
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bicyclic sulfonium pincer ligand with a thiatriptycenium backbone-synthesis and applications in π-acid catalysis†
Mohammad
Zafar
 ,
Donia
Shamali
,
Nitsan
Barel
,
David
Danovich
and
Yuri
Tulchinsky
*
,
Donia
Shamali
,
Nitsan
Barel
,
David
Danovich
and
Yuri
Tulchinsky
*
Institute of Chemistry, the Hebrew University of Jerusalem, Jerusalem, Israel. E-mail: yuri.tulchinsky@mail.huji.ac.il
First published on 10th February 2025
Abstract
A bicyclic PSP-pincer ligand featuring a sulfonium cation at the bridgehead position of its rigid triptycene-like scaffold was synthesized and metallated with a bis-cationic Pt(II) center. The resulting tris-cationic Pt(II) complex exhibits excellent photostability and improved catalytic performance compared to its analogue with the previously reported sulfonium pincer ligand.
Triptycenes, bicyclic molecules featuring a rigid paddlewheel-shaped skeleton, were first prepared in the early 1940s,1 and have since found extensive applications as key structural elements in materials chemistry,2 catalysis,3 and supramolecular systems,4 including sensors and molecular machines. Replacing the apical methyne group in these molecules with boron,5 nitrogen6 or phosphorus7 atoms results in the corresponding aza-, bora-, and phosphatriptycene derivatives, which have also been prepared, though they remain much less explored (Scheme 1a). Within this series, only the triptycene and the phosphatriptycene were shown to engage their apical atom in coordinative bonding of transition metals.8 Specifically, chelation-assisted activation of the bridgehead Csp3–H bond in triptycene-based PCP pincer ligands, first prepared and extensively studied by Gelman,9 resulted in highly thermally robust complexes (Scheme 1b). Among these, Ir(III)-triptycene complexes exhibited an excellent catalytic performance in transfer hydrogenation of ketones and transfer dehydrogenation of alkanes, as demonstrated by Gelman9a and Brookhart.10 Phosphatriptycenes, on the other hand, have seen little use as ligands in homogeneous catalysis, despite being known for 50 years.7a Due to a significant ring strain imposed by their bicyclic scaffold, the apical P atom in these compounds is highly pyramidalized, which increases the s character of its lone pair. Consequently, phosphatriptycenes are somewhat weaker electron donors compared to the non-cyclic phosphines, although assessment of their Tollman electronic parameter in a Rh(acac)CO system (Scheme 1c) suggested only a small difference (7 cm−1 at most).11 Indeed, when a phosphatriptycene ligand was employed in Pd-catalyzed reactions benefitting from an electron-poor metal center, it offered only a modest improvement over other weak donors, such as trifurylphosphine and triphenylarsine.12
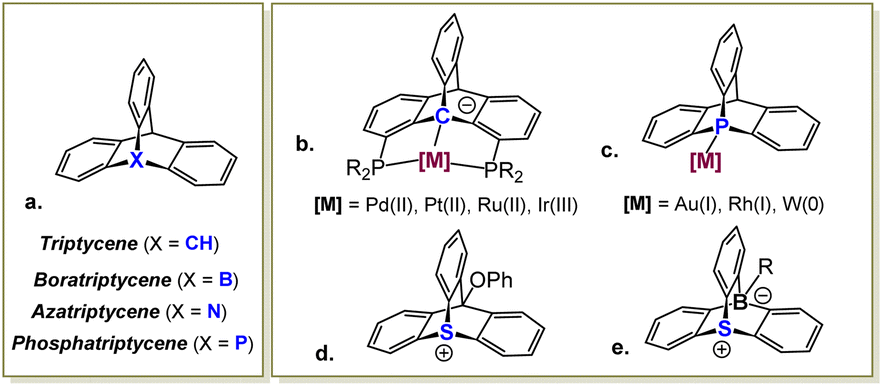 | ||
| Scheme 1 General structure of a triptycene molecule and its heteroatom-based analogues (a), transition metal complexes of triptycene (b) and phosphatriptycene (c), the only previously reported thiatriptycenium cation (d), and a zwitterionic 9-sulfonium-10-boratriptycene (e). | ||
A while ago, we introduced a new class of positively-charged pincer ligands13 based on sulfonium cations, including ligand L1 (Scheme 2a), which provided the first example of an aromatic sulfonium cation coordinated to transition metals (Rh(I) and Pt(II)).14 Computational analysis of these complexes proved the sulfonium centers in these ligands to be highly π-acidic. Yet, L1 was not suitable for the use as an ancillary ligand in π-acid catalysis due to the excessive flexibility of its backbone, which resulted in a hemilabile behaviour15 and an easy dissociation of the metal–sulfonium bond.
