
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAerobic oxidation-induced virtual radical coupling of mixed-metal PtRh2 trinuclear complexes accompanied by the formation of a Rh–Rh bond†
Amiru
Yokoyama
,
Hiroshi
Nakajima
and
Takanori
Nishioka
 *
*
Department of Chemistry, Graduate School of Science, Osaka Metropolitan University, Osaka 558-8585, Japan. E-mail: nishioka@omu.ac.jp
First published on 31st January 2025
Abstract
A mixed-metal PtIIRhI2 trinuclear complex with triply bridging sulfide ligands, generated by the reaction of a Rh(I) nbd complex with a bis(hydrosulfido) Pt(II) complex bearing a bisNHC ligand, is oxidised under aerobic conditions to give a diamagnetic hexanuclear cluster accompanied by the formation of a RhII–RhII bond.
The formation and cleavage of metal–metal bonds in cluster compounds contributes to their reactions, for example, by reserving electrons and providing reaction sites on their cluster cores. To evaluate metal–metal bonding in such cluster compounds, traditional counting of cluster valence electrons (CVEs)1 based on the 18-electron rule is still useful, especially for late transition metals. The 48-CVE trinuclear complexes, consisting of three metal-cyclopentadienyl or its derivative ligand units and two triply bridging sulfide ligands, have three metal–metal bonds.2 Reduction of the complexes leads to the metal–metal bond cleavage,2a,b,3 and one of the resulting 50-CVE complexes acts as a precatalyst for the electrochemical reduction of CO2 to give oxalate as the C–C coupling product.4
We have focused on the intermetallic interaction of such trinuclear sulfido complexes bearing metal-N-heterocyclic carbene (NHC) ligand units and reported the reaction of a trinuclear complex with Ag(I) ions to give a heptanuclear Pt3–Ag–Pt3 cluster with the formation of four Ag–Pt bonds.5 All metal ions in the heptanuclear complex with 106 CVEs fulfil a closed-shell configuration with six Pt–Pt and four Ag–Pt bonds, which compensates for 20 electrons, giving a total of 126 electrons for the seven closed-shell metal ions.
For the evaluation of metal–metal bonds, the number of d-electrons of metal centres are also important. Some of transition metal complexes with an odd number of d-electrons tend to dimerise giving diamagnetic dinuclear complexes with a intermetallic bond represented by a dimanganese decacarbonyl complex, in which the Mn–Mn bond was cleaved by reduction (Scheme 1(a)).6 Oxidation of transition metal complexes also leads the formation of an intermetallic bond, such as a diplatimun tetrakis(pyridinethiolato) complex with a PtIII–PtIII bond, which was obtained by oxidation of the diplatinum(II) complex (Scheme 1(b)).7
