
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enabling Ca plating and stripping by electrolyte manipulation in low-volatility solvents for Ca metal batteries†
Kohei
Shibuya
 ab,
Kazuaki
Kisu
*c,
Arunkumar
Dorai
d,
Yukiya
Shinoda
a,
Takara
Shinohara
ab and
Shin-ichi
Orimo
*ae
ab,
Kazuaki
Kisu
*c,
Arunkumar
Dorai
d,
Yukiya
Shinoda
a,
Takara
Shinohara
ab and
Shin-ichi
Orimo
*ae
aInstitute for Materials Research (IMR), Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan. E-mail: shin-ichi.orimo.a6@tohoku.ac.jp
bIchikawa Research Centre, Sumitomo Metal Mining Co. Ltd., Nakakokubun 3-18-5, Ichikawa, Chiba 272-8588, Japan
cCollege of Engineering, Shibaura Institute of Technology, Toyosu 3-7-5 Koto-ku, Tokyo 135-8548, Japan. E-mail: kkisu@shibaura-it.ac.jp
dInstitute of Multidisciplinary Research for Advanced Materials Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan
eAdvanced Institute for Materials Research (AIMR), Tohoku University, Katahira 2-1-1, Aoba-ku, Sendai 980-8577, Japan
First published on 20th May 2025
Abstract
Efficient Ca plating/stripping and the development of low-volatility electrolytes remain key challenges for the broad-scale application of Ca metal batteries. In this study, we demonstrate that CaBr2 addition modifies the electrolyte environment, enabling highly reversible Ca plating/stripping in electrolytes based on low-volatility diglyme as a solvent previously considered nonfunctional.
Ca metal batteries have gained considerable attention as a next-generation energy storage technology owing to the natural abundance, low reduction potential (−2.87 V vs. standard hydrogen electrode), and high volumetric capacity (2072 mA h cm−3) of Ca.1 However, the practical implementation of Ca metal batteries is restricted because of the limitations in designing practical electrolytes that exhibit low volatility and allow efficient Ca plating/stripping.2
Recently, electrolytes containing weakly coordinating anions, such as Ca(B[hfip]4)23,4 and Ca(CB11H12)2,5,6 have gained increasing attention for enabling reversible Ca plating/stripping at room temperature. Ca(CB11H12)2 is effective because of its fluorine-free nature, which prevents the formation of the Ca2+ diffusion–blocking CaF2. In conventional electrolyte systems based on Ca(CB11H12)2, 1,2-dimethoxyethane (DME) and tetrahydrofuran (THF) have been used as solvents because of their ability to weakly coordinate Ca2+ cations and resulting efficient desolvation. However, their low boiling points (DME: 82 °C, THF: 66 °C) lead to volatility and safety concerns. Despite these issues, no studies have yet explored the use of Ca(CB11H12)2 with low-volatility solvents that exhibit reversible Ca plating/stripping.
To overcome these limitations, diglyme (G2) has been proposed as an alternative solvent, exhibiting a high boiling point (162 °C) while maintaining properties similar to those of DME (known as monoglyme, G1) and THF. However, the extended ether chain of G2 enhances Ca2+ solvation and may hinder desolvation. Studies on similar ether-based multivalent electrolytes7,8 suggest that this effect increases the energy barrier for Ca2+ desolvation, resulting in increased overpotentials and sluggish electrochemical kinetics. Therefore, optimising solvation dynamics in G2-based electrolytes is crucial for efficient Ca plating/stripping.
