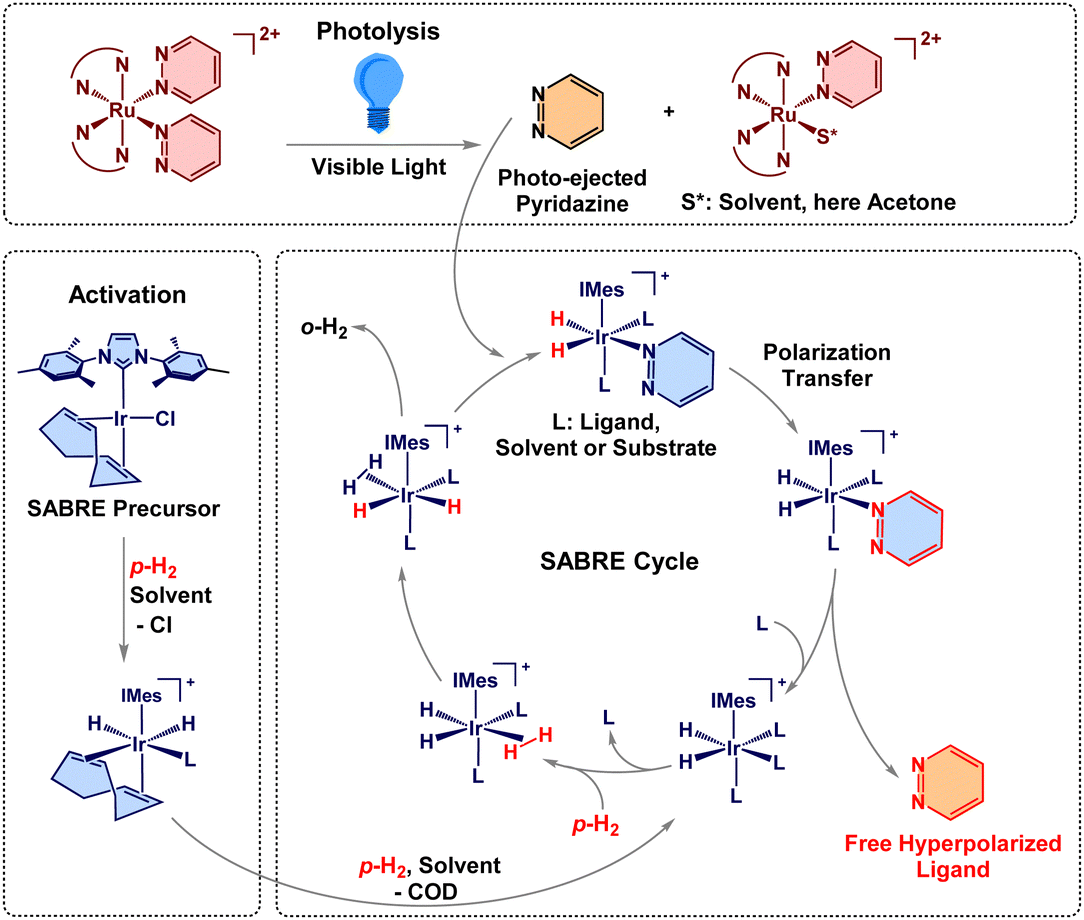 | ||
| Scheme 1 The photoactivated SABRE process with 1.7,9–12 Red bonds and chemical symbols represent spin-polarization and o-H2 is ortho-hydrogen released from the IrIMes catalyst. | ||
Light activation presents an interesting addition to SABRE, offering simple and controllable initialization of hyperpolarization across various sample formats. In NMR, in situ light irradiation is also of growing interest.15–17 So far, photo modulation has been limited to para-hydrogenation or photoisomerization following SABRE.18–22 The light activation shown in this work can provide on-demand release of desirable target molecules and be incorporated into the SABRE cycle. Here, we chose the amply studied Ru(II) polypyridyl complexes—well known for efficient ligand photoejection triggered by visible light.23–28
This work uses two Ru(II) polypyridyl complexes, Ru(bpy)2(pyridazine)22+ (1) and Ru(tpy)(biq)(pyrazine)2+ (2), bpy = 2,2′-bipyridine, tpy = 2,2′,2′′-terpyridine, biq = 2,2′-biquinoline.23,29 These chromophores undergo efficient photoejection of their monodentate ligands under visible light excitation. Additionally, photolyzed 1 has demonstrated DNA binding and 2 is established as both cytotoxic and photo-cytotoxic.7,29 These properties make Ru(II) molecules interesting candidates for photoactivated chemotherapy.30 The freely diffusing ligand (pyridazine or pyrazine) can be readily incorporated into the SABRE cycle to be hyperpolarized, Scheme 1. These initial target molecules were selected as both are already well-established SABRE substrates.31,32 The light-triggered motif enables the early acquisition of hyperpolarized signals immediately upon ligand release. The system's simplicity and broad applicability is further underscored by acquiring data using a low-field benchtop NMR spectrometer operating at 1.4 T (60 MHz). SABRE acquisition allows for greater specificity of ligand detection than might be achieved through alternate methods of spectroscopy, such as UV-Visible spectroscopy. Also, magnetic resonance techniques (NMR and MRI) are free of penetration depth limitations, which allows for detection in opaque samples such as tissue. The simplicity of SABRE compared to other hyperpolarization techniques, combined with the use of a cryogen-free benchtop spectrometer in this work, exemplifies a more affordable, straightforward, and user-friendly approach, expanding the possibilities for adoption by a wider audience.