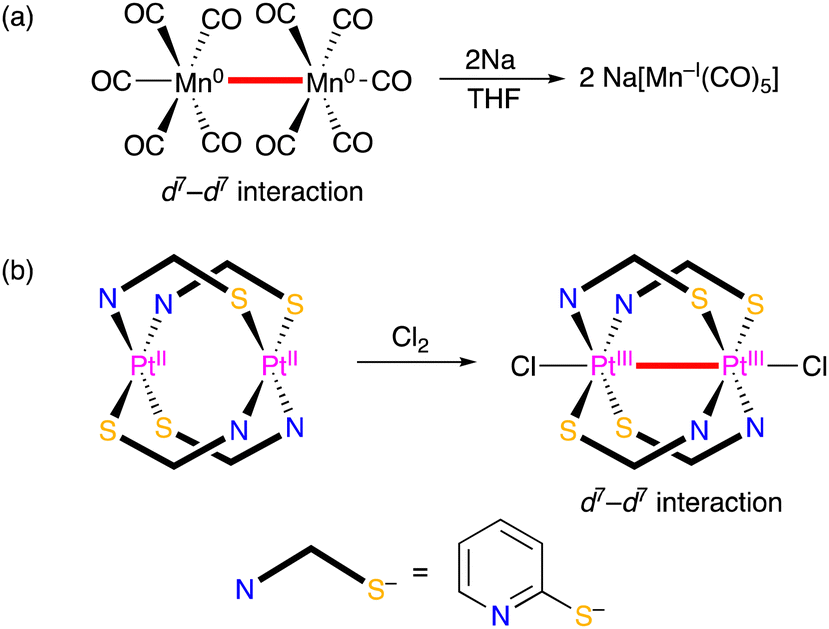 | ||
| Scheme 1 Examples for intermetallic bond (a) cleavage and (b) formation of transition metal complexes with redox reactions. | ||
Our previous work suggests that the incorporation of NHC ligands with the strong σ-donation ability into 48-CVE trinuclear complexes increases the electron densities of the trinuclear sulfide cores resulting in enhancement of their reactivity.5,8d,e Based on this fact, we planned to synthesize a 48-CVE trinuclear complex composed of one platinum(II)-bis-N-heterocyclic carbene (bisNHC) and two rhodium(I)-norbornadiene (nbd) units bridged by two sulfide ligands to obtain a more electron-rich metal-sulfide core. In this study, we demonstrate the formation of an unprecedented hexanuclear mixed-metal cluster, which has an additional metal–metal bond connecting the two PtRh2 trinuclear moieties, by oxidative coupling of the 48-CVE PtRh2 trinuclear complex. The redox behaviour of the hexanuclear cluster accompanied with the formation and cleavage of the intermetallic bond is also reported.
When a CHCl3 solution of a bis(hydrosulfido) platinum complex bearing a methylene-bridged bisNHC ligand was added to a reddish orange solution of rhodium nbd chloride dimer, the colour of the solution immediately changed to dark brown, and then a green reaction mixture was obtained. This colour change suggests that an intermediate was formed in the early stage of the reaction. X-ray diffraction study of the green product revealed a hexanuclear structure consisting of two trinuclear moieties connected by a Rh–Rh bond (Fig. 1). Due to the formation of the Rh–Rh bond, each of two trinuclear units is unsymmetric about the PtRh2 plane. Although the Rh–Rh bond between the trinuclear units (3.1042(6) Å) is relatively long, the 1H NMR spectrum of the hexanuclear cluster ([1]2+) in CD3OD exhibited four sharp signals of the 4- and 5-NHC protons and several nbd signals (Fig. S1 in ESI†), indicating that the structure of [1]2+, which is composed of the two unsymmetric trinuclear units, remains intact even in solution. Since the formal oxidation states of the metal centres in each trinuclear unit in [1]2+ should be Pt(II), Rh(I) and Rh(II), the RhII–RhII bond connecting the