Solvation manipulation using dual salt additives, explored in Mg and Ca systems, is a promising approach.9–11 Among the potential additives, Br− anions demonstrate a substantial effectiveness for modifying the solvation structure.6,12 Br− anions weaken excessive solvation interactions, facilitating desolvation and accelerating cation transport across the electrode interface.13 Additionally, Br− anions suppress the generation of insulating byproducts, such as CaCO3, thereby promoting the development of an ion-conductive solid electrolyte interphase (SEI) and enhancing long-term electrochemical stability.14,15
In this study, we selected CaBr2 as an additive because of its adequate solubility in G2, verified electrochemical activity, and compatibility with low-volatility electrolyte systems. Regarding other calcium halides, CaI2 offers the advantages of a low Ca2+ diffusion barrier and favourable SEI potential but is poorly soluble in ethers,16 whereas CaCl2 was found to be partially soluble but showed no Ca plating/stripping behaviour in our preliminary experiments. Hence, we prepared G2-based electrolytes containing 0.01, 0.03, or 0.05 M CaBr2 and 0.4 M Ca(CB11H12)2 to investigate their electrochemical performance and determine the mechanisms underlying solvation structure modulation and SEI formation. Higher concentrations were not used because of the solubility limit of CaBr2 (0.06 M). The details of experimental methods are provided in the ESI.†
The Ca plating/stripping performance of the Ca(CB11H12)2-based electrolytes with CaBr2 was investigated through cyclic voltammetry (CV; Fig. 1a, b and Fig. S1, S2). Notably, the addition of a small amount of CaBr2 (0.01–0.05 M) drastically improved Ca plating/stripping behaviour, whereas no distinct peaks were observed in the absence of CaBr2 (Fig. 1a inset). These results suggest that the strong coordination between G2 and Ca2+ inhibits Ca plating/stripping.17 To examine electrolyte durability during Ca plating/stripping, we evaluated the overvoltage behaviour through galvanostatic cycling tests (Fig. 1c). At 0.01 M CaBr2, the voltage gradually rose to 1 V, suggesting resistive-phase build-up due to electrolyte decomposition. In contrast, for 0.03 and 0.05 M CaBr2, the overvoltage increase was substantially suppressed compared with that observed at 0.01 M CaBr2. The final cycle overvoltage was 0.8 V at 0.03 M CaBr2 and 0.6 V at 0.05 M CaBr2, indicating an improvement in electrochemical performance. These enhancements were attributed to solvation structure modifications induced by Br− anions and changes in the SEI composition. Ionic conductivity measurements (Fig. S3, ESI†) showed minimal impact from CaBr2 addition, indicating a limited contribution to the performance enhancement.
 | ||
| Fig. 1 (a) Cyclic voltammograms of Ca plating/stripping for 0.4 M Ca(CB11H12)2 with 0.05 M CaBr2 in diglyme (G2) at the fourth cycle. Data for conditioning cycles are shown in Fig. S1 (ESI†). Inset: Presents the cyclic voltammogram recorded without CaBr2. (b) Geometry of the CB11H12− anion. (c) Galvanostatic Ca plating/stripping cycling performance of Ca/Ca symmetric cells at a current density of 5 μA cm−2. Insets: Show enlarged voltage profiles at intermediate and final cycles. | ||
The coordination environment of Ca2+ was investigated through nuclear magnetic resonance (NMR) and Raman spectroscopies. The 43Ca NMR spectra featured sharp peaks at all CaBr2 concentrations, indicating a fast exchange between coordination states and highly symmetrical average structure (Fig. 2a). The chemical shift change from −28.4 ppm at 0 M to −13.3 ppm at 0.05 M suggested a concomitant decrease in the shielding of the Ca nucleus. This trend was attributed to the substitution of coordinating G2 molecules by Br− anions, which promoted the formation of contact ion pairs (CIPs) in place of solvent-separated ion pairs (SSIPs).18,19 Separately, the addition of CaBr2 had no noticeable effect on the 1H and 11B NMR spectra (Fig. S4, ESI†), which indicated that the interaction of this additive with the CB11H12− anion was minimal.
