
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReductive alkylation of azoarenes to N-alkylated hydrazines enabled by hexafluoroisopropanol†
Siddhartha Kumar
Senapati
and
Animesh
Das
 *
*
Department of Chemistry, Indian Institute of Technology Guwahati, Guwahati-781039, Assam, India. E-mail: adas@iitg.ac.in
First published on 29th March 2025
Abstract
A one-pot tandem approach to N-alkyl-N,N′-diarylhydrazines was developed using a sequence of reduction of azoarene to hydrazoarene followed by reductive alkylation by an aldehyde. The mild reaction conditions suppress N–N cleaved products and selectively provide trisubstituted hydrazine derivatives. The mechanistic study demonstrates that the iminium formation step is likely to be the rate-limiting step.
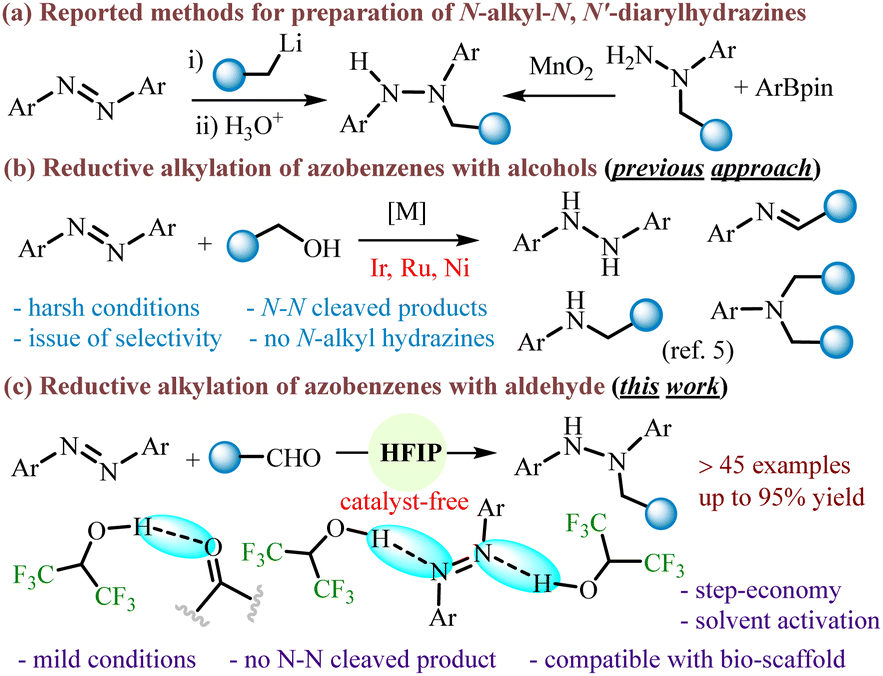
Hydrazine derivatives are an important class of compounds that are widely utilized in the pharmaceutical, agrochemical, polymer and dye industries.1 They also have useful synthetic applications in the synthesis of nitrogen-containing compounds and coupling reactions.2 Nevertheless, the synthesis of target-based N-alkyl-N,N′-diarylhydrazines is difficult, which seriously restricts their bioactivity and application research. The reported methods are the addition of alkyllithium to the N
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of an azobenzene under cryogenic conditions or the oxidative coupling of N-alkyl-N-arylhydrazines with aryl borates (Scheme 1a).3 Therefore, it is deemed desirable to develop a mild and efficient method for the construction of N-alkyl-N,N′-diarylhydrazines without the use of reactive organolithium reagents, expensive borates, and external oxidants.
N bond of an azobenzene under cryogenic conditions or the oxidative coupling of N-alkyl-N-arylhydrazines with aryl borates (Scheme 1a).3 Therefore, it is deemed desirable to develop a mild and efficient method for the construction of N-alkyl-N,N′-diarylhydrazines without the use of reactive organolithium reagents, expensive borates, and external oxidants.