two trinuclear units is necessary for the diamagnetism. These facts suggest that the initial step in the reaction of the bis(hydrosulfido) platinum and rhodium nbd complexes is the formation of the 48-CVE PtIIRh2I trinuclear complex ([2]). The 48-CVE complex [2] is oxidised by molecular oxygen to afford the paramagnetic PtIIRhIRhII trinuclear complex radical ([2]˙+), which immediately dimerises with another 48-CVE complex [2] to hexanuclear complex radical [1]˙+, followed by rapid oxidation to the final product [1]2+ (Scheme 2). Supporting this fact, the same reaction under an inert atmosphere only gave a brown reaction mixture, the colour of which changed to green when the mixture was exposed to air. These reactions were monitored using 1H NMR spectroscopy (Fig. S4 in ESI†), and the resulting green solution contained mainly [1]2+, as confirmed by 1H NMR spectroscopy (Fig. S4(e) in ESI†) and ESI-MS (Fig. S5 in ESI†). The reactions of the bis(hydrosulfido) platinum complexes bearing a bisNHC ligand with other metal sources to give trinuclear complexes have been reported.5,8
 | ||
| Fig. 1 (a) Structure of the cationic moiety of mixed-metal hexanuclear cluster [1]Cl2. Hydrogen atoms are omitted for clarity. (b) Metal-sulfide core of [1]Cl2 with intermetallic distances (Å). | ||
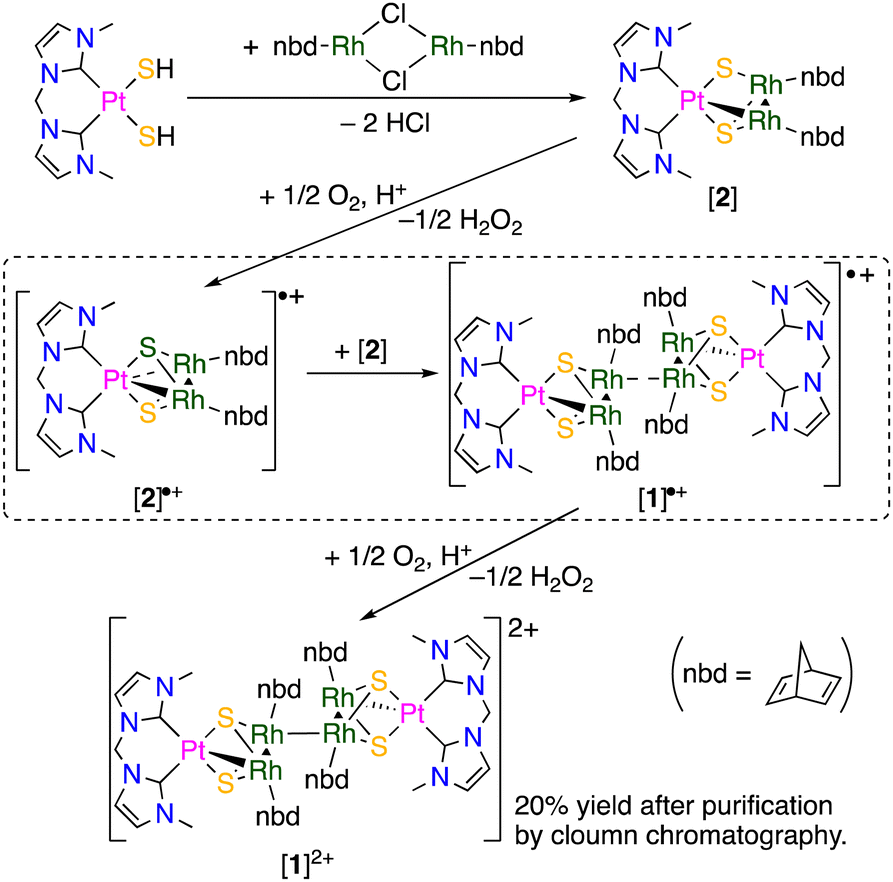 | ||
| Scheme 2 Formation and oxidative coupling of trinuclear precursor [2] affording hexanuclear cluster [1]2+via the formation of one-electron oxidised trinuclear intermediate [2]˙+ and hexanuclear precursor [1]˙+. | ||
To investigate the intermediates and the product, DFT calculations were performed for [1]2+, [2] and complex radical [2]˙+. The HOMO−2 of [1]2+ indicates a weak interunit bonding interaction between the two Rh centres of the trinuclear moieties (Fig. 2). The HOMO of the 48-CVE complex [2] and the SOMO of the complex radical [2]˙+ are similar to each other and are predominantly Rh–Rh antibonding (Fig. 3). By one-electron oxidation of [2] to [2]˙+, one electron is removed from the HOMO of [2], resulting in a significant shrinkage of the Rh–Rh bond from 3.112 to 2.933 Å, which shows two intermetallic bonds in [2] and two and half intermetallic bonds in [2]˙+. The spin density of [2]˙+ is delocalised on the two Rh centres (Fig. 4). The two complex monocations share the unpaired electrons on the Rh centres, leading to the formation of an interunit Rh–Rh bond to afford the diamagnetic hexanuclear cluster [1]2+.
 | ||
| Fig. 2 (a) Optimised structure and (b) HOMO−2 of [1]2+. Pt: beige, Rh: green, S: yellow, N: blue, C: grey and H: white. | ||
 | ||
| Fig. 3 Optimised structures of (a) 48-CVE complex [2], (b) one-electron oxidised complex radical [2]˙+, (c) HOMO of [2] and (d) SOMO of [2]˙+. Pt: beige, Rh: green, S: yellow, N: blue, C: grey and H: white. | ||
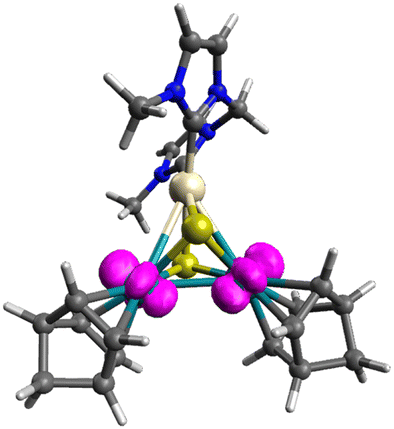 | ||
| Fig. 4 Spin density of [2]˙+ (magenta). Pt: beige, Rh: green, S: yellow, N: blue, C: grey and H: white. | ||
The backward reaction of the hexanuclear cluster to the trinuclear complex by reduction was examined using cyclic voltammetry. The hexanuclear cluster [1]2+ was reduced around −1.2 V vs. Fc+/Fc, and a reoxidation wave appeared around −0.4 V (Fig. 5). When the scan was reversed before the reduction wave, the reoxidation wave was not observed. This fact clearly indicates that the two-electron reduced hexanuclear cluster [1] (Scheme 3(a)) is unstable and the subsequent dissociation reaction proceeds to give the 48-CVE complex [2] (Scheme 3(b)). The reoxidation is probably due to the oxidation of the trinuclear 48-CVE complex [2] to the paramagnetic complex radical [2]˙+ (Scheme 3(c)). However there is no rereduction wave corresponding to the reduction of the paramagnetic complex [2]˙+ in the repeated scans (Fig. 5). This fact means that the paramagnetic complex is unstable and readily gives the hexanuclear cluster [1]˙+ by the reaction with [2] (Scheme 3(d)). The reoxidation potential of [1]˙+ should be more negative and the complex immediately oxidised to [1]2+(Scheme 3(e)). The large difference in the potentials of the reduction and reoxidation waves can be attributed to the dissociation and association between the hexanuclear and trinuclear clusters. The hexanuclear cluster is presumably stabilised against reduction by the interunit Rh–Rh bond formation. Each of other previously reported 48-CVE MM2′ complexes, [{M(bisNHC)}(M′Cp*)2(μ3-S)2]2+ (M = Pd, Pt; M′ = Rh, Ir), exhibit two reversible redox couples corresponding to [48-CVE]/[49-CVE] and [49-CVE]/[50-CVE] in CV measurements.8b,c These observations indicate that the paramagnetic species of these complexes are relatively stable and no dimerisation proceeds probably due to the bulkier Cp* ligands.
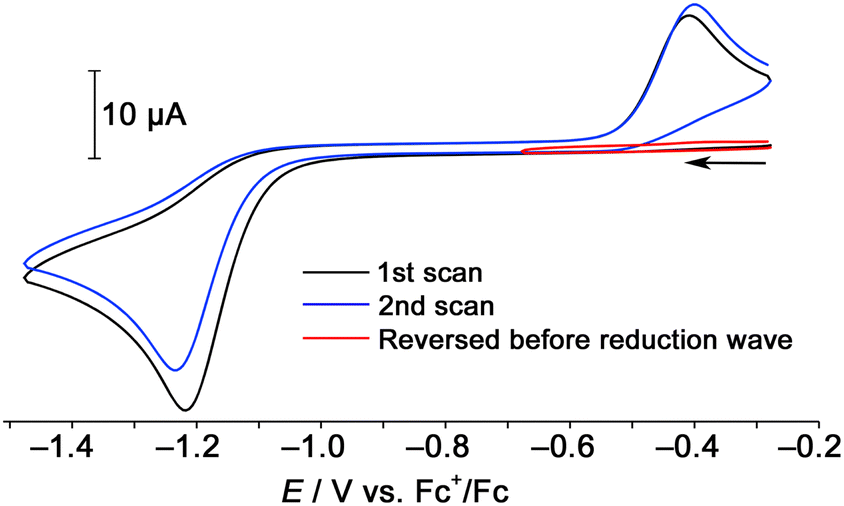 | ||
| Fig. 5 Cyclic voltammograms of [1]Cl2 in CH3CN with 0.10 mol L−1 of nBu4NPF6 (Scan rate: 100 mV s−1). | ||
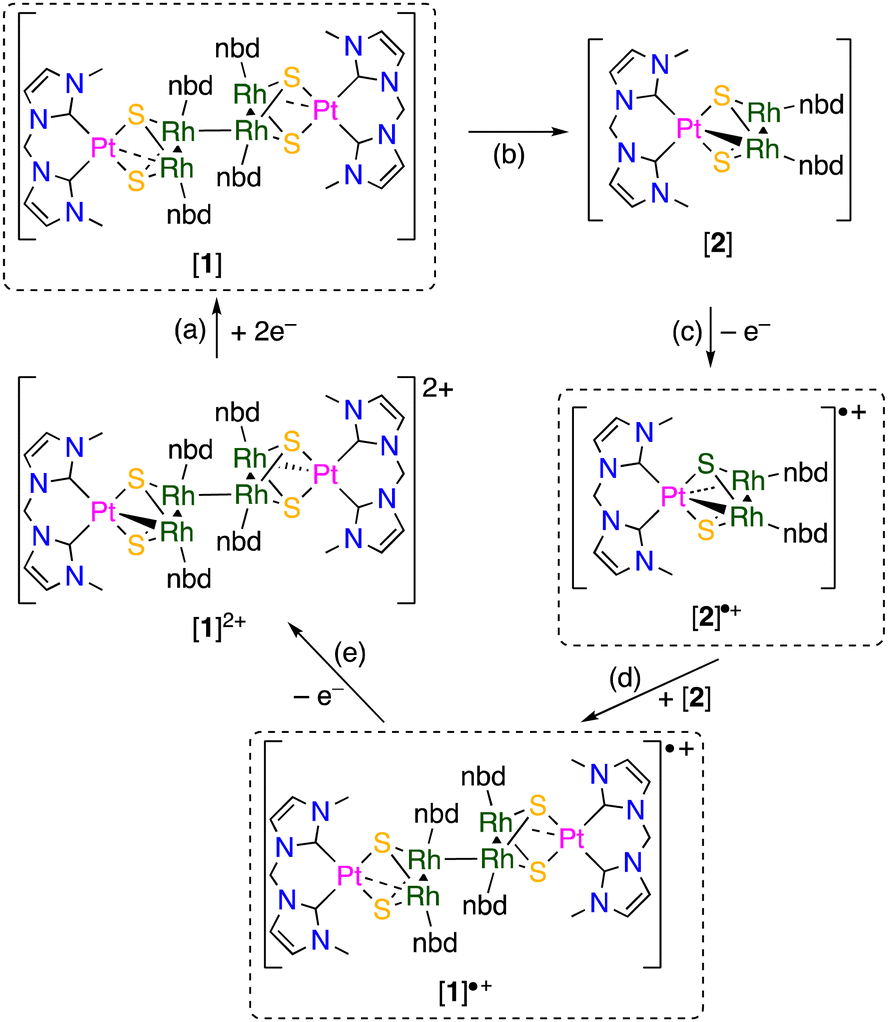 | ||
| Scheme 3 ECECE process on reduction and reoxidation scans of [1]Cl2; (a) two-electron reduction affording a neutral hexanuclear complex, (b) subsequent reaction giving trinuclear 48-CVE complex [2], (c) reoxidation of the 48-CVE complex affording the complex radical [2]˙+, (d) reaction of [2]˙+ with [2] and (e) reformation of [1]2+ by one-electron oxidation of [1]˙+. | ||
To clarify the reductive dissociation and oxidative coupling between the hexanuclear [1]2+ and trinuclear [2] complexes, the chemical reduction of [1]2+ and aerobic reoxidation were investigated. The 1H NMR spectrum of the mixture of [1]Cl2 and 2 eq. of decamethylcobaltocene in CD3CN under an inert atmosphere showed two doublet for the 4,5-NHC protons, broad signals for the nbd ligands and a singlet for the bridging methylene of the bisNHC ligand, which can be assigned to the averaged signals for the 48-CVE trinuclear complex due to the dynamic behaviour of the bisNHC ligand (Fig. 6(a)).8c,d After exposing the mixture to air, the colour of the mixture immediately changed to green. The 1H NMR spectrum of the reoxidation product exhibited the signals of the hexanuclear cluster (Fig. 6(b)). These results clearly indicate that the hexanuclear cluster [1]2+ dissociates into the 48-CVE trinuclear complexes [2] via two-electron reduction, and [2] is oxidised in air to give the hexanuclear cluster, which is consistent with the observations from the CV measurements.
