
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmphoteric tetrazole-substituted eleven-ring-fused acene derivatives with multiple fluorescent protonation states†
Ying-Hsuan
Liu‡
,
Cory
Ruchlin‡
 ,
Heorhii V.
Humeniuk
and
Dmytro. F.
Perepichka
*
,
Heorhii V.
Humeniuk
and
Dmytro. F.
Perepichka
*
Department of Chemistry, McGill University, Montreal, Quebec H3A 0B8, Canada. E-mail: dmytro.perepichka@mcgill.ca
First published on 6th May 2025
Abstract
We synthesized novel tetrazole-substituted diacenaphthoanthracenediimides 2 by azide cycloaddition to the corresponding cyano-substituted precursors. Reversible protonation/deprotonation of the tetrazole moieties provides distinct fluorescent species with photoluminescence quantum yields of 12–34%. The facile deprotonation of 2 enables its processing in non-halogenated solvents (alcohol).
The design of π-electron functional materials is critical for advancing organic optoelectronic applications.1–3 In addition to numerous π-core modifications, substituent effects are widely used in tailoring the optoelectronic properties of such materials.4–7 In particular, the electron-withdrawing cyano group (Hammett constant σp = 0.66)8 is commonly employed to induce donor–acceptor interactions in thermally activated delayed fluorescence (TADF)-based emitters9,10 or lower the LUMO energies in n-type organic semiconductors.11 Converting cyano groups into 1H-tetrazoles (Tz) offers a facile strategy to expand the functionality of CN-substituted materials.12 As an electron-withdrawing moiety, Tz (σp = 0.56)8 is slightly weaker than CN, but its hydrogen bonding abilities and amphoteric acid/base properties bring additional functionality to such derivatives.13–15 Surprisingly, Tz is almost completely unexplored in the design of optoelectronic materials. Only a few Tz-based fluorescent probes for the detection of F−, CN− and metal ions and bioimaging have been reported.16–19
Tz derivatives are commonly used as intermediates in the synthesis of 1,3,4-oxadiazole-based semiconductors.20–22 Tz-terminated donor–acceptor dyads have been studied in dye-sensitized solar cells but showed no advantages compared to COOH-terminated dyes.23,24 Recently, Tz-substituted donor–acceptor molecules have been compared with CN- and oxadiazole-substituted analogues as TADF emitters.25
Exploring tetracyanoanthracenediacenaphthalimides (1) as n-type semiconductors,26 we speculated that converting the four cyano groups in these compounds into tetrazoles would offer new functionality while retaining sufficient electron deficiency. In this communication, we report the synthesis of novel tetrakis(1H-tetrazole)diacenaphthoanthracenediimides (2) via the [2+3] cycloaddition of 1 with in situ generated hydrazoic acid (Fig. 1a). The protonation/deprotonation of the Tz groups in 2 enables solubility in alcohols (Fig. 1b). This amphoteric behavior allows transitions between dicationic, neutral, and dianionic protonation states, each with distinct absorption and emission properties. The photoluminescence quantum yields (PLQYs) of neutral 2a (12%) increase significantly upon both protonation (30%) and deprotonation (34%), promising potential applications as fluorescent probes across a wide pH range.
 | ||
| Fig. 1 (a) Synthetic route from 1 to 2 and (b) the protonation/deprotonation transformations of 2a2+, 2a, and 2a2−. The photographs at the bottom show insoluble 2a suspended in pure 2-propanol, becoming soluble upon the addition of a few drops of TFA or TEA. | ||
Cyanated precursors 1a,b have been synthesized via a Knoevenagel condensation of substituted acenaphthalimide diketones with tetrakis(cyanomethyl)benzene (the details are given in the ESI†).26,272a and b were obtained through a [2+3] cycloaddition using sodium azide under acetic acid-catalyzed microwave conditions (Fig. 1a). This approach provides a safer alternative to the direct addition of (explosive) HN3. The long branched 2-hexyldodecyl (2a) and the cyclohexyl (2b) chains were used to provide film-forming properties and to improve solubility in polar solvents for spectroscopic studies, respectively. The lower yield of 2a compared to the long-chain substituted 2b is likely due to the use of crude insoluble 1a as a starting material. Both Tz derivatives are obtained as stable red powders with low-to-moderate solubility in polar organic solvents (e.g., MeCN, DMF, DMSO, etc.). Thermogravimetric analysis (TGA, Fig. S1, ESI†) shows the stability of the synthesized Tz derivatives up to 230 °C, at which point a well-defined decomposition step, corresponding to the release of eight N2 molecules, is observed. Their photostability is tested in aerated acetonitrile solution under 390 nm illumination (6.5 W) showing moderate photostability (t1/2 = 293 min) comparable to that of other fluorescent dyes such as coumarin 153 (t1/2 = 55 min), fluorescein (t1/2 = 178 min) and rhodamine 101 (t1/2 = 468 min, Fig. S2, ESI†).