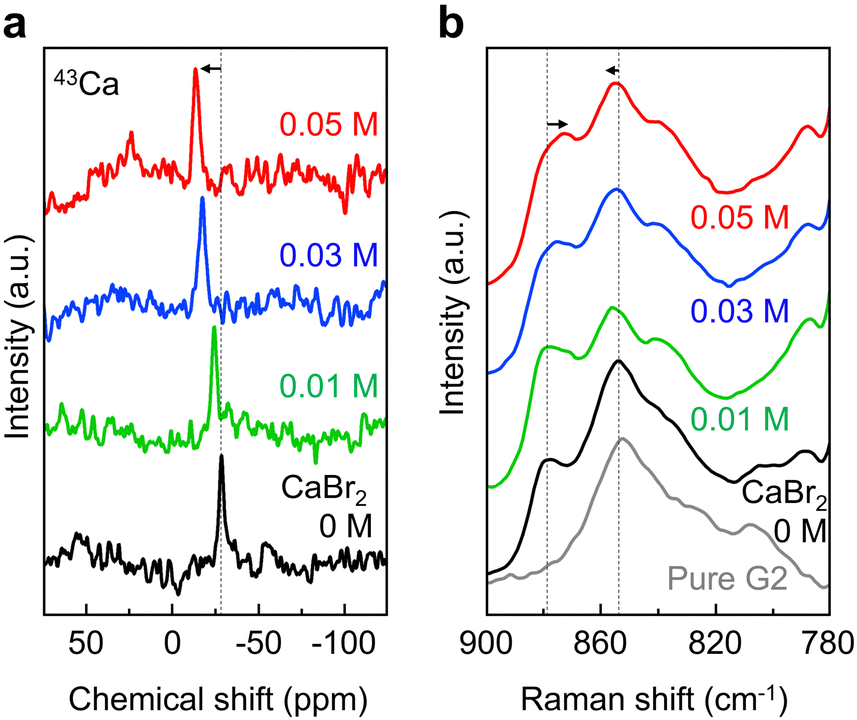 | ||
| Fig. 2 Coordination environment investigation of Ca2+ in 0.4 M Ca(CB11H12)2 with 0, 0.01, 0.03, and 0.05 M CaBr2 in G2. (a) 43Ca NMR and (b) Raman spectra. | ||
The results of Raman spectroscopy analysis further supported this structural evolution. For pure G2, characteristic vibrational bands appeared at 852, 826, and 808 cm−1 (Fig. 2b and Fig. S5, ESI†). Upon the addition of Ca(CB11H12)2, new peaks emerged at 877.5 and 835 cm−1, corresponding to Ca2+–G2 coordination, as previously reported for G2-based K-ion systems with symmetric solvation structures.20 As the CaBr2 concentration increased from 0 to 0.05 M, the 877.5 cm−1 band shifted to 872.8 cm−1, which indicated a weakening of the Ca2+–G2 interaction, probably due to the progressive replacement of G2 molecules by Br− anions.18 This vibrational shift was consistent with the NMR spectroscopy-observed solvation change and confirmed the progressive formation of CIPs in the presence of Br− anions. In addition to the red shift, a minimal blue shift was observed in the C–O–C symmetric stretching region. The corresponding band shifted from 854.2 cm−1 (free G2) to 854.4 cm−1 (0 M CaBr2) and further to 855.4 cm−1 (0.05 M CaBr2). The initial shift reflected the coordination of Ca2+ to G2, and the subsequent blue shift was attributed to the increased rigidity or symmetry in the Ca2+–G2 coordination environment induced by Br−. This interpretation is supported by the results of theoretical studies on glyme–metal ion complexes, where similar frequency shifts were correlated with changes in coordination structure and bond rigidity.21
To investigate the SEI formation changes induced by CaBr2 addition, the Ca metal surface after 30 min of electrolyte soaking was analysed using ex situ X-ray photoelectron spectroscopy (XPS; Fig. 3a). The deconvoluted C 1s spectra featured three peaks attributable to alkyl carbon (C–C) at 284.5 eV, oxygen-bound carbon (C–O) at 286.5 eV, and electron-withdrawing group–associated carbon (COO) at 288.0 eV. At 0.05 M CaBr2, the contents of C–O and COO species were lower than those at 0 M, which suggested a reduction in the amount of organic materials and the suppressed formation of G2-derived decomposition products. The O 1s spectra featured a dominant peak at 531 eV (C–O) and additional peaks at 533 eV (O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and 530 eV (Ca–O). At 0.05 M CaBr2, the Ca–O peak gained intensity, whereas the O–CO peak lost intensity, which indicated CaO formation and the suppression of organic species formation. Similarly, the Ca 2p spectra displayed a consistent trend, revealing an increase in the peak intensities for CaO (346.5 and 350 eV) and a decrease in those for CaCO3 (347 and 350.5 