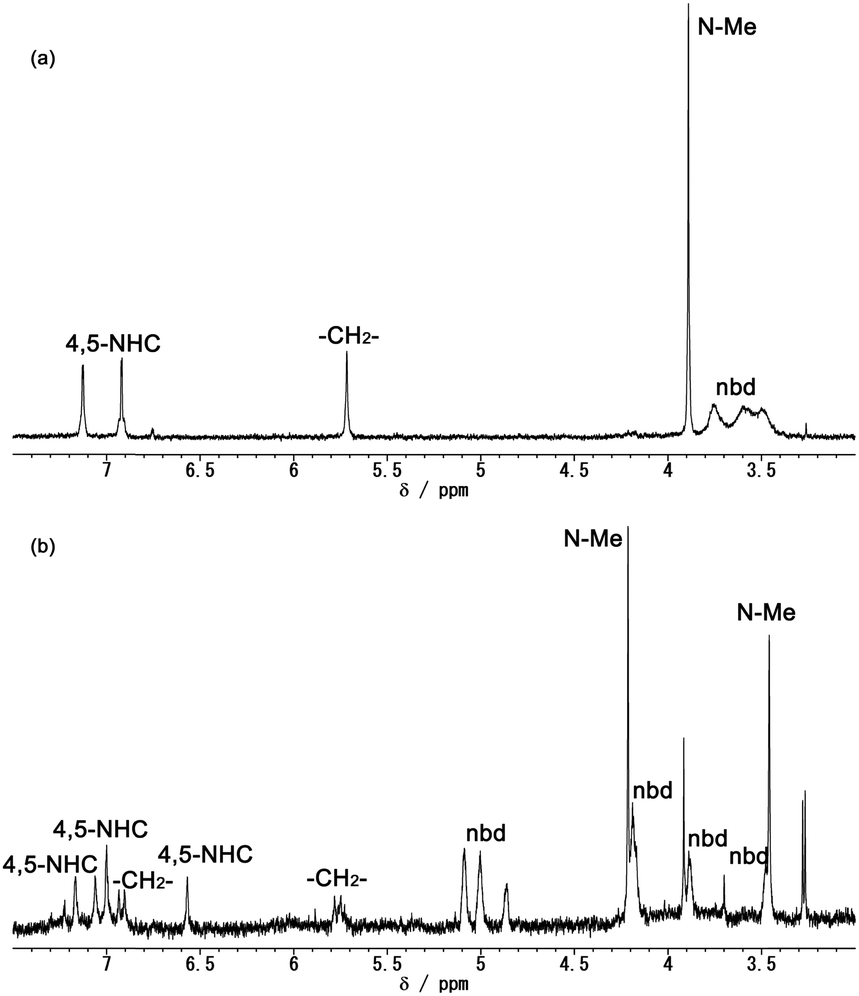 | ||
| Fig. 6 1H NMR spectra of the reaction mixture of [1]Cl2 and 2 eq. of decamethylcobaltocene (a) under an inert atmosphere and (b) after exposing to air. (CD3CN, 400 MHz, 293 K) (see Fig. S8 in ESI† for the whole spectra). | ||
In conclusion, we have demonstrated unprecedented virtual radical coupling of mixed-metal trinuclear complexes via interunit metal–metal bond formation to afford a hexanuclear cluster induced by aerobic oxidation. The hexanuclear cluster is stabilised against reduction by the coupling of trinuclear complex radicals and dissociates into two trinuclear complexes upon reduction. The reduced species appears to be sufficiently reactive to provide reaction sites on the metal core for small molecules. The investigation for reactions of organic halides and the reduced species at the triply bridging sulfide ligands to afford C–S coupling products is in progress for novel applications of such cluster compounds as catalysts.
A. Y. performed the experiments and collected and analysed the data with supervision from T. N.; T. N. conceived the idea, designed the experiments and wrote the manuscript; H. N. discussed the data and reviewed and corrected the manuscript.
This work was supported by JSPS KAKENHI Grant Number 22K05146.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for the hexanuclear complex has been deposited at the CCDC under 2411823 and can be obtained from https://www.ccdc.cam.ac.uk.Conflicts of interest
There are no conflicts to declare.Notes and references
- P. Atkins, T. Overton, J. Rourke, M. Weller and F. Armstrong, Shriver & Atkins Inorganic Chemistry, Oxford University Press, Oxford, UK, 2006, pp. 557–558 Search PubMed.
- (a) C. R. Pulliam, J. B. Thoden, A. M. Stacy, B. Spencer, M. H. Englert and L. F. Dahl, J. Am. Chem. Soc., 1991, 113, 7398–7410 CrossRef CAS; (b) A. Venturelli and T. B. Rauchfuss, J. Am. Chem. Soc., 1994, 116, 4824–4831 CrossRef CAS; (c) T. Nishioka and K. Isobe, Chem. Lett., 1994, 1661–1664 CrossRef CAS.
- (a) M. Sorai, A. Kosaki, H. Suga, S. Seki, T. Yoshida and S. Otsuka, Bull. Chem. Soc. Jpn., 1971, 44, 2364–2371 CrossRef CAS; (b) A. B. Rives, Y. Xiao-Zeng and R. F. Fenske, Inorg. Chem., 1982, 21, 2286–2294 CrossRef CAS.
- (a) Y. Kushi, H. Nagao, T. Nishioka, K. Isobe and K. Tanaka, Chem. Lett., 1994, 2175–2178 CrossRef CAS; (b) Y. Kushi, H. Nagao, T. Nishioka, K. Isobe and K. Tanaka, J. Chem. Soc., Chem. Commun., 1995, 1223–1224 RSC; (c) K. Tanaka, Y. Kushi, K. Tsuge, K. Toyohara, T. Nishioka and K. Isobe, Inorg. Chem., 1998, 37, 120–126 CrossRef CAS PubMed.
- N. Yabune, H. Nakajima and T. Nishioka, Dalton Trans., 2020, 49, 7680–7683 RSC.
- P. Atkins, T. Overton, J. Rourke, M. Weller and F. Armstrong, Shriver & Atkins Inorganic Chemistry, Oxford University Press, Oxford, UK, 2006, p. 546 Search PubMed.
- K. Umakoshi, I. Kinoshita, A. Ichimura and S. Ooi, Inorg. Chem., 1987, 26, 3551–3556 CrossRef CAS.
- (a) Y. Maeda, H. Hashimoto and T. Nishioka, Dalton, 2012, 41, 12038–12048 RSC; (b) Y. Maeda, H. Hashimoto, I. Kinoshita and T. Nishioka, Inorg. Chem., 2014, 53, 661–663 CrossRef CAS PubMed; (c) Y. Maeda, H. Hashimoto, I. Kinoshita and T. Nishioka, Inorg. Chem., 2015, 54, 448–459 CrossRef CAS PubMed; (d) N. Yabune, H. Nakajima and T. Nishioka, RSC Adv., 2024, 14, 29355–29367 RSC; (e) N. Yabune, H. Nakajima and T. Nishioka, Dalton Trans., 2021, 50, 12079–12082 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data, experimental details, NMR and mass spectra, cyclic voltammograms and DFT calculations. CCDC 2411823. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00112a |
| This journal is © The Royal Society of Chemistry 2025 |