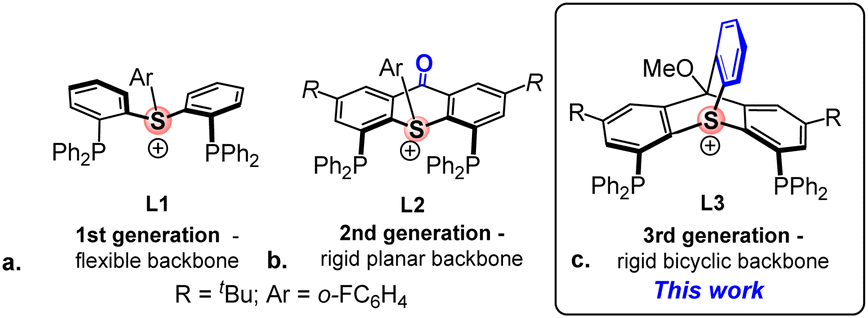 | ||
| Scheme 2 1st, 2nd, and 3d generations of the aromatic sulfonium pincer ligands ((a), (b), and (c), respectively). | ||
To address this issue, we designed the 2nd generation sulfonium ligand, L2, where the phenyl rings of the backbone are bridged by a carbonyl group (Scheme 2b). The rigidity of the resulting thioxanthone-based backbone proved imperative for stabilizing the highly electrophilic tris-cationic sulfonium–Pt(II) complex, which showed an excellent performance as a π-acid catalyst in carbophilic cycloisomerization reactions.16 Unfortunately, L2 was found to be extremely light-sensitive, both as a free ligand and when coordinated to Pt(II). This outcome was hardly surprising, given that aromatic sulfonium cations are known to decompose upon irradiation into diaryl sulfides and highly reactive aryl cations, a property which made them widely employed as photoacids in nanolithography.17
It then occurred to us that placing the sulfonium center at the bridgehead position of a triptycene-type framework might significantly enhance the photostability of our ligand, while also imparting the necessary rigidity to its backbone to prevent the S–Pt bond dissociation. We now describe the synthesis of such a bicyclic compound, the 3rd generation sulfonium ligand, L3 (Scheme 2c), and report its improved performance in π-acid catalysis.
To date, only a single cationic trypticene analogue with a sulfur atom at its bridgehead position was reported. This compound (Scheme 1d) was synthesized via a formal [4+2] cycloaddition of a benzyne synthon to a thioxanthone.18 We therefore hypothesized that an analogous bicyclic scaffold could also be prepared from a bis-phosphineoxide-functionalized thioxanthone derivative 1 (Scheme 3a), which we had reporter earlier.16 Indeed, treating 1 with 2-(trimethylsilyl)phenyl triflate in presence of TBAF led to the formation of a cycloaddition product, [2](OTf). This transformation was immediately apparent from the absence of the characteristic carbonyl stretch in IR at ca. 1630 cm−1, as well as the appearance of 4 new 1H NMR signals (vide infra), attributed to the newly added arene moiety (Fig. S1, ESI†). Concurrently, the lowest field signal in 13C NMR corresponding to the carbonyl carbon of 1 shifted nearly 100 ppm upfield (as verified by the 1H–13C HMBC, see Fig. S7, ESI†). Protecting the tertiary alcohol group in [2](OTf) as a methyl ether, followed by phosphine oxide reduction, afforded the desired bicyclic ligand [4](OTf) (Scheme 3a). For the subsequent catalytic studies two anion exchanged analogues, [4](BF4) and [4](PF6) were also prepared.