In acetonitrile solution, 2a exhibits a characteristic vibronically structured absorption band at λabsmax = 544 nm and an emission band at λemimax = 582 nm, with a moderate Stokes shift of 38 nm (0.15 eV) and a PLQY of 12% (Fig. 2 and Table 1). The absorption/emission bands of 2 are broader and red-shifted compared to 1 by 50–100 nm, probably because of a stronger donor character of the anthracene core without CN substituents (Fig. S3, ESI†). The addition of triethylamine (TEA, pKMeCNBH+ ∼ 18.5) results in a redshift to λabsmax = 568 nm, due to the deprotonation of 2a (pKMeCNa of unsubstituted tetrazole ∼14.5). Given the structure of the molecule, we attribute this absorption to the dianion 2a2− with one deprotonated Tz unit on each side of the anthracene core. As expected, almost no changes were observed upon addition of AcOH (pKMeCNa ∼ 23.5) to a solution of 2a in MeCN, confirming its neutral state. However, the addition of a stronger trifluoroacetic acid (TFA, pKMeCNa ∼ 12.7) leads to a significant blue shift to λabsmax = 515 nm, which plausibly corresponds to the dication (2a2+). These changes are reversible, and the original spectrum of 2a can be recuperated upon neutralization of acidic/basic solutions (Fig. S6, ESI†). Time-dependent density functional theory (TD-DFT) calculations show a very similar progressive red shift from 2a2+ (514 nm) to 2a (539 nm) and 2a2− (584 nm, Tables S1–S4 and Fig. S14, ESI†).
 | ||
| Fig. 2 (a) Absorption and (b) emission spectra of 2a2+, 2a, and 2a2− in the indicated solutions with or without acids and bases. The spectra are normalized at the S0 → S1 transition band. | ||
Interestingly, no further red/blue shifts were observed upon addition of stronger base diazabicycloundecene (DBU, pKMeCNBH+ ∼ 24.3) or trifluoromethanesulfonic acid (TfOH, pKMeCNa ∼ 0.7), suggesting no further deprotonation/protonation under these conditions. This thermodynamic instability of the tetraanionic/tetracationic species could be rationalized by the electrostatic repulsion of the adjacent Tz rings. A blue shifted absorption has been observed under highly acidic conditions (TfOH in the TFA solvent) possibly due to the formation of 2a4+, but the change was not fully reversible (Fig. S5, ESI†). Treating 2a solution in THF with a much stronger base (lithium diisopropylamide, LDA) resulted in decomposition.
Titrations of 2a were performed in acetonitrile, monitoring the absorption spectra at each step. Deprotonation by DBU (Fig. 3, see Fig. S7 for titration with TEA, ESI†) resulted in a sharp transition from the neutral 2a spectrum to that of 2a2− at ∼2.5 equiv. of base added. The imperfect isosbestic points (inset) as well as the buffering activity observed early in the titration curve are attributed to the formation of monoanions in equilibrium with the neutral and dianion species. The lack of selective formation of monoanions can be rationalized by the large spatial separation of the two charged Tz rings (∼10 Å), resulting in a small difference in their pKa values. Acid titration with TFA (Fig. S7, ESI†) showed a similar sharp transition to a dication after ∼2 equiv. of acid were added. However, the spectral evolution is affected by the slow aggregation of 2a2+ (Fig. S8, ESI†).
 | ||
| Fig. 3 (a) Absorption spectra of 2a in CH3CN (5 × 10−6 M) during titration with DBU. The inset shows the enhanced view of the imperfect isosbestic points. (b) Degree of deprotonation determined from absorption spectra versus equivalents of DBU added. (c) Aromatic region of the 500 Hz 1H NMR spectrum of 2a in CD3CN (1.5 × 10−3 M) under titration with DBU. | ||
1H NMR was used to track the titration of 2a with DBU in CD3CN by the chemical shifts of the aromatic protons (Fig. 3c). The spectrum of neutral 2a shows three peaks (Ha, Hb, and Hc) which are more shielded and severely broadened due to aggregation (the compound immediately precipitates upon addition of TFA). Upon addition of 0.5 equiv. of DBU, we observed the formation of a new set of peaks, presumably due to disaggregated anionic species. Approaching two equivalents of the added base, the peaks are sharpened and shifted slightly upfield (by ∼0.1 ppm for Hb) due to the increased negative electron density on the Tz rings.