Ligand-releasing photolysis using 455 nm visible light is well documented for both 1 and 2 in water. To ensure that our methodology would rapidly generate free ligands in the current solvent system (acetone-d6), we first conducted in situ photochemical 1H NMR experiments at high-field (9.4 T, 400 MHz); see the ESI,† for complete details. Ligand loss in acetone-d6 occurred rapidly under LED excitation inside the NMR spectrometer (see Fig. S1 and S2, ESI†). This represents the first ligand ejection from 1, forming Ru(bpy)2(pyridazine)2+ featuring one coordinated pyridazine ligand. This is demonstrated by quantitative NMR of the photolysis of 1, as even 400 s of photolysis would yield only 48% free pyridazine, relative to the starting concentration of 1 (not two equivalents or 200%), see ESI.† This behaviour echoes prior work in water, where the second photolysis event, the second ligand ejection, was sluggish and occurred over 150 minutes of light excitation.7
After confirming rapid ligand-loss photolysis for 1 and 2 in acetone-d6, SABRE experiments were pursued using the experiment and sequence presented in Fig. 1. Visible light irradiation and p-H2 bubbling were performed simultaneously in a 6.5 mT polarization transfer field (PTF). 6.5 mT was used because this magnetic field establishes level anticrossings matching the nuclear-spin energy levels to allow for optimized polarization flow from hydrides to protons on pyrazine and pyridazine.6,12,13,31–34 The SABRE experiments used a specialized gas bubbling system to facilitate p-H2 dissolution through a solution inside a 5 mm NMR tube. The sample was positioned within the solenoid coil set to the designated PTF and the 30 s p-H2 bubbling time was utilized. Upon termination of p-H2 bubbling, the sample was transferred to the 1.4 T NMR, and the signal was acquired using a 90° RF pulse. We note that the temperature in the bore of the utilized Oxford XPulse benchtop spectrometer was 40 °C and could not be altered, warming the samples to 21 °C immediately following the transfer, as measured by a methanol-d4 thermometer, see ESI.† The temperature during hyperpolarization was maintained at 21 °C to decrease evaporation of the solvent, acetone-d6, and to maintain a consistent concentration, despite possibly non-optimized hyperpolarization.31
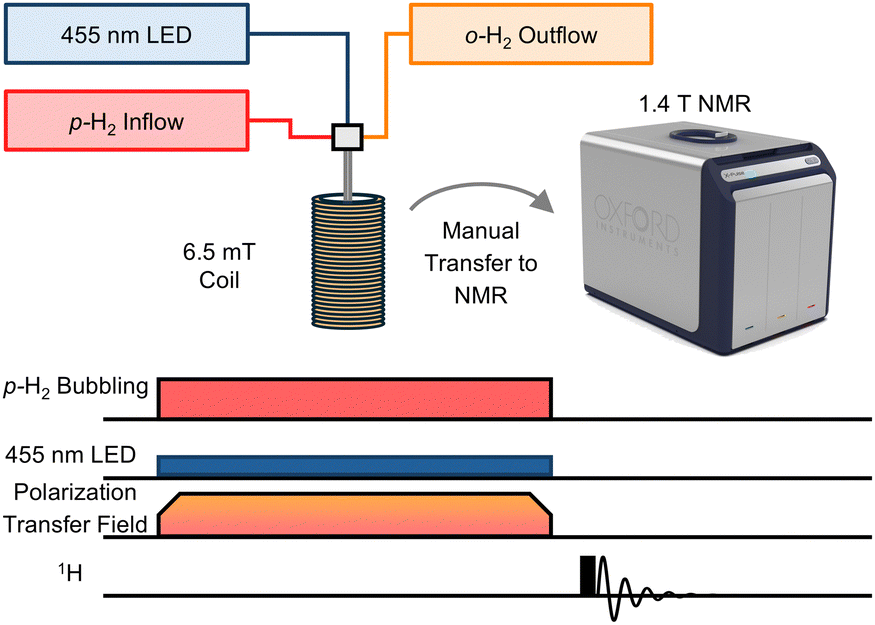 | ||
| Fig. 1 The photo-induced SABRE experiment and signal acquisition sequence. The 455 nm excitation light and p-H2 are applied simultaneously to the sample while in the PTF of 6.5 mT for 30 s. Next, the sample is manually transferred (approximately 1–2 s) to the NMR, and the signal is acquired. The image of the 1.4 T NMR spectrometer is courtesy of Oxford Instruments. | ||
Preliminary hyperpolarization experiments were performed without light; only p-H2 was added to the solution within the 6.5 mT PTF. As expected, no significant signals were observed, indicating little to no free ligand present in the absence of light irradiation, see Fig. 2. Slightly more thermally-induced ligand release was measured in 2 relative to 1 during the dark scans, despite attempts to prepare samples without room light. We note that the absorbance of 1 is blue-shifted relative to 2.7 Additionally, prior work with 2 has demonstrated limited long-term thermal stability in solution; 63% of the complex remained after 24 hours at 37 °C dissolved in water, as opposed to the 88% of 1 remaining after 72 hours under identical experimental conditions.7,29 Measurements performed here in protonated acetone, see ESI,† showed that 98% of 1 remained after 24 h at 40 °C in the dark and only 18% of 2 remained after 21 h under the same conditions. We conclude that the 40 °C NMR operating temperature leads to more thermalized ligand loss in 2 relative to 1 but does not significantly influence results achieved via photoactivation as detailed below.
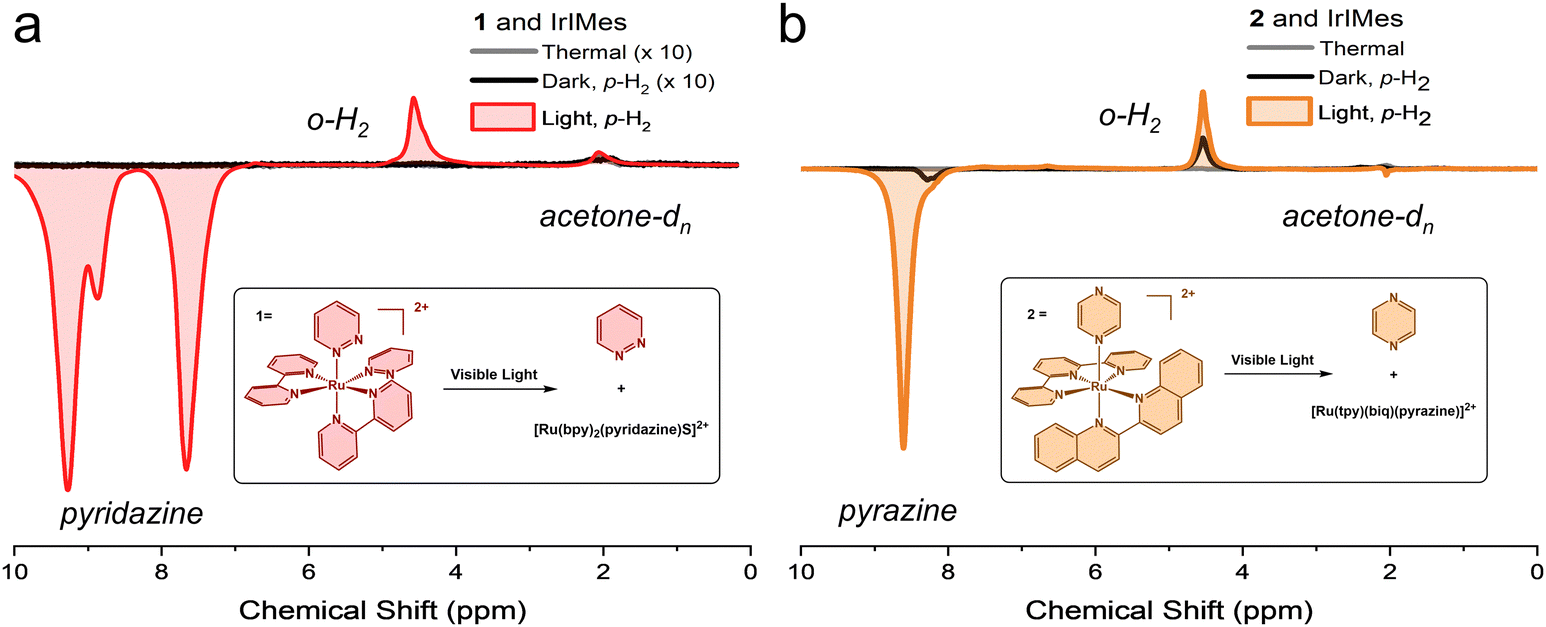 | ||
| Fig. 2 A. Complex 1 and the SABRE catalyst, shown with the thermal scan in the absence of p-H2, the dark scan with p-H2 added in the 6.5 mT PTF, and the light scan with 455 nm light and p-H2 added in the 6.5 mT PTF. The thermal scan, grey, and the dark scan, black, are magnified 10-fold. B. Complex 2 and the SABRE catalyst here shown with the thermal scan without p-H2, the dark scan with p-H2 added in the 6.5 mT PTF, and the light scan with 455 nm light and p-H2 added in the 6.5 mT PTF. | ||
Upon visible light excitation, 1H NMR signal enhancement increased dramatically, resulting in an enhancement of 3229-fold (p = 1.6%) and 457-fold (p = 0.22%) at 1.4 T with the photo-released ligands from the prodrugs 1 and 2, respectively, as compared to an external standard, see ESI.† In contrast, minimal signal enhancement occurred in the dark under identical experimental conditions, as shown in Fig. 2. The PTF utilized here is optimized for maximum hyperpolarization transfer to the substrate, and typically results in emissive peaks.31,32 The unexpected line shape from acetone-dn (n < 6) at 2.05 ppm in Fig. 2b may indicate IrIMes catalysed hydrogen–deuterium exchange (1H/2H) occurring with the solvent, acetone-d6.35 No evidence suggests that the bpy ligand from 1 is released, in accordance with previous work.24 Hyperpolarization was observed on both the free substrate with pyridazine at 9.27 and 7.66 ppm (Fig. 2a) and pyrazine at 8.59 ppm (Fig. 2b) as well as the IrIMes-bound substrate, with coordinated pyridazine and pyrazine at 8.87 ppm (Fig. 2a) at 8.20 ppm (Fig. 2b), respectively. Measurements for pyridazine are derived from the peak at 7.66 ppm to avoid the contribution of bound hyperpolarized pyridazine, see ESI.† Hyperpolarization was also observed at the o-H2 peak at 4.5 ppm. However, there was no evidence of a partially negative line (PNL), and minor signal enhancement was also measurable on the Ir-hydride peak(s).36,37
These data were achieved without extensive activation. The typical sample undergoes two cycles of measurements in the dark, followed by up to six cycles of measurements in the presence of light. There is likely a buildup of substrate and p-H2 on the catalyst, further increasing the polarization of the free ligand. Notably, the ratio of free substrate to the IrIMes catalyst at the scan with the highest hyperpolarization never exceeds 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating relatively efficient polarization transfer. This demonstrates the utility of light activation in presenting substrates to a SABRE catalyst on demand, where hyperpolarization can easily be activated and measured.
:1, indicating relatively efficient polarization transfer. This demonstrates the utility of light activation in presenting substrates to a SABRE catalyst on demand, where hyperpolarization can easily be activated and measured.
In conclusion, the findings demonstrate the viability of light-initiated or phototriggered SABRE, offering more control over the SABRE process. We have shown substantial 1H NMR signal enhancements, several thousand times that of the thermal signal (over 3200-fold for free pyridazine derived from 1 and over 400-fold for free pyrazine derived from 2). This approach uses visible light in conjunction with a 1.4 T benchtop NMR spectrometer, offering a widely applicable modality for generating hyperpolarized NMR signals of photo-released compounds. This photochemically-activated approach enables facile sample preparation and may become useful for the in situ detection of photo-released drugs, albeit requiring the addition of p-H2. The procedure rapidly generates hyperpolarization without the need for specialized NMR pulse sequences.