eV) as the CaBr2 concentration increased. As CaCO3 hinders Ca2+ ion movement,22,23 this compositional shift suggests potential improvements in ion transport. Similar trends were observed after galvanostatic Ca plating/stripping cycling (Fig. S6, ESI†), in line with the results shown in Fig. 1c. In the absence of Br−, post-CV XPS analysis indicated the loss of polymeric components and emergence of CaCO3 and CaO (Fig. S7, ESI†), suggesting that the SEI had a limited stability. The B 1s spectra exhibited a dominant peak at 188.4 eV corresponding to the B–H moieties of the CB11H12− anion, with no detectable peak at ∼191 eV corresponding to B–O species;6 this finding confirmed the chemical stability of the CB11H12− anion on the Ca metal surface. This anion may be encapsulated within organic components and polymeric phases to afford a gel-like polymer electrolyte. The Br 3d spectrum showed peaks exclusively at 0.05 M CaBr2, suggesting the formation of a Br-containing SEI. As shown in Fig. 3b, surface atomic composition analysis revealed a decrease in the atomic concentration of C and an increase in those of Ca and Br, indicating a transition from an organic-rich SEI to an inorganic-rich SEI.
O) and 530 eV (Ca–O). At 0.05 M CaBr2, the Ca–O peak gained intensity, whereas the O–CO peak lost intensity, which indicated CaO formation and the suppression of organic species formation. Similarly, the Ca 2p spectra displayed a consistent trend, revealing an increase in the peak intensities for CaO (346.5 and 350 eV) and a decrease in those for CaCO3 (347 and 350.5 eV) as the CaBr2 concentration increased. As CaCO3 hinders Ca2+ ion movement,22,23 this compositional shift suggests potential improvements in ion transport. Similar trends were observed after galvanostatic Ca plating/stripping cycling (Fig. S6, ESI†), in line with the results shown in Fig. 1c. In the absence of Br−, post-CV XPS analysis indicated the loss of polymeric components and emergence of CaCO3 and CaO (Fig. S7, ESI†), suggesting that the SEI had a limited stability. The B 1s spectra exhibited a dominant peak at 188.4 eV corresponding to the B–H moieties of the CB11H12− anion, with no detectable peak at ∼191 eV corresponding to B–O species;6 this finding confirmed the chemical stability of the CB11H12− anion on the Ca metal surface. This anion may be encapsulated within organic components and polymeric phases to afford a gel-like polymer electrolyte. The Br 3d spectrum showed peaks exclusively at 0.05 M CaBr2, suggesting the formation of a Br-containing SEI. As shown in Fig. 3b, surface atomic composition analysis revealed a decrease in the atomic concentration of C and an increase in those of Ca and Br, indicating a transition from an organic-rich SEI to an inorganic-rich SEI.
