
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron appended Ru-NHC catalyzed selective deoxygenative hydrogenation of tertiary amides and desulfurization of thioamides†
Raju Sharmaa,
Gowtham Kenguvab,
Rambabu Dandelab and
Prosenjit Daw *a
*a
aDepartment of Chemical Sciences, Indian Institute of Science Education and Research Berhampur, Transit Campus, (Govt. ITI Building), Engg. School Junction, Berhampur 760010, Odisha, India. E-mail: pdaw@iiserbpr.ac.in
bDepartment of Industrial and Engineering Chemistry, Institute of Chemical Technology, Bhubaneswar 751013 Odisha, India
First published on 15th May 2025
Abstract
Herein, we disclosed a selective hydrogenation of tertiary amides and desulfurization of thioamides to amines with phenylsilane as a reductant, using a bifunctional Ru-NHC complex 1 appended with a Lewis acid in the secondary coordination sphere. The catalytic process was further utilized to synthesize biologically relevant amine drug molecules in good yields.
Amine functionality is prevalent as a key motif in many biologically active natural products and has important applications in pharmaceuticals and agrochemicals.1,2 Amides are naturally abundant or are easily synthesized from their carboxylic acid derivatives.3 In this regard, the selective deoxygenative hydrogenation of amides offers an attractive route for amine synthesis.4,5 However, with higher chemical inertness and competitive C–O/C–N cleavage of the amide bond, selective hydrogenative (C–O) cleavage is highly challenging. Thus, to activate and selectively deoxygenative hydrogenation of amides, Lewis acid additives are usually required along with the reducing agents. Several transition and main group metals, viz. Ru,6 Ir,7,8 Mn,4 and Mg,9 have been explored for the selective hydrogenation of secondary and tertiary amides using external Lewis acid additives in the presence of H2 or silanes as a reductant. Furthermore, without the aid of an external Lewis acid, a few transition metal complexes were also found to catalyze the deoxygenative hydrogenation of various tertiary amides to amines using silanes as reductants.10–12 In addition to that, some metal-free approaches have been reported for deoxygenative hydrogenation of various tertiary amides that rely on the use of a Lewis acidic boron catalyst and stoichiometric amounts of silane reagents or ammonia borane as the hydride source13–17 (Scheme 1). Although the reported homogeneous catalysts or metal-free approaches were found to be effective towards amide deoxygenative hydrogenation, a higher pressure of H2 gas or higher silane concentration along with Lewis acid additives are required. Thus, the development of an active molecular catalyst appended with Lewis acidic functionality using a mild reductant is highly desirable for the selective amide deoxygenative hydrogenation to amines under additive-free conditions.
 | ||
| Scheme 1 Lewis acid-assisted amide hydrogenation. | ||
The design of homogeneous systems appended with Lewis acidic boranes in the secondary coordination sphere (SCS) has gained significant attention due to their unique ability to capture and activate various incoming substrate molecules during catalysis.18 Several transition metal complexes with appended Lewis acidic arms in SCS (9-BBN, -Bpin, -BCy2) have been reported and utilized in various catalytic transformations like hydrazine activation,19,20 CO2 reduction,21 alkyne hydrogenation,22 nitrile activation,23 etc.
In this work, we report the synthesis of a Ru-NHC complex 1 appended with pinacolatoboron as a Lewis acidic arm in the SCS, which was used as a catalyst for the selective hydrogenation of various tertiary amides and thioamides to amines in high yields using phenylsilane as a reductant. To the best of our knowledge, a well-characterized molecular catalyst with an appended Lewis acidic boron arm in SCS has not yet been utilized for the selective deoxygenative hydrogenation of tertiary amides and thioamides to amines.
Ligand L1 was synthesized using piperidine-substituted pyridine imidazole with Br(CH2)3Bpin in dry acetonitrile (Scheme 2 and Fig. S27–S31, ESI†).24 The molecular structure of L1 is depicted in Fig. 1. The treatment of L1 with Ag2O followed by the addition of [Ru(p-cymene)Cl2]2 in dry acetonitrile yielded complex 1 as a light yellow solid in 90% yield (Scheme 2). The formation of 1 was confirmed using 1H, 13C, 11B and ESI-HRMS spectroscopic techniques (Fig. S44–S49, ESI†). Furthermore, treatment of complex 1 with 1.5 equiv. of KPF6 in dry CH2Cl2 readily afforded the complex 1-PF6 as a light brown solid (Fig. S52–S59, ESI†). The single crystal data of 1-PF6 features a piano stool geometry around the cationic Ru-center where the appended boron arm remains as a pendant (Fig. 1). 1-BF4 was synthesized by the treatment of 1 with AgBF4 in acetonitrile (Fig. S75–S80, ESI†).
 | ||
| Scheme 2 Synthesis of metal complexes. | ||
 | ||
| Fig. 1 Crystal structure of L1 and 1-PF6. Hydrogen atoms and counter anion are omitted for clarity. | ||
The catalytic reactivity of complex 1 was examined towards selective hydrogenation (C–O cleavage) of N,N-dimethylbenzamide (4a) to the corresponding amine (5a) (Table 1). The product 5a was observed in 97% NMR yield using 1 (1 mol %), PhSiH3 (1.1 equiv. w.r.t. 4a), and toluene (1 mL) at 80 °C for 36 h (entry 1, Table 1). The selectivity of the reaction was monitored by NMR and GC-MS analysis, where no C–N bond cleavage products (dimethylamine or benzyl alcohol) of 4a were observed (Fig. S81, ESI†). A decreased yield of 5a was observed by lowering the phenylsilane concentration, temperature and reaction time (entries 2–5). Varying other silanes and solvents resulted in a lower yield of 5a (entries 6–8, Table 1 and details in Table S1, ESI†). No 5a formation was observed in the absence of a catalyst (entry 9), while using 1-PF6 and 1-BF4, 94% and 96% NMR yield of 5a were detected (entries 10 and 11, Fig. S82 and S91, ESI†).
|
|||
|---|---|---|---|
| Entry | Catalyst (1 mol%) | Silanes (equiv.) | Yieldb (%) |
| a Reaction conditions: 4a (0.503 mmol, 1 equiv.), catalyst (1 mol%), silane (x equiv. w.r.t. 4a), solvent (1 mL), 80 °C, 36 h.b Product 5a was detected by GC-MS and its yields were calculated by 1H NMR spectroscopy using mesitylene as the IS.c Reaction was performed at 60 °C.d Reaction was performed for 24 h.e THF as solvent. NR = no reaction. | |||
| 1 | 1 | PhSiH3 (1.1) | 97 |
| 2 | 1 | PhSiH3 (1) | 85 |
| 3 | 1 | PhSiH3 (0.8) | 70 |
| 4c | 1 | PhSiH3 (1.1) | 75 |
| 5d | 1 | PhSiH3 (1.1) | 88 |
| 6 | 1 | Ph2SiH2 (1.1) | 55 |
| 7 | 1 | Et3SiH (1.1) | NR |
| 8e | 1 | PhSiH3 (1.1) | 61 |
| 9 | — | PhSiH3 (1.1) | NR |
| 10 | 1-PF6 | PhSiH3 (1.1) | 94 |
| 11 | 1-BF4 | PhSiH3 (1.1) | 96 |

With the optimized conditions (Table 1, entry 1), the reduction of various tertiary amides was tested (Table 2). Benzamide derivatives with electron-donating groups (–Me, –OMe) afforded good isolated yields (5b–5c). N-Benzoyl morpholine or piperidine substrates with electron-donating and -withdrawing substituents (–OMe, –Me, –F, –CF3) yielded the desired amines in 77–86% isolated yield (5d–5i). N-Methyl-N-phenylbenzamide (4j) with both electron-donating and -withdrawing groups (–Et, –F) yielded 5j–5l in a moderate yield. Further, amides bearing a benzylic phenyl, heterocyclic ring, alkene, strained 3-membered cyclopropane ring, aliphatic amide and cyclic lactam 5–8 membered rings were found to be compatible and the desired amines were isolated in good yields (5m–5u).

Interestingly, 5v–5w was isolated in good to moderate yield via selective cleavage of both aliphatic and aromatic diamides 4v and 4w (Table 2b). The deoxygenative hydrogenation of secondary amide was also examined using complex 1, where the NMR yield of 58% was achieved using N-phenylbenzamide as the substrate (Fig. S171–S173, ESI†). Furthermore, to demonstrate the practical applicability of the developed reaction protocol, a few biologically active tertiary amine drug molecules such as antimycotic agent, butenafine (5x); fenpiprane (5y) having antiallergic and antispasmodic activity; thrombotic stroke agent, ticlopidine (5z); and antihistamine agent, meclizine (5z′) were prepared with good isolated yields under the optimized reaction conditions using catalyst 1 (Table 2c). A scale-up reaction of 4a (3 mmol) was performed under the optimized reaction conditions, where 98% NMR yield of 5a formation was achieved (Fig. S210, ESI†).
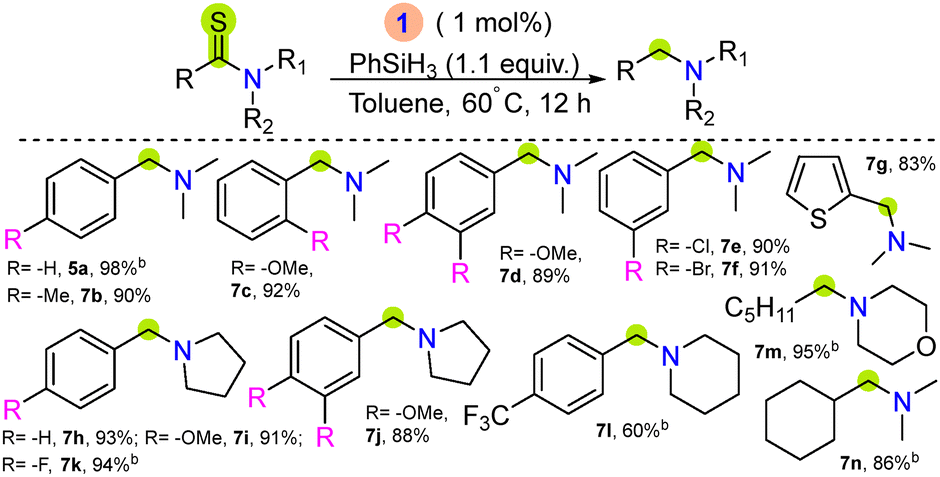
Furthermore, the catalytic efficiency of complex 1 was examined towards desulfurizative hydrogenation of tertiary thioamides. Thioamides are isosteres of amides found in many natural products and are versatile precursors for the synthesis of organic amines.25,26 The reductive desulfurization of thioamides using transition metals with silanes or H2 gas also offers an attractive pathway for amine generation, and a few reports have been disclosed.27–29 The major challenges for such catalytic conversion include competitive C–S/C–N cleavage and catalyst poisoning in the presence of sulfur-containing molecules.27 The optimal reaction conditions were achieved using 1 mol% of complex 1, N,N-dimethylbenzothioamide, 6a (0.503 mmol), 1.1 equiv. PhSiH3 in dry toluene at 60 °C for 12 hours (Table S2, entry 5, and Fig. S92, ESI†) afforded 98% NMR yield of 5a. Subsequently, the substrate scope for various tertiary thioamides was studied, where electron-donating and -withdrawing, heterocyclic, and aliphatic functionalized substrates afforded the corresponding amines in high yield under the optimized reaction conditions (Table 3). The hydrogenation of primary thioamide was also examined using 1, where dehydrosulfurizative product nitrile was observed, and no selective amine product was detected (Table S3 and Fig. S3B, ESI†).