This work was supported by the US Department of Energy, Office of Science, Genomic Science Program under Award Numbers DE-SC0023334 and DE-SC0025315. ECG and DH gratefully acknowledge the National Institutes of Health (Grant GM107586) for the support of this research. This work was performed in part by the Molecular Education, Technology and Research Innovation Center (METRIC) at NC State University, which is supported by the State of North Carolina.
Data availability
The data supporting this article, including experimental details, synthesis of 1 and 2, NMR analyses, thermal analyses, etc., have been included in the ESI.†Conflicts of interest
T. T. declares a stake of ownership in Vizma Life Sciences (VLS). VLS is developing products related to the research being reported. T. T. serves on the Scientific Advisory Board (SAB) of VLS. The terms of this arrangement have been reviewed and approved by NC State University in accordance with its policy on objectivity in research.Notes and references
- C. R. Bowers and D. P. Weitekamp, J. Am. Chem. Soc., 1987, 109, 5541–5542 CrossRef CAS.
- C. R. Bowers and D. P. Weitekamp, Phys. Rev. Lett., 1986, 57, 2645–2648 CrossRef CAS PubMed.
- T. C. Eisenschmid, R. U. Kirss, P. P. Deutsch, S. I. Hommeltoft, R. Eisenberg, J. Bargon, R. G. Lawler and A. L. Balch, J. Am. Chem. Soc., 1987, 109, 8089–8091 CrossRef CAS.
- T. B. R. Robertson, N. Gilbert, O. B. Sutcliffe and R. E. Mewis, Chem. Phys. Chem., 2021, 22, 1059–1064 CrossRef CAS PubMed.
- J. Eills, D. Budker, S. Cavagnero, E. Y. Chekmenev, S. J. Elliott, S. Jannin, A. Lesage, J. Matysik, T. Meersmann, T. Prisner, J. A. Reimer, H. Yang and I. V. Koptyug, Chem. Rev., 2023, 123(4), 1417–1551 CrossRef CAS PubMed.
- R. W. Adams, J. A. Aguilar, K. D. Atkinson, M. J. Cowley, P. I. P. Elliott, S. B. Duckett, G. G. R. Green, I. G. Khazal, J. López-Serrano and D. C. Williamson, Science, 2009, 323, 1708–1711 Search PubMed.
- D. Havrylyuk, M. Deshpande, S. Parkin and E. C. Glazer, Chem. Commun., 2018, 54, 12487–12490 RSC.
- D. A. Barskiy, S. Knecht, A. V. Yurkovskaya and K. L. Ivanov, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 114–115, 33–70 Search PubMed.
- R. W. Adams, S. B. Duckett, R. A. Green, D. C. Williamson and G. G. R. Green, J. Chem. Phys., 2009, 131, 194505 Search PubMed.
- J. H. Ardenkjær-Larsen, B. Fridlund, A. Gram, G. Hansson, L. Hansson, M. H. Lerche, R. Servin, M. Thaning and K. Golman, PNAS, 2003, 100, 10158–10163 CrossRef PubMed.
- M. Czarnota, A. Mames, M. Pietrzak, S. Jopa, F. Theiß, G. Buntkowsky and T. Ratajczyk, Angew. Chem., 2024, 136, e202309188 CrossRef.
- J. F. P. Colell, A. W. J. Logan, Z. Zhou, R. V. Shchepin, D. A. Barskiy, G. X. Ortiz, Q. Wang, S. J. Malcolmson, E. Y. Chekmenev, W. S. Warren and T. Theis, J. Phys. Chem. C, 2017, 121, 6626–6634 Search PubMed.
- M. J. Cowley, R. W. Adams, K. D. Atkinson, M. C. R. Cockett, S. B. Duckett, G. G. R. Green, J. A. B. Lohman, R. Kerssebaum, D. Kilgour and R. E. Mewis, J. Am. Chem. Soc., 2011, 133, 6134–6137 CrossRef CAS PubMed.
- K. MacCulloch, P. Tomhon, A. Browning, E. Akeroyd, S. Lehmkuhl, E. Y. Chekmenev and T. Theis, Magn. Reson. Chem., 2021, 59, 1225–1235 CrossRef CAS PubMed.
- K. Bentley, M. D. Hareram, G.-W. Wang, A. A. V. Millman, I. Perez-Ortega, L. M. Nichols, C. C. Bories, L. E. Walker, A. W. Woodward, A. P. Golovanov, L. S. Natrajan and I. Larrosa, J. Am. Chem. Soc., 2025, 147(6), 5035–5042 CrossRef CAS PubMed.
- T. J. N. Hooper, R. de Oliveira-Silva and D. Sakellariou, J. Mat. Chem. A, 2025, 13, 933–939 RSC.
- N. M. Ivanov, A. I. Slivkov and W. T. S. Huck, Angew. Chem., Int. Ed., 2025, 64, e202415614 CrossRef CAS PubMed.
- B. Eguillor, P. J. Caldwell, M. C. R. Cockett, S. B. Duckett, R. O. John, J. M. Lynam, C. J. Sleigh and I. Wilson, J. Am. Chem. Soc., 2012, 134, 18257–18265 CrossRef CAS PubMed.
- B. Procacci, S. B. Duckett, M. W. George, M. W. D. Hanson-Heine, R. Horvath, R. N. Perutz, X.-Z. Sun, K. Q. Vuong and J. A. Welch, Organometallics, 2018, 37, 855–868 CrossRef CAS.
- A. Thomas, M. Haake, F.-W. Grevels and J. Bargon, Angew. Chem., Int. Ed. Engl., 1994, 33, 755–757 CrossRef.
- A. S. Kiryutin, V. P. Kozinenko and A. V. Yurkovskaya, ChemPhotoChem, 2024, 8, e202300151 Search PubMed.
- E. E. Brown, I. Mandzhieva, P. M. TomHon, T. Theis and F. N. Castellano, ACS Cent. Sci., 2022, 8, 1548–1556 CrossRef CAS PubMed.
- J. V. Caspar and T. J. Meyer, Inorg. Chem., 1983, 22, 2444–2453 CrossRef CAS.
- B. Durham, J. L. Walsh, C. L. Carter and T. J. Meyer, Inorg. Chem., 1980, 19, 860–865 CrossRef CAS.
- B. Bosnich and F. P. Dwyer, Aust. J. Chem., 1966, 19, 2229–2233 CrossRef CAS.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649–9651 RSC.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803–4810 CrossRef CAS.
- E. Borfecchia, C. Garino, D. Gianolio, L. Salassa, R. Gobetto and C. Lamberti, Catal. Today, 2014, 229, 34–45 CrossRef CAS.
- D. Havrylyuk, K. Stevens, S. Parkin and E. C. Glazer, Inorg. Chem., 2020, 59, 1006–1013 CrossRef CAS PubMed.
- N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690–10701 RSC.
- K. M. Appleby, R. E. Mewis, A. M. Olaru, G. G. R. Green, I. J. S. Fairlamb and S. B. Duckett, Chem. Sci., 2015, 6, 3981–3993 RSC.
- I. Reile, R. L. E. G. Aspers, J.-M. Tyburn, J. G. Kempf, M. C. Feiters, F. P. J. T. Rutjes and M. Tessari, Angew. Chem., Int. Ed., 2017, 56, 9174–9177 CrossRef CAS PubMed.
- A. N. Pravdivtsev, A. V. Yurkovskaya, H.-M. Vieth, K. L. Ivanov and R. Kaptein, Chem. Phys. Chem., 2013, 14, 3327–3331 CrossRef CAS PubMed.
- T. Theis, M. L. Truong, A. M. Coffey, R. V. Shchepin, K. W. Waddell, F. Shi, B. M. Goodson, W. S. Warren and E. Y. Chekmenev, J. Am. Chem. Soc., 2015, 137, 1404–1407 CrossRef CAS PubMed.
- K. X. Moreno, K. Nasr, M. Milne, A. D. Sherry and W. J. Goux, J. Magn. Reson., 2015, 257, 15–23 CrossRef CAS PubMed.
- A. S. Kiryutin, G. Sauer, A. V. Yurkovskaya, H.-H. Limbach, K. L. Ivanov and G. Buntkowsky, J. Phys. Chem. C, 2017, 121, 9879–9888 CrossRef CAS.
- V. V. Zhivonitko, K. Sorochkina, K. Chernichenko, B. Kótai, T. Földes, I. Pápai, V.-V. Telkki, T. Repo and I. Koptyug, Phys. Chem. Chem. Phys., 2016, 18, 27784–27795 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR analyses, thermal analyses, etc., have been included in the ESI. See DOI: https://doi.org/10.1039/d4cc06807f |
| This journal is © The Royal Society of Chemistry 2025 |