 | ||
| Scheme 1 Alkylation of azoarenes and synthesis of N-alkyl-N,N′-diarylhydrazines. | ||
Direct NN functionalization of azobenzenes is one of the promising synthetic approaches to synthesize N,N′-diaryl hydrazine derivatives and various N-heterocycles. However, unlike highly reactive alkyl azodicarboxylates with a strongly electrophilic NN double bond, azobenzenes typically exist in a stable and nonpolar trans form, showing low NN bond reactivity.4 In a recent study, transition-metal catalyzed reductive alkylation of azobenzenes with alcohols is attempted for the construction of N-alkyl-N,N′-diarylhydrazines (Scheme 1b).5 However, the process failed to obtain the desired hydrazine and led to the formation of N–N cleaved products. The over-reduction of azoarenes to amine derivatives renders the process less efficient for the synthesis of N-alkylated hydrazine. Besides, the methods require the use of transition metal catalysts, harsh conditions, and lack of tolerance with hydrogenation-sensitive functional groups. Therefore, we were interested to develop a mild and efficient method for the construction of substituted hydrazine derivatives under catalyst-free conditions with a wide range of substrate scopes.
The use of 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) has recently gained attention because of their unique physical and chemical properties (such as low nucleophilicity, high ionizing power, strong hydrogen-bond donor ability and cation stabilization).6 This is believed to be considered as an ideal reaction medium in promoting various reactions under mild conditions including unsaturated carbon–carbon bond functionalization,6f C–H functionalization,6d,e hydroarylation,7N-alkylation,8C-alkylation reaction9 and aza-Piancatelli cyclization.10 Nevertheless, the use of HFIP only as an activator has received limited attention in reduction chemistry.
Herein, we envisaged that the strong H-bond donor properties of HFIP can not only aid in the hydrogenation of azoarenes to hydrazoarenes but also in the reductive N-alkylation of the resulting compounds (Scheme 1c). Our investigation initiated with the reaction of azobenzene (1a, 0.5 mmol), benzaldehyde (2a, 0.5 mmol), and Hantzsch ester (HE) (1 mmol) in HFIP (0.1 mL) at 60 °C. After 2 h, N-alkylated hydrazine 3a was obtained in 22% yield, along with 32% intermediate hydrogenated product hydrazobenzene (1a′, Table 1, entry 1). To improve the yield of desired product 3a, the equivalency of HFIP was gradually increased (entry 2). When HFIP was used in 0.5 mL, product 3a was obtained in 92% yield (entry 3). A decrease in the yield of 3a has been observed when the reaction was examined with a mixture of solvents (entries 4–6). The yield of 3a was also decreased by reducing the reaction time (entry 7) or by reducing the reaction temperature (entry 8). Then, the reaction was examined with –CF3 functionalized thiourea as a hydrogen-bond donor catalyst in DCE, providing the product 3a in 60% yield, indicating that the hydrogen-bonding donor property is important in this transformation (entry 9). We have examined the reaction by replacing HFIP with a relatively weak hydrogen bond donor solvent, isopropanol, which failed to provide any desired product (entry 10). This reiterates the significance of the role of HFIP in the current transformation as a strong H-bond donor solvent. We next intended to explore the scope of the N-alkylation reaction with a variety of aldehydes 2a–t, 2aa–ah using 1a as the standard substrate (Table 2). The substrates having both electron-donating (e.g., –Me, –OMe, –OBn) and electron-withdrawing groups (e.g., –Cl, –CF3) at the para-position in the aryl moiety were efficiently reacted to obtain the desired products 3a–3f in 83–92% yields. Slightly higher yields were realized for electron-donating groups. With boronic acid containing aldehyde, the desired product 3i was formed with 81% yield. Since, the present reaction conditions tolerate the –B(OH)2 group functional groups, it can be useful for further functionalization in Pd-catalyzed Suzuki–Miyaura cross-coupling reaction. Furthermore, meta- and ortho-substituent bearing aldehydes afforded the corresponding hydrazine products 3j–3k, 3l–3n in moderate to good yields. Notably, the retention of reducible-functional groups (4-Cl, 4-OBn, 4-CF3, 4-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh, 4-O-allyl, 3-Br, 2-Br, 2-CO2Me, 2-NO2) in the final products showcases the excellent chemoselectivity of the present reductive protocol. Interestingly, 1-pyrene aldehyde also led to the products 3o in 83% yields. When 1,4-benzenedialdehyde was used as an alkylating agent the poly N-alkylated hydrazine 3p was readily obtained with notable selectivity. Besides aromatic aldehydes, the scope of the reaction was expanded to heteroaromatic aldehydes and a variety of aliphatic aldehydes and cyclic ketones, yielding 3q–3t and 3u. Considering the efficiency of the present protocol, the scope of the reaction was further expanded using highly functionalized substrates with complex molecular settings.
CPh, 4-O-allyl, 3-Br, 2-Br, 2-CO2Me, 2-NO2) in the final products showcases the excellent chemoselectivity of the present reductive protocol. Interestingly, 1-pyrene aldehyde also led to the products 3o in 83% yields. When 1,4-benzenedialdehyde was used as an alkylating agent the poly N-alkylated hydrazine 3p was readily obtained with notable selectivity. Besides aromatic aldehydes, the scope of the reaction was expanded to heteroaromatic aldehydes and a variety of aliphatic aldehydes and cyclic ketones, yielding 3q–3t and 3u. Considering the efficiency of the present protocol, the scope of the reaction was further expanded using highly functionalized substrates with complex molecular settings.
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (0.5 mmol), HE (1 mmol) in HFIP (0.5 mL) at 60 °C for 2 h under Ar, isolated yields. b With HBD catalyst 1,3-bis(3,5-bis(trifluoromethyl)phenyl)thiourea (15 mol%) in DCE (0.5 mL). | ||
| 1 | HFIP (0.1 mL) | 22%, (32% of 1a′) |
| 2 | HFIP (0.2 mL, 0.3 mL, 0.4 mL) | 41%, 65%, 81% |
| 3 | None | 92% |
| 4 | HFIP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCE (1:3, 0.5 mL) :DCE (1:3, 0.5 mL) |
25% |
| 5 | HFIP:CH3OH (1:3, 0.5 mL) |
21% |
| 6 | HFIP:toluene (1:3, 0.5 mL) |
18% |
| 7 | With 0.5 h, 1 h, 1.5 h | 26%, 48%, 74% |
| 8 | At 25 °C, at 45 °C | 0%, 14% |
| 9b | With HBD catalyst | 60% |
| 10 | With isopropanol (0.5 mL) | 0% |
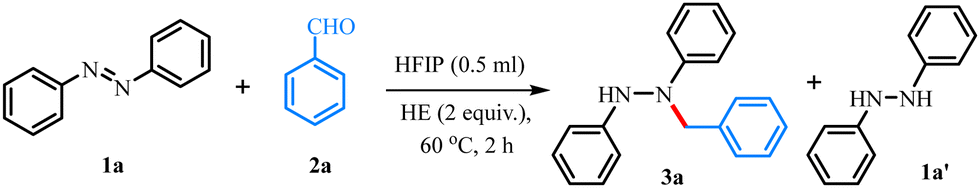
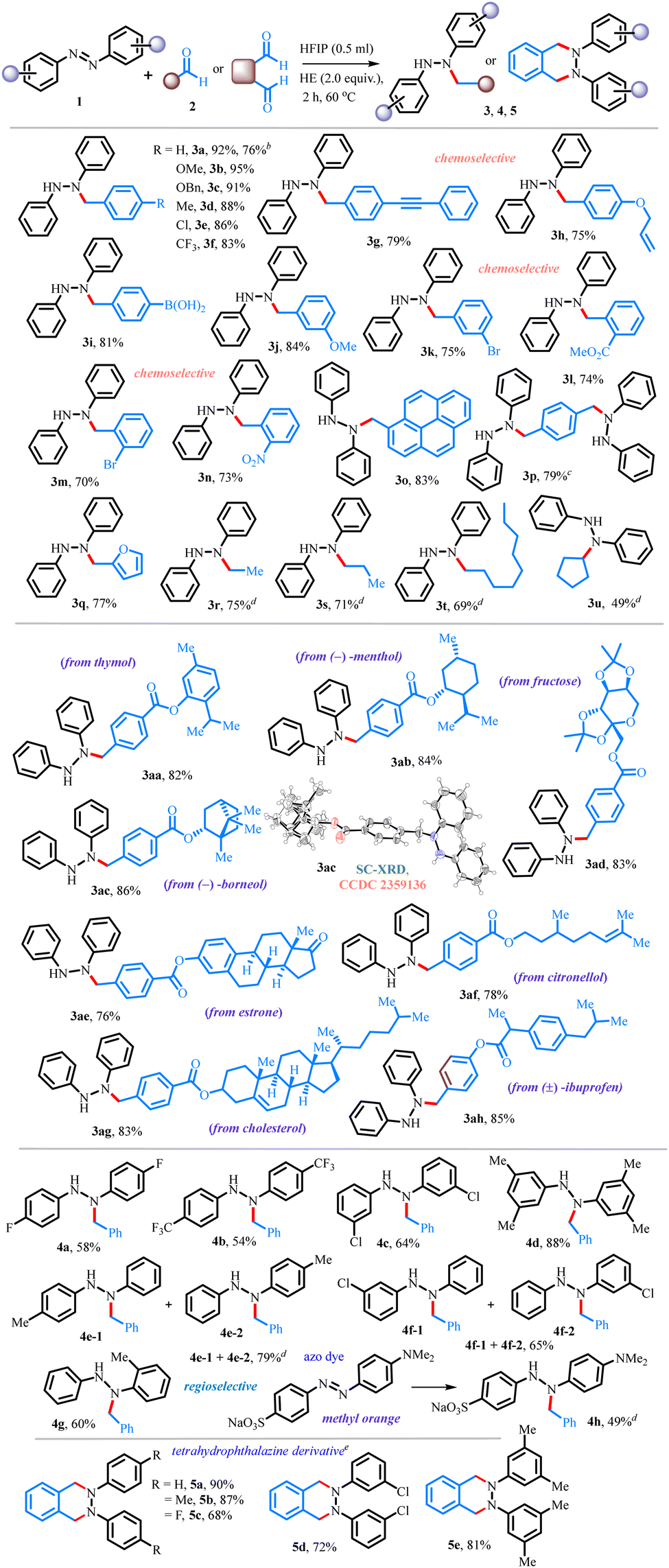
A range of biologically relevant motifs (BRM) were tested and were independently subjected to the optimized reaction conditions, affording the corresponding hydrazine derivatives containing thymol (3aa, 82%), (−)-menthol (3ab, 84%), (−)-borneol (3ac, 86%), fructose (3ad, 83%), estrone (3ae, 76%), citronellol (3af, 78%), cholesterol (3ag, 83%), and (±)-ibuprofen (3ah, 85%) moieties. The structure of the product 3ac was further confirmed by single-crystal X-ray diffraction analyses. This suggests that there are no structural (chemical and stereochemical) changes of the complex architecture in the entity. To further demonstrate the generality and practicability of this reaction, a range of azobenzenes 1b–1g were tested. Substituted symmetrical azobenzenes having p-F (1b), p-CF3 (1c), m-Cl (1d) and m,m′-(CH3)2 (1e) all efficiently provided the corresponding alkylated products 4a–4d in 54–88% yields. On the other hand, unsymmetrical azobenzenes 1f and 1g provided an inseparable mixture of the alkylated products 4e and 4f in 81% and 65%, respectively, whereas ortho-substituted azobenzene 1h afforded a single regioisomeric product 4g in 60% yield. The methodology was further extended towards commercially used azo dyes such as methyl orange, obtaining N-alkylated product 4h in 49% yield. It's worth mentioning that when the reaction was examined with phthalaldehyde under standard conditions, biologically relevant tetrahydrophthalazine derivatives 5a–5e were obtained in 68–90% yields. Additionally, the reaction was shown to work effectively on a 2 mmol-scale (see ESI,† Scheme S8), and the method displays the convenient regeneration of the Hantzsch ester by re-reducing the aromatized by-product (see ESI,† Schemes S9 and S10).
Furthermore, the synthesized molecules can be easily transformed into useful molecules (Scheme 2). For example, N–N bond cleavage of various hydrazine derivatives 3a, 3aa, 3ac and 3ag proceeded smoothly using Zn in AcOH, delivering the secondary amines 6a–6d in moderate to good yields. It is worth mentioning that the current reductive approach is applicable for the synthesis of secondary amines 6b–6d with complex molecular settings. Compound 5a can also be easily transformed into CXCR4 antagonist 71211 (7, 85% yield) via N–N bond cleavage. Furthermore, compound 5a was converted to 2,3-diphenyl-2,3-dihydro phthalazine-1,4-dione 8 (77%) under oxidative conditions by a cyclometalated ruthenium catalyst and periodate.12
 | ||
| Scheme 2 Post synthetic modification and reductive N–N bond cleavages. | ||
The mechanism of reaction was studied noting the hydrogen bond donor properties of HFIP (Fig. 1). Close investigation of the nature of binding in solution shows that there is a weak hydrogen bond interaction between –OH of HFIP and the N-atom of the azobenzene moiety. It was confirmed by the 1H NMR with a downfield shift (from δ = 3.21 to 3.61 ppm) of the signal of –OH protons in HFIP along with a weak upfield shift (from δ = 4.39 to 4.31 ppm) of the signal of the –CH proton in HFIP (Fig. S1, ESI†). The signals corresponding to ortho-aromatic protons in 1a were slightly shifted in the upfield (Δδ 0.2 ppm).13 Then, interaction of HFIP with the freshly distilled aldehyde and carbonyl group in HE was also confirmed by 1H and 13C NMR, and IR spectroscopic experiments (Fig. S2–S4, ESI†). Upon addition of aldehyde 2f,14 the hydroxyl peak of HFIP was shifted downfield by nearly 0.52 ppm in the 1H NMR, and the carbonyl value moved downfield (Δδ 2.76 ppm) in the 13C NMR. Furthermore, the carbonyl band shifted to a lower frequency (from 1708 to 1694 cm−1) in the IR spectrum. Experimental observation suggests that there is a weak hydrogen bonding interaction between HFIP and the aldehyde group.14 Likewise, interaction of HFIP with the carbonyl group in HE was also validated by 13C NMR with a downfield shift (Δδ 0.7 ppm) (Fig. S5 and S6, ESI†).
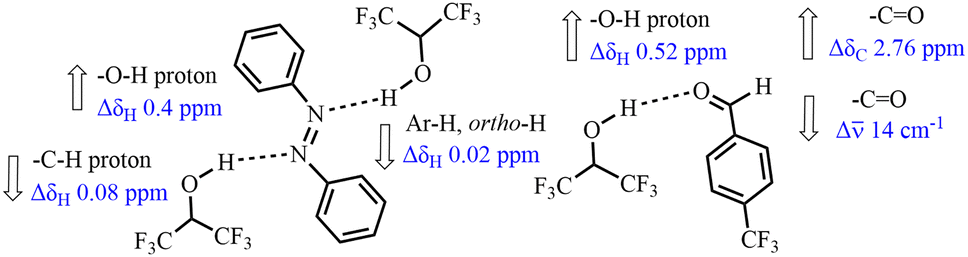 | ||
| Fig. 1 Interaction of HFIP with 1a and aldehyde 2f by NMR and IR studies. | ||
Next, a few control experiments were carried out to gain mechanistic insights into the synthesis of hydrazine derivatives (see ESI,† Section S8.4). The reaction of azobenzene 1a in the absence of aldehyde under standard conditions afforded 1,2-diphenyl hydrazine 1a′ in 98% yield. For the subsequent step, the reaction was conducted with the formed 1a′, and aldehyde 2a under standard conditions and the desired product 3a was obtained in 87% yield (Scheme S11, ESI†), from which it can be inferred that the reaction possibly occurs in two steps i.e., reduction of azobenzene, followed by reductive N-alkylation of hydrazobenzene to N-alkylated hydrazine. Then, the electronic effect of the substrate on the rate of the reaction was examined (Scheme S12 and S13, ESI†). A competitive reaction suggested that the electron-donating group bearing a substituent (–Me) on the azobenzene enhanced the rate of the reaction with respect to an electron-withdrawing group (p-F). Likewise, an electron-donating group (p-OMe) on the benzaldehyde enhanced the rate of the reaction with respect to an electron-withdrawing group (p-Cl). In contrast, there is no considerable effect of electronics on the rate of the reaction in the azobenzene hydrogenation step (see ESI,† Scheme S12). Furthermore, we have investigated the kinetic Hammett correlation study using various para-substituted benzaldehydes under standard reaction conditions (see ESI,† Section S8.7). The reaction rates are in the order OMe > Me > H > Cl. The least-squares fit to the plot of [log(kX/kH)] vs. Hammett σ+ constant gave a straight line with a ρ+ value of −1.3. A negative ρ+ value suggests that in the transition state, electrons are flowing away, which is stabilised by the electron-donating substituents. The effect of temperature on the rate of condensation reaction was examined by using Eyring analysis (see ESI,† Section S8.8). The activation parameters were found to be ΔH‡ = 29.2 ± 1.6 kcal mol−1 and ΔS‡ = +12.02 ± 0.9 cal K−1 mol−1. The positive entropy of activation suggests that the involvement of a dissociative process is the turnover-limiting step. Based on the thermodynamic activation parameters, kinetic Hammett studies, and competitive reaction on the substrate, one can postulate that the iminium formation step is likely to be the rate-limiting step.
On the basis of the above experimental findings, a proposed pathway for the formation of N-alkylated hydrazine has been demonstrated in Scheme 3. First, the azobenzene 1a is activated by the hydroxyl group of HFIP via hydrogen bonding interaction, which is then reduced by Hantzsch ester to provide hydrazobenzene 1a′. The in situ generated hydrazobenzene can react with aldehyde to form hemiaminal species 1C, followed by condensation leading to the formation of iminium intermediate 1D. Then, this was readily reduced by Hantzsch ester to provide the N-alkylated hydrazine 3a.
 | ||
| Scheme 3 Proposed pathway for the formation of N-alkylated hydrazine. | ||
In summary, catalyst-free HFIP-promoted one-pot tandem reduction of azoarene with subsequent reductive alkylation using aldehydes to form N-alkylated hydrazine derivatives has been demonstrated. The protocol shows notable selectivity as the formation of N–N cleaved product amines and/or imines is completely suppressed. A wide range of functional group tolerance and structural features encountered in pharmaceuticals was demonstrated. This study demonstrates the general utility of HFIP in reduction chemistry.
A. D. gratefully acknowledges the SERB, DST (CRG/2022/01606) and SR/FST/CS-II/2017/23c) for the financial support.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) U. Ragnarsson, Chem. Soc. Rev., 2001, 30, 205–213 RSC
; (b) N. Raziq, M. Saeed, M. S. Ali, M. Shahid, M. Lateef and S. Zafar, Nat. Prod. Res., 2022, 36, 961–966 CrossRef CAS PubMed
-
(a) S. Balgotra, P. K. Verma, R. A. Vishwakarma and S. D. Sawant, Catal. Rev., 2020, 62, 406–479 CrossRef CAS
-
(a) A. R. Katritzky, J. Wu and S. V. Verin, Synthesis, 1995, 651–653 CrossRef CAS
- F. A. Jerca and V. V. Jerca, Nat. Rev. Chem., 2022, 6, 51–69 CrossRef PubMed
-
(a) S. Bansal, R. G. Gonnade and B. Punji, Catal. Sci. Technol., 2023, 13, 2705–2713 RSC
-
(a) H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood IV and J. Aubé, Chem. Rev., 2022, 122, 12544–12747 CrossRef CAS PubMed
- Selected examples:
(a) C. Qi, V. Gandon and D. Lebœuf, Angew. Chem., Int. Ed., 2018, 57, 14245–14249 CrossRef CAS
- Selected examples:
(a) Z. Zhang, X. Zhang, Y. Liu, X. Wang and X. Zhang, J. Org. Chem., 2023, 88, 14189–14192 CrossRef CAS
-
(a) S. Wang, G. Force, R. Guillot, J. F. Carpentier, Y. Sarazin, C. Bour, V. Gandon and D. Lebœuf, ACS Catal., 2020, 10, 10794–10802 CrossRef CAS
- D. Leboeuf, L. Marin, B. Michelet, A. Perez-Luna, R. Guillot, E. Schulz and V. Gandon, Chem. – Eur. J., 2016, 22, 16165–16171 CrossRef CAS PubMed
- W. Q. Zhan, Z. X. Liang, A. Z. Zhu, S. Kurtkaya, H. Shim, J. P. Snyder and D. C. Liotta, J. Med. Chem., 2007, 50, 5655–5664 CrossRef CAS
- M. Konwar, T. Das and A. Das, Org. Lett., 2024, 26, 1184–1189 CrossRef CAS PubMed
- Yu. A. Mikheeva and Yu. A. Ershov, Russ. J. Phys. Chem. A, 2018, 92, 1499–1507 CrossRef
-
(a) J. Gaster, S. Kozuch and D. Pappo, Angew. Chem., Int. Ed., 2017, 56, 5912–5915 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2359136. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc00425j |
| This journal is © The Royal Society of Chemistry 2025 |