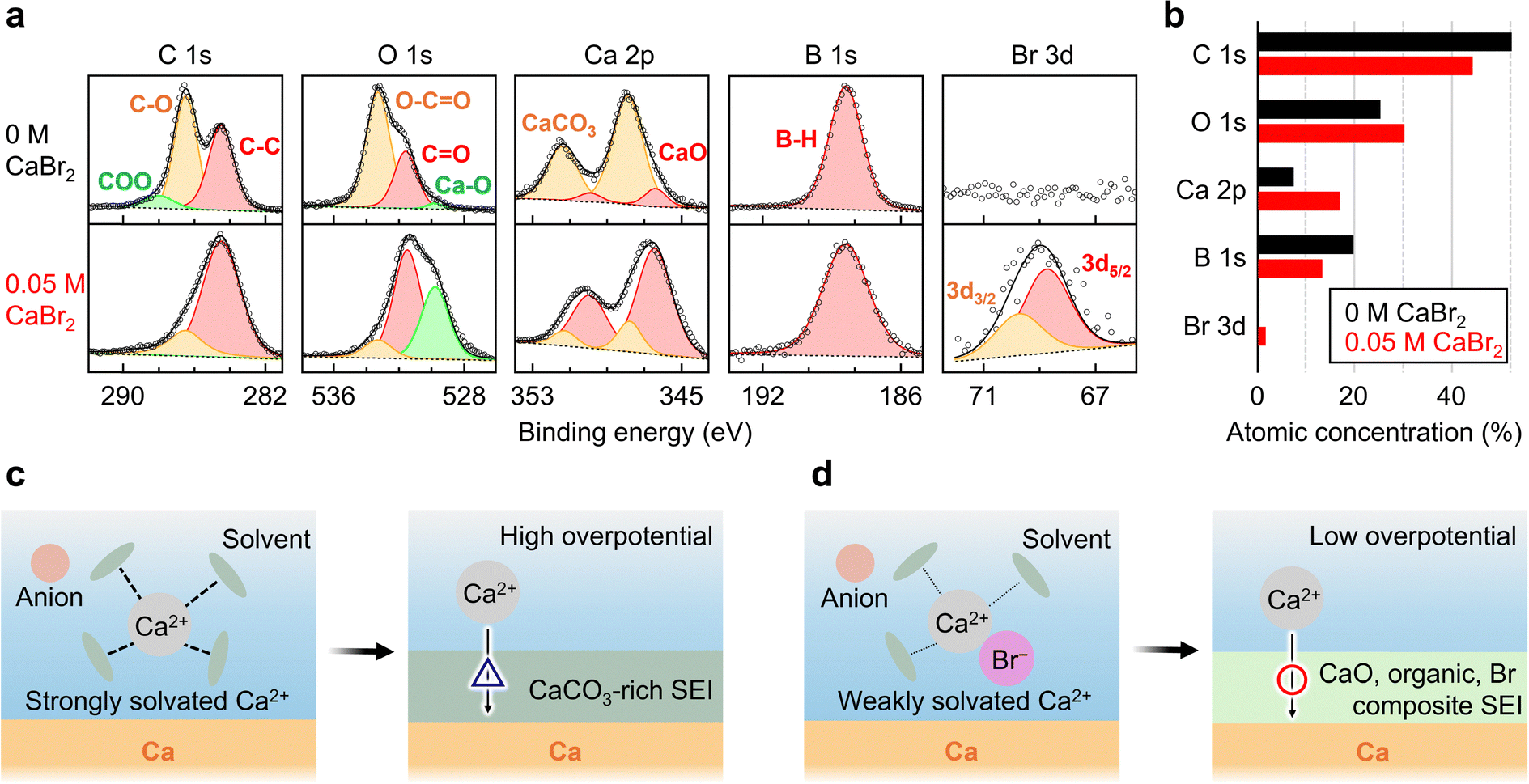 | ||
| Fig. 3 (a) C 1s, O 1s, Ca 2p, B 1s, and Br 3d spectra of Ca metal soaked for 30 min in 0.4 M Ca(CB11H12)2 with 0 and 0.05 M CaBr2 in G2. (b) Atomic concentration (atom%) of the passivation layer components on the Ca metal surface. (c) and (d) Schematics of solvation structure and SEI formation in electrolytes (c) without and (d) with CaBr2. | ||
Based on these results, the schematic illustrations in Fig. 3c and d provide a plausible explanation for the solvation structures and SEI formation mechanisms in the presence and absence of CaBr2. In the absence of CaBr2, the Ca metal surface forms a CaCO3-rich SEI, which impedes Ca2+ conduction and results in a high overpotential. This behaviour can be attributed to the predominant presence of weakly coordinating CB11H12− anions, which may promote the formation of SSIPs. During the desolvation process, solvent decomposition is expected to prevail over anion interactions, leading to the excessive formation of CaCO3. Conversely, the introduction of CaBr2 substantially