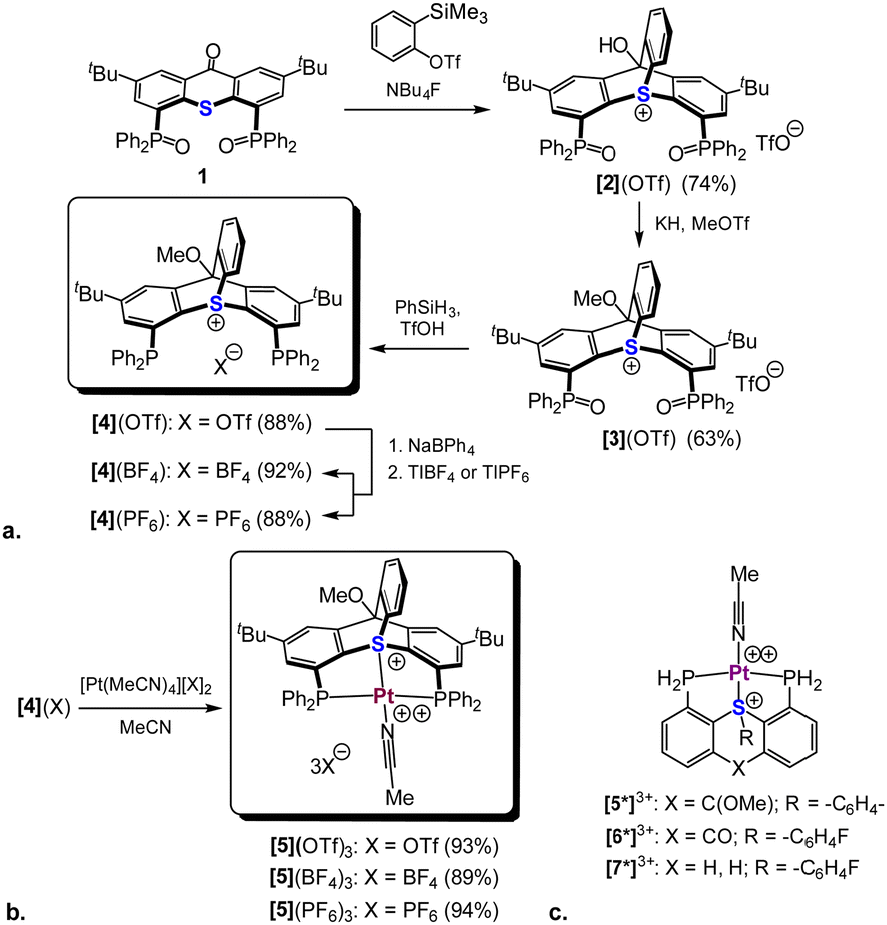 | ||
| Scheme 3 Synthesis of the thiatriptycenium-based ligands [4](OTf), [4](BF4), and [4](PF6) (a) the corresponding tris-cationic Pt(II) complexes (b), and model Pt(II) complexes of sulfonium ligands L3 ([5]3+), L2 ([6]3+), and L1 ([7]3+) (c). | ||
The 1H NMR of [4](OTf) revealed the benzyne-derived arene group (protons a–d) as a set of four distinct signals at δ = 8.05, 7.58, 7.09, and 6.05 ppm showing strong cross-correlations in 1H–1H COSY spectrum (Fig. 1a). In addition, the bridgehead carbon resonating at δ = 88.6 ppm in 13C NMR showed a clear correlation in 1H–13C HMBC with both the methoxy protons (δ = 4.38 ppm) and the aromatic protons e (δ = 8.05 ppm) of [4](OTf) (Fig. S14, ESI†).