We were able to solubilize 2a in water with the addition of 1% DMSO. Titration of this solution was monitored by pH and absorption spectroscopy (Fig. S9, ESI†), leading to an estimated pKa of ∼6.7 (cf.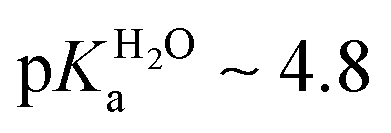 for 1-phenyltetrazole).
for 1-phenyltetrazole).
The emission spectra show a similar trend of blue/red shifts as the lowest energy absorption band: λemimax = 545 nm (2a2+), 580 nm (2a), and 613 nm (2a2−). The PLQY of 2a increases upon both the protonation (2a2+, PLQY 30%) and deprotonation (2a2−, PLQY 34%). Such retention of luminescence efficiency can be explained by the electronic decoupling of the Tz substituents from the π-core. Indeed, the DFT optimization of 2 in different protonation states indicates that the Tz substituents are nearly perpendicular to the π-core, with the HOMO/LUMO delocalized over the π-core (Fig. S12 and S13, ESI†).
Time-correlated single photon counting (TCSPC) measurements were performed on 2a, 2a2+, and 2a2− in acetonitrile solutions. 2a2− exhibited a classical monoexponential decay of the excited state, while 2a and 2a2+ required fitting with biexponential decays, possibly due to aggregation of the less soluble dicationic and neutral species (Table 1 and Fig. S7, ESI†).
Facile ionization of the Tz substituents allows processing of these large polycyclic aromatic hydrocarbons from non-chlorinated solvents (alcohols).28 Indeed, long-chain substituted 2b can be spin-coated from a 2-propanol solution with a few drops of TEA, producing uniform red films. The TEA base is readily removed by drying in a vacuum, producing neutral 2b as supported by comparing its absorption spectrum with that in solution (Fig. 4). Solvent-annealing the films in AcOH vapor does not significantly shift the spectrum, suggesting a neutral state of 2b. The emission band of the film (λemimax =704 nm) is significantly red-shifted from that in solution (λabsmax = 612 nm), suggesting exciton delocalization in the solid state. Protonation/deprotonation switching in the solid films can be initiated by their exposure to TFA and TEA vapor, respectively, resulting in similar spectral changes to those observed in solution (Fig. S11, ESI†).
 | ||
| Fig. 4 Absorption (solid) and emission (dashed) spectra of 2b in acetonitrile solutions and vacuum-dried and AcOH-treated films. The photograph shows the as-cast film. | ||
In conclusion, we synthesized novel Tz-substituted polycyclic aromatic hydrocarbons 2a/2b from the corresponding cyano-substituted precursors. The amphoteric Tz substituents enable solubilization of these large polycyclic (11 fused rings) aromatic hydrocarbons in alcohols and even in aqueous media (with 1% DMSO). Changing the acidity of the medium, we observed three spectroscopically distinct states attributed to the dicationic, neutral and dianionic species of 2. The absorption and emission bands undergo a progressive red shift with increasing electron density on the anthracene core (i.e., from dication to dianion), which can be explained in terms of donor–acceptor interactions between the dicarboxyimide termini and the anthracene core. Interestingly, the fluorescence is maintained (PLQY = 12–34%) across all protonation states, which is probably due to electronic decoupling between the conjugated core and (nearly orthogonal) Tz substituents. These properties highlight the potential of Tz functionalization as a possible approach for design of bioimaging and pH sensing of fluorescent dyes. Given the reasonable thermal (Tdec >200 °C) and photostability displayed by these derivatives, we anticipate that the electron-withdrawing properties and easy ionization of Tz-substituents will enrich the design toolbox of organic electronic materials.
This work was funded by the NSERC of Canada and FRQNT. The authors thank Digital Alliance Canada for providing access to computational resources. C. R. acknowledges the doctoral scholarship from FRQNT. H. V. H. acknowledges the postdoctoral scholarship from FRQNT.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- Z. B. Henson, K. Müllen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS PubMed.
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 Search PubMed.