In order to get insight into the reaction mechanism, a series of control experiments were performed. To check the homogeneity of the catalytic process, a mercury drop experiment was performed (300 equiv. of Hg w.r.t. 1), where a 90% NMR yield of amine 5a was observed (Scheme 3 and Fig. S9, ESI†). Furthermore, to understand the role of the appended Lewis acidic pinacolatoboron and Lewis basic piperidine arm in the SCS, two new ruthenium complexes 2 and 3 were synthesized (Scheme 3 and Fig. S61–S66, S68–S72, ESI†) and utilized for the deoxygenative hydrogenation of 4a under the optimized reaction conditions, which afforded 90% and 38% of 5a, respectively (Scheme 3 and Fig. S4, S5, ESI†). Furthermore, a catalytic reaction was performed using catalyst 3 and external alkyl–Bpin additives, Ph(CH2)2Bpin (5 mol%) under the optimized reaction conditions, where only 37% of 5a was observed (Scheme 3 and Fig. S6, ESI†) whereas only Ph(CH2)2Bpin (5 mol%), 4a and PhSiH3 (1.1 equiv.) (without any metal complex) showed no amine 5a formation (Scheme 3 and Fig. S7, ESI†).
 | ||
| Scheme 3 Control experiments. | ||
Furthermore, treatment of catalyst 1 and PhSiH3 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at 50 °C for 1 hour depicted a singlet peak at –11.08 ppm in C6D6 and –12.25 ppm in CDCl3 in 1H NMR corresponding to the Ru–H intermediate (A), additionally a peak at m/z = 633.2906 was also observed in ESI-HRMS analysis (Fig. S10–S12, ESI†). Furthermore, to examine the active role of Ru–H an NMR scale stoichiometric reaction was performed where an in situ generated intermediate (A) (1 and PhSiH3 (1:1) in C6D6) was treated with amide 4a (1 equiv.), and heating for 1 hour at 80 °C, almost full formation of amine 5a was observed in 1H NMR (Fig. S13, ESI†). Additionally, intermediate (A) was generated using 1 mol% of 1 and PhSiH3 in an NMR tube and was transferred into a reaction containing amide 4a (1 mmol) and PhSiH3 (0.5 mmol) in toluene and heated at 80 °C for 36 hours, affording a 43% NMR yield of amine 5a (Fig. S14, ESI†). These results depicted that intermediate (A) might act as an active intermediate during the catalytic process. A variable temperature (from 25 °C to −40 °C) NMR experiment was performed with a 1:10 mixture of 1 and amide 4a in acetone-d6; however, no significant change was observed in 1H and 11B NMR. However, variable temperature NMR studies (25 °C to −20 °C) with a 1:20 mixture of 1 and aliphatic amide, N,N-dimethylacetamide (DMA), revealed a minor peak at 22.48 ppm in 11B NMR upon lowering the temperature to −20 °C (Fig. S217, ESI†). Furthermore, using 1 with an excess of DMA, a peak at 22.29 ppm was observed in 11B NMR at room temperature (Fig. S218, ESI†),30 and under optimized reaction conditions, 87% NMR yield of N,N-dimethylethanamine was observed (Fig. S220, ESI†). Moreover, a strong interaction was observed using a mixture of 1 and dimethylaminopyridine (DMAP) (1:5), where a significant up-field shift in 1H NMR was observed for methylene (–CH2) protons (0.91 to 0.47 ppm) adjacent to the –Bpin arm upon decreasing the temperature, whereas 11B NMR showed an up-field shift from 32.30 to 12.80 ppm (Fig. S16 and S17, ESI†).31 These results depicted that the appended Lewis acidic boron arm in SCS of complex 1 can act as a directing group for substrate binding and activation during catalysis. An ESI-HRMS control experiment was performed using a 1:1:1 mixture of 1, PhSiH3, and 4a in toluene at 60 °C, where the formation of amine 5a was observed within 10 minutes, and the Ru-H intermediate (A) (m/z = 633.2906) was detected. Additionally, a peak at m/z = 802.3511 corresponding to amine 5a bound Ru-complex [1-Cl− + 5a]+ was also observed (Fig. S18, ESI†). 1,3-Diphenyldisiloxane, (PhSiH2)2O, and polysiloxanes were identified in ESI-HRMS as by-products during the catalysis. The treatment of (PhSiH2)2O and 4a in the presence of catalyst 1 afforded 35% of 5a under the optimized catalytic conditions, depicting that (PhSiH2)2O also acts as an active hydride source (Fig. S19 and S216, ESI†).32,33
:1) at 50 °C for 1 hour depicted a singlet peak at –11.08 ppm in C6D6 and –12.25 ppm in CDCl3 in 1H NMR corresponding to the Ru–H intermediate (A), additionally a peak at m/z = 633.2906 was also observed in ESI-HRMS analysis (Fig. S10–S12, ESI†). Furthermore, to examine the active role of Ru–H an NMR scale stoichiometric reaction was performed where an in situ generated intermediate (A) (1 and PhSiH3 (1:1) in C6D6) was treated with amide 4a (1 equiv.), and heating for 1 hour at 80 °C, almost full formation of amine 5a was observed in 1H NMR (Fig. S13, ESI†). Additionally, intermediate (A) was generated using 1 mol% of 1 and PhSiH3 in an NMR tube and was transferred into a reaction containing amide 4a (1 mmol) and PhSiH3 (0.5 mmol) in toluene and heated at 80 °C for 36 hours, affording a 43% NMR yield of amine 5a (Fig. S14, ESI†). These results depicted that intermediate (A) might act as an active intermediate during the catalytic process. A variable temperature (from 25 °C to −40 °C) NMR experiment was performed with a 1:10 mixture of 1 and amide 4a in acetone-d6; however, no significant change was observed in 1H and 11B NMR. However, variable temperature NMR studies (25 °C to −20 °C) with a 1:20 mixture of 1 and aliphatic amide, N,N-dimethylacetamide (DMA), revealed a minor peak at 22.48 ppm in 11B NMR upon lowering the temperature to −20 °C (Fig. S217, ESI†). Furthermore, using 1 with an excess of DMA, a peak at 22.29 ppm was observed in 11B NMR at room temperature (Fig. S218, ESI†),30 and under optimized reaction conditions, 87% NMR yield of N,N-dimethylethanamine was observed (Fig. S220, ESI†). Moreover, a strong interaction was observed using a mixture of 1 and dimethylaminopyridine (DMAP) (1:5), where a significant up-field shift in 1H NMR was observed for methylene (–CH2) protons (0.91 to 0.47 ppm) adjacent to the –Bpin arm upon decreasing the temperature, whereas 11B NMR showed an up-field shift from 32.30 to 12.80 ppm (Fig. S16 and S17, ESI†).31 These results depicted that the appended Lewis acidic boron arm in SCS of complex 1 can act as a directing group for substrate binding and activation during catalysis. An ESI-HRMS control experiment was performed using a 1:1:1 mixture of 1, PhSiH3, and 4a in toluene at 60 °C, where the formation of amine 5a was observed within 10 minutes, and the Ru-H intermediate (A) (m/z = 633.2906) was detected. Additionally, a peak at m/z = 802.3511 corresponding to amine 5a bound Ru-complex [1-Cl− + 5a]+ was also observed (Fig. S18, ESI†). 1,3-Diphenyldisiloxane, (PhSiH2)2O, and polysiloxanes were identified in ESI-HRMS as by-products during the catalysis. The treatment of (PhSiH2)2O and 4a in the presence of catalyst 1 afforded 35% of 5a under the optimized catalytic conditions, depicting that (PhSiH2)2O also acts as an active hydride source (Fig. S19 and S216, ESI†).32,33
Based on control experiments and previous reports on Lewis acid-assisted amide deoxygenative hydrogenation,4,6,7,13,15–17 a plausible mechanism was proposed (Fig. S221, ESI†). The [Ru–H] (A) might be the active species during the catalytic cycle and facilitate hydride insertion into the amide, which can be activated by the appended boron Lewis acid arm of the catalyst13,17 along with the silane intermediate. The phenylsilane can regenerate the active [Ru–H] (A) via a σ-bond metathesis pathway33 and affords disiloxane, which might further act as a hydride source with the formation of polysiloxanes during the catalytic cycle, and subsequent hydride transfer from (A) affords the desired amine product.
In summary, a bifunctional Ru-NHC complex 1 was synthesized and utilized as an efficient catalyst for the selective hydrogenation of various tertiary amides and thioamides to amines under ambient conditions. The catalytic protocol was also utilized to synthesize biologically relevant amine drug molecules in good yield. The presence of Lewis acid in the SCS of complex 1 was found to enhance the catalytic activity via substrate activation.
P. D. thanks IISER Berhampur, Central Advance Instrument Facility (CAIF), and ANRF, SERB, DST India for the core research grant (CRG/2023/001880) for financial support. RS thanks IISER Berhampur for the fellowship. R. D. and G. K. assisted with SCXRD studies.
Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for L1 and 1-PF6 have been deposited in the CCDC, and the CCDC numbers are 2346559 and 2349798, respectively.†Conflicts of interest
There are no conflicts to declare.References
- K. Eller, et al., Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, 2015, pp. 1–55 Search PubMed.
- A. Ricci, Modern Amination Methods, Wiley-VCH, Weinheim, 2000 Search PubMed.
- R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 7453–7465 CrossRef CAS.
- Y. Q. Zou, et al., ACS Catal., 2018, 8, 8014–8019 CrossRef CAS PubMed.
- A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477–5510 CrossRef CAS PubMed.
- J. Coetzee, et al., Chem. – Eur. J., 2013, 19, 11039–11050 CrossRef CAS PubMed.
- M. L. Yuan, et al., ACS Catal., 2016, 6, 3665–3669 CrossRef CAS.
- F. Han, et al., Sci. China: Chem., 2023, 66, 1094–1100 CrossRef CAS.
- N. L. Lampland, et al., ACS Catal., 2015, 5, 4219–4226 CrossRef CAS.
- S. Park and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 640–653 CrossRef CAS PubMed.
- S. Pisiewicz, et al., Chem. – Eur. J., 2014, 2345–2349 CAS.
- C. M. Macaulay, et al., Dalton Trans., 2019, 48, 9581–9587 RSC.
- D. Mukherjee, et al., Angew. Chem., Int. Ed., 2016, 55, 13326–13329 CrossRef CAS PubMed.
- R. C. Chadwick, et al., J. Org. Chem., 2014, 79, 7728–7733 CrossRef CAS PubMed.
- A. Chardon, et al., Chem. – Eur. J., 2017, 23, 2005–2009 CrossRef CAS PubMed.
- Y. Pan, et al., Adv. Synth. Catal., 2019, 361, 2301–2308 CrossRef CAS.
- M. T. Peruzzi, et al., Chem. Commun., 2018, 54, 5855–5858 RSC.
- M. W. Drover, Chem. Soc. Rev., 2022, 51, 1861–1880 RSC.
- J. J. Kiernicki, et al., Inorg. Chem., 2019, 58, 1147–1154 CrossRef CAS PubMed.
- O. Tutusaus, et al., J. Am. Chem. Soc., 2013, 135, 3403–3406 CrossRef CAS PubMed.
- E. E. Norwine, et al., J. Am. Chem. Soc., 2022, 144, 15038–15046 CrossRef CAS PubMed.
- K. N. T. Tseng, et al., J. Am. Chem. Soc., 2016, 138, 10378–10381 CrossRef CAS PubMed.
- M. L. Clapson, et al., Chem. – Eur. J., 2023, 29, e202203763 CrossRef CAS PubMed.
- M. Toure, et al., Dalton Trans., 2015, 44, 7139–7143 RSC.
- R. N. Hurd and G. Delamater, Chem. Rev., 1961, 61, 45–86 CrossRef CAS.
- T. N. Hansen and C. A. Olsen, Chem. – Eur. J., 2024, 30, e202303770 CrossRef CAS PubMed.
- Z. Wang, et al., Angew. Chem., Int. Ed., 2023, 62, e202215963 CrossRef CAS PubMed.
- J. Luo, et al., J. Am. Chem. Soc., 2020, 142, 21628–21633 CrossRef CAS PubMed.
- K. Fukumoto, et al., Organometallics, 2013, 32, 2889–2892 CrossRef CAS.
- J. Li, et al., J. Am. Chem. Soc., 2020, 142, 13011–13020 CrossRef CAS PubMed.
- F. Lima, et al., Angew. Chem., Int. Ed., 2016, 55, 14085–14089 CrossRef CAS PubMed.
- J. A. Buonomo, et al., Synthesis, 2018, 50, 278–281 CrossRef CAS PubMed.
- P. Pandey and J. K. Bera, Chem. Commun., 2021, 57, 9204–9207 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2346559 and 2349798. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00399g |
| This journal is © The Royal Society of Chemistry 2025 |