 | ||
| Fig. 1 Comparision of 1H–1H COSY spectra (aromatic region only) of the thiatriptycenium ligand [4](OTf) (a) and its Pt(II)(MeCN) complex [5](OTf)3 (b). | ||
The bicyclic backbone of [4](OTf) was further confirmed by XRD (Fig. 2a). For the best of our knowledge, [4](OTf) represents the first crystallographyically characterized sulfur-based trypticene analog, which we suggest to call thiatriptycenium. Similarly to phosphatroptycenes, the apical S atom in [4](OTf) is significantly more pyramidalized compared to that in a simple triphenylsulfonium cation19 or in its phosphine-functionalized analog L114 (Σangles = 291.26° vs. 314.39°, respectively). On the other hand, the C–S bond lengths of 1.788 Å (av.), typical to other sulfonium cations, appear unaffected by the ring strain. In fact, similar geometric features were observed in the zwitterionic 9-sulfonium-10-boratriptycenes recently introduced by Chardon20 (Scheme 1e).
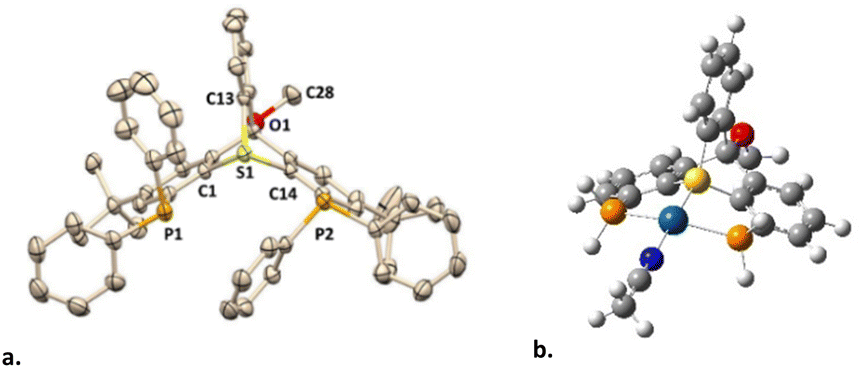 | ||
| Fig. 2 The XRD structure of ligand [4](OTf), H atoms and counteranion are omitted for clarity (a) and the optimized geometry of complex [5*]3+ (b). | ||
Prior to complexation of [4](OTf), we tested the photostability of this ligand by exposing its CDCl3 solution in an NMR tube to ambient daylight. To our delight, this compound showed no noticeable decomposition even after 5 days, in a stark contrast to the 2nd generation sulfonium-based ligand, which fully decomposed within 3 h of a similar light exposure (Fig. S42a and b, respectively, ESI†)!
Reaction of ligand [4](OTf) with an equimolar amount of the [Pt(CH3CN)4](OTf)2 precursor in MeCN resulted in a clean formation of a single product, [5](OTf)3 (Scheme 3b). This compound was identified as a symmetric complex with a κ3-P,S,P coordination mode based on a single peak in the 31P NMR at δ = 18.1 ppm (1JPt–P = 2198 Hz) and the characteristic virtual triplet pattern of the ipso-carbons21 in the 13C NMR spectrum (Fig. S28 and S30, respectively, ESI†). Most notably, in the 1H NMR the signal attributed to proton d, adjacent to the sulfonium nucleus, shifted from δ = 6.05 to 8.09 ppm, compared to the free ligand (Fig. 1b), a downfield shift considerably larger than those observed for the rest of the aromatic protons (0.3–0.5 ppm). Such a pronounced deshielding strongly supports coordination of the sulfur atom to a highly electrophilic Pt2+ center in this compound. The fourth coordination site in [5](OTf)3 (assuming a typical square planar geometry of the Pt(II) center) is most likely occupied by a weakly coordinated MeCN. Although anion coordination cannot be completely ruled out, it appears less likely, based on the 19F NMR of this complex, which shows only a single sharp peak at δ = −78 ppm, characteristic of a free triflate anion (Fig. S29, ESI†). Furthermore, analogues of [5](OTf)3 with non-coordinative anions, which were prepared for comparison (Scheme 3b), exhibited nearly identical 31P and 1H NMR spectra.
Despite multiple attempts, no XRD-quality single crystals of complex [5](OTf)3 (or its anion-exchanged analogues) could be grownprompting us to resort to DFT calculations (ωB97X-D3/Def2-TZVP). The Pt-coordination sphere of the optimized model complex [5*]3+ (Scheme 3c and Fig. 2b) closely matched the one previously calculated for the analogous model complex [6*]3+ of the 2nd generation ligand16 (see Table S2 for a detailed comparison, ESI†). Given the excellent agreement between the optimized geometry of the latter and the XRD structure of [6](BF4)3 (Scheme 4a), we believe that the true geometry of [5](OTf)3 is also reliably represented by its optimized model.
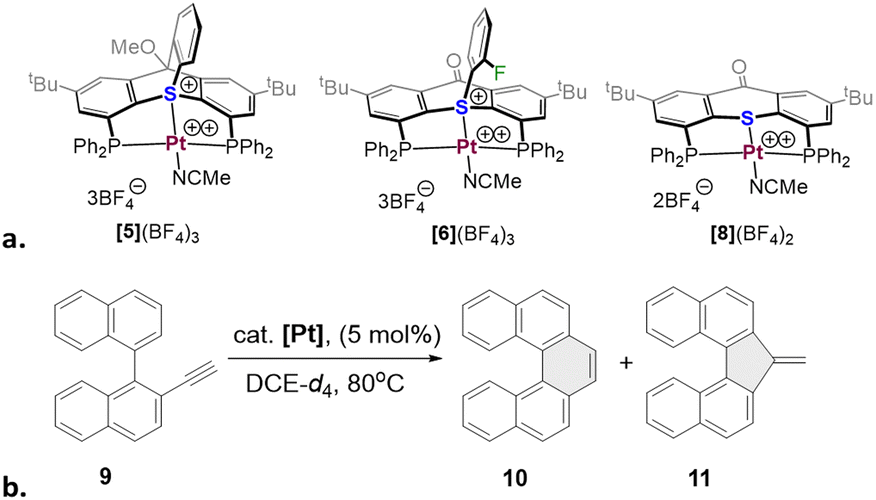 | ||
| Scheme 4 Structures of Pt(II) catalysts with thiatriptycenium-, sulfonium-, and thioether-based pincer ligands (left to right), (a) and the benchmark catalytic cycloisomerization of 2-ethynyl-1,1′-binapthalene 9 (b). | ||
Using this geometry, we employed computational methods to quantitatively compare the electronic properties of the three generations of our sulfonium ligands, including an optimized model complex [7*]3+ (Scheme 1c), representing a synthetically inaccessible15 putative Pt(MeCN) complex of ligand L1 (Table S3, ESI†). While the EDA NOCV revealed no significant differences between the three model complexes ([5*]3+, [6*]3+, and [7*]3+) in terms of type and strength of L–M orbital interactions, it indicated that ligand L3 is the weakest σ-donor and the strongest π-acceptor in this series. As a result, a positive charge on the Pt atom found by NBO was slightly higher in [5*]3+ (+ 0.421), than in its counterparts (+ 0.407 and +0.395 in [6*]3+ and [7*]3+, respectively).
Encouraged by these computational results, we proceeded to the catalytic studies, focusing on complex [5](BF4)3. This complex was selected since it shares the anions with the analogous sulfonium and thioether complexes we reported earlier ([6](BF4)3 and [8](BF4)2, Scheme 4a), and therefore is best suited for evaluating the catalytic efficiency of the thiatriptycenium ligand. For this we employed the same benchmark reaction and substrate (9), as studied before with the 2nd generation sulfonium ligand (Scheme 4b and Table 1).
| Entry | Catalyst (5 mol%) | Time (min or h) | Conversion (%) | Isolated yield (%) (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11 ratio) :11 ratio) |
|---|---|---|---|---|
| a Data we reported previously (see ref. 16). b Without protection from light. | ||||
| 1 | [5](BF4)3 | 3 h | 100 | 92 (100:0) |
| 2 | [6](BF4)3 | 6 ha | 100a | 97 (90:10)a |
| 3 | [8](BF4)2 | 24 ha | 7a | n.d.a |
| 4 | [6](BF4)3 | 24 hb | 63b | n.d.b |
| 5 | [5](OTf)3 | 50 min | 100 | 88 (90:10) |
| 6 | [5](PF6)3 | 6 h | 40 | n.d. |
Gratifyingly, complex [5](BF4)3 proved to be an excellent catalyst for this reaction, achieving a full conversion within 3 h and producing the pentahelicene 10 as the sole product in a high yield (92%). With an isolated yield only slightly lower than that reported for [6](BF4)3, complex [5](BF4)3 offers full regioselectivity and reduces the overall reaction time into half (Table 1, entries 1 and 2). Moreover, as evident from Fig. 3, the initial reaction rate with [5](BF4)3 is nearly 7 times faster, reaching 50% conversion in only ∼15 min compared to ∼105 min, required for a similar conversion with complex [6](BF4)3.
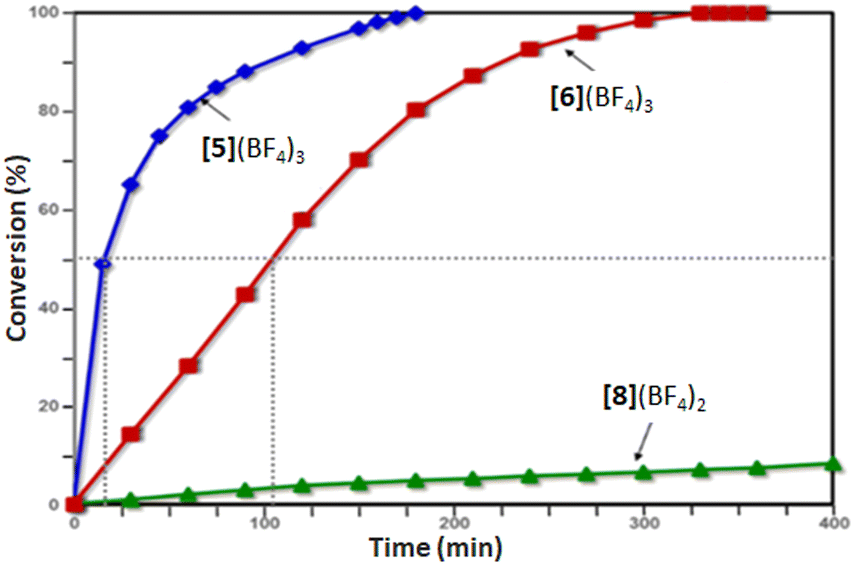 | ||
| Fig. 3 Ligand effect on the rate of Pt-catalyzed cycloisomerization of 9. The dashed lines highlight the difference in time required to achieve 50% conversion with the 2nd and 3rd generation ligands. | ||
Next, we evaluated the light sensitivity of [6](BF4)3 by comparing its catalytic performance in this reaction under light-protected and ambient daylight conditions. The results revealed nearly identical reaction kinetics and product distribution (single product) in both cases (Fig. S49a, ESI†). In contrast, the performance of [6](BF4)3 under ambient light was strongly compromised, achieving only 63% conversion after 24 hours (Table 1, entry 4).
To study the influence of the anions, known to have non-negligible effects on the reactivity of cationic π-acid catalysts as shown in several experimental22 and theoretical23 studies, we employed complexes [5](OTf)3 and [5](PF6)3. Indeed, with [5](OTf)3 the reaction was complete in less than an hour; yet, this rate acceleration was impaired by formation of the 5-exo isomer 11, as a minor product. The [5](PF6)3 analogue, on the other hand, did not affect the regioselectivity, but drastically slowed down the reaction progress (Table 1, entries 5, 6 and Fig. S49b, ESI†).
To evaluate the substrate scope of this reaction, the reactivity of complex [5](BF4)3 was tested on a representative set of three other o-alkynyl biaryls, 12a–c (Scheme 5a). As with substrate 9, only the corresponding 6-endo products, 13a–c, were obtained. While the electron-rich and electronically-neutral substrates (12a and 12b, respectively) afforded the products in good yields, presence of an electron-withdrawing substituent in 12c limited the conversion to only 40% after 24 h (Table 2, entries 1–3).
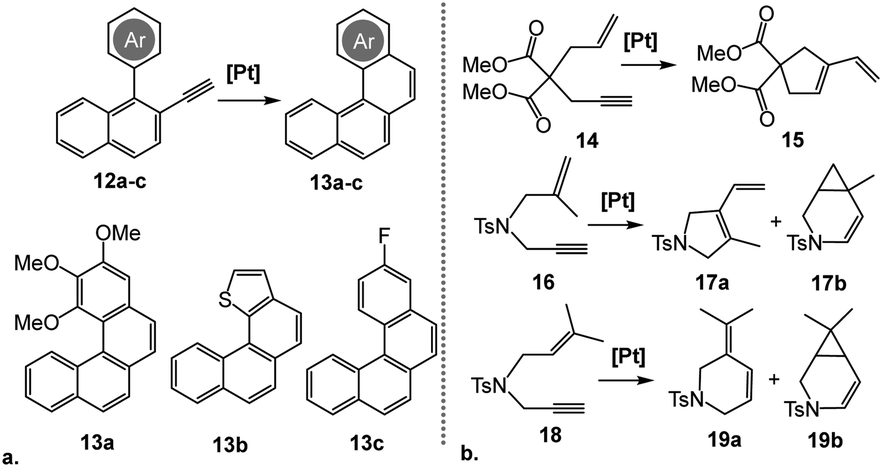 | ||
| Scheme 5 Catalytic cycloisomerization of representative o-alkynylated biaryls 12a–c (a) and enyne substrates 14, 16 and 18 (b). Reaction conditions: 5 mol% [Pt], DCE-d4, 80 °C. | ||
| Entry | Products | Time (min or h) | Conversion (%) | Isolated yield (%) (product ratio) |
|---|---|---|---|---|
| 1 | 13a | 50 min | 100 | 88 |
| 2 | 13b | 6 h | 100 | 82 |
| 3 | 13c | 24 h | 40 | n.d. |
| 4 | 15 | 55 min | 100 | 92 |
| 5 | 17a,b | 1 h | 100 | 86 (44:56) |
| 6 | 19a,b | 45 min | 100 | 84 (76:24) |
We also tested the performance of [5](BF4)3 in several enyne cyclization reactions carried out under the same conditions (Scheme 5b), all of which reached a full conversion within an hour. Cycloisomerization of a dimethyl-malonate substrate 14 proceeded smoothly, yielding exclusively the 5-exo product 15 (Table 2, entry 4). Conversely, the tosylamide-based substrates featuring gem-disubstituted and trisubstituted alkenes, 16 and 18, respectively, formed mixtures of the corresponding 5- or 6-membered cyclic dienes, 17a or 19a, with the cyclopropane-annulated products, 17b or 19b (Table 2, entries 5 and 6).
All in all, the thiatrypticenium-based ligand L3 demonstrated high activity in Pt(II)-catalyzed cycloisomerizations, being on par with the α-cationic phosphines and arsines studied by Alcarazo,24 but perhaps leaving room for improvement compared to certain Z-type antimony(V)-based ligands reported by Gabbai.25 We believe that the rigidity and stability of this novel ligand framework provides a promising platform for further structural fine-tuning, that might lead to even greater performance in π-acid catalysis.
This research was supported by the Israel Science Foundation (Grant 2079/22), for which we express our sincere gratitude.
Data availability
Synthetic protocols, multinuclear NMR spectra, crystallographic information, and computational data is available in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- P. D. Bartlett, et al., J. Am. Chem. Soc., 1942, 64, 2649–2653 CrossRef CAS.
- (a) L. Zhao, et al., Chem. Lett., 2010, 39, 658–667 CrossRef CAS; (b) J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301–3315 RSC; (c) M.-J. Gu, et al., Org. Biomol. Chem., 2021, 19, 10047–10067 RSC.
- (a) F. K.-C. Leung, et al., ACS Omega, 2017, 2, 1930–1937 CrossRef CAS PubMed; (b) H. Ube, et al., Nat. Commun., 2017, 8, 14296 CrossRef CAS PubMed.
- (a) C.-F. Chen, Chem. Commun., 2011, 47, 1674–1688 RSC; (b) C.-F. Chen and Y. Han, Acc. Chem. Res., 2018, 51, 2093–2106 CrossRef CAS PubMed.
- A. Chardon, et al., Angew. Chem., Int. Ed., 2020, 59, 12402–12406 CrossRef CAS PubMed.
- D. J. Kreil and V. R. Sandel, J. Chem. Eng. Data, 1976, 21, 132–133 CrossRef CAS.
- (a) C. Jongsma, et al., Tetrahedron, 1974, 30, 3465–3469 CrossRef CAS; (b) L. Hu, et al., J. Org. Chem., 2019, 84, 11268–11274 CrossRef CAS PubMed; (c) D. Mahaut, et al., J. Phys. Chem. A, 2022, 126, 2794–2801 CrossRef CAS PubMed.
- H. Gildenast, et al., Dalton Trans., 2022, 51, 7828–7837 RSC.
- (a) C. Azerraf and D. Gelman, Chem. – Eur. J., 2008, 14, 10364–10368 CrossRef CAS PubMed; (b) C. Azerraf and D. Gelman, Organometallics, 2009, 28, 6578–6584 CrossRef CAS; (c) I. Kisets and D. Gelman, Organometallics, 2018, 37, 526–529 CrossRef CAS.
- D. Bézier and M. Brookhart, ACS Catal., 2014, 4, 3411–3420 CrossRef.
- L. Hu, et al., Dalton Trans., 2021, 50, 4772–4777 RSC.
- T. Agou, et al., Chem. Lett., 2004, 33, 1028–1029 Search PubMed.
- M. Zafar, et al., Chem. Commun., 2024, 60, 9871–9906 RSC.
- R. Li, et al., Chem. Sci., 2022, 13, 4770–4778 RSC.
- R. Li, et al., Organometallics, 2023, 42, 246–258 CrossRef CAS.
- R. Li, et al., Angew. Chem., Int. Ed., 2024, 63, e202314997 CrossRef CAS PubMed.
- F. Dumur, Polymers, 2023, 15, 4202 CrossRef CAS PubMed.
- L. Zhang, et al., Org. Biomol. Chem., 2017, 15, 7181–7189 RSC.
- T. M. Klapotke, et al., Z. Naturforsch., 2009, 64b, 467–469 Search PubMed.
- D. M. A. Osi, et al., Angew. Chem., Int. Ed., 2022, 61, e202112342 CrossRef PubMed.
- V. Subramaniyan, et al., Inorg. Chem., 2022, 62, 123–136 CrossRef PubMed.
- (a) Z. Lu, et al., Chem. Rev., 2021, 121, 8452–8477 CrossRef CAS PubMed; (b) J. Schießl, et al., Adv. Synth. Catal., 2018, 360, 2493–2502 CrossRef.
- G. Ciancaleoni, et al., ACS Catal., 2015, 5, 803–814 CrossRef CAS.
- (a) C. J. Rugen and M. Alcarazo, Synlett, 2022, 16–26 CAS; (b) J. W. Dube, et al., J. Am. Chem. Soc., 2016, 138, 6869–6877 CrossRef CAS PubMed.
- D. You and F. P. Gabbaï, Trends Chem., 2019, 1, 485–496 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2416301. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00310e |
| This journal is © The Royal Society of Chemistry 2025 |