- Y.-H. Liu and D. F. Perepichka, J. Mater. Chem. C, 2021, 9, 12448–12461 Search PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- H. S. Vogelbaum and G. Sauvé, Synth. Met., 2017, 223, 107–121 CrossRef CAS.
- X. Deng, Y. Gao, F. Jiang, Y. Zhang, J. Zhu and M. Wang, ACS Appl. Mater. Interfaces, 2024, 16, 32481–32489 CrossRef CAS PubMed.
- A. D. Hendsbee, J.-P. Sun, W. K. Law, H. Yan, I. G. Hill, D. M. Spasyuk and G. C. Welch, Chem. Mater., 2016, 28, 7098–7109 Search PubMed.
- C. Hansch, A. Leo and R. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 Search PubMed.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 Search PubMed.
- Y. Li, E. Huang, X. Guo and K. Feng, Mater. Chem. Front., 2023, 7, 3803–3819 RSC.
- R. Vishwakarma, C. Gadipelly and L. K. Mannepalli, ChemistrySelect, 2022, 7, e202200706 CrossRef CAS.
- V. A. Ostrovskii, E. A. Popova and R. E. Trifonov, Adv. Heterocycl. Chem., 2017, 123, 1–62 Search PubMed.
- M. V. Kaplanskiy, O. E. Faizullina and R. E. Trifonov, J. Phys. Chem. A, 2023, 127, 5572–5579 Search PubMed.
- G. Zhang, X. Hao, Y. Zou, S. Liu, J. Wei, Z. Dong and Z. Ye, J. Mater. Chem. A, 2024, 12, 33249–33256 Search PubMed.
- K. Ponnuvel, V. Padmini and R. Sribalan, Sens. Actuators, B, 2016, 222, 605–611 Search PubMed.
- R. S. Fernandes, J. Kumari, D. Sriram and N. Dey, ACS Appl. Bio Mater., 2023, 6, 4158–4167 Search PubMed.
- J.-Y. Li, X.-Q. Zhou, Y. Zhou, Y. Fang and C. Yao, Spectrochim. Acta, Part A, 2013, 102, 66–70 Search PubMed.
- W.-H. Ding, W. Cao, X.-J. Zheng, W.-J. Ding, J.-P. Qiao and L.-P. Jin, Dalton Trans., 2014, 43, 6429–6435 Search PubMed.
- S. Janietz, J. Barche, A. Wedel and D. Sainova, Macromol. Chem. Phys., 2004, 205, 187–198 CrossRef CAS.
- M. A. Reddy, A. Thomas, K. Srinivas, V. J. Rao, K. Bhanuprakash, B. Sridhar, A. Kumar, M. N. Kamalasanan and R. Srivastava, J. Mater. Chem., 2009, 19, 6172–6184 Search PubMed.
- H.-S. Wang, M.-S. Su and K.-H. Wei, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3331–3339 Search PubMed.
- Z. J. Chermahini, A. N. Chermahini, H. A. Dabbagh and A. Teimouri, J. Energy Chem., 2015, 24, 770–778 CrossRef.
- S. Ramasamy, M. Boopathy, S. Johnsanthoshkumar and K. Subramanian, Polymer, 2017, 115, 128–136 CrossRef CAS.
- C. Leonhardt, A. Mauri, I. Garin, N. W. Rosemann, W. Wenzel, U. Lemmer, M. Kozlowska and S. Bräse, Chem. – Eur. J., 2024, 30, e202401682 CrossRef CAS PubMed.
- Y.-H. Liu, P. Ghamari, M. Wei, C. Ruchlin, D. Cui, F. Rosei and D. F. Perepichka, Chem. Mater., 2024, 36, 11618–11627 CrossRef CAS.
- Z. Wu, W. Liu, X. Yang, W. Li, L. Zhao, K. Chi, X. Xiao, Y. Yan, W. Zeng, Y. Liu, H. Chen and Y. Zhao, Angew. Chem., Int. Ed., 2023, 62, e202307695 Search PubMed.
- C. R. Harding, J. Cann, A. Laventure, M. Sadeghianlemraski, M. Abd-Ellah, K. R. Rao, B. S. Gelfand, H. Aziz, L. Kaake, C. Risko and G. C. Welch, Mater. Horiz., 2020, 7, 2959–2969 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, 1H NMR spectra, absorption/emission spectra, TGA curves, and DFT calculations. See DOI: https://doi.org/10.1039/d5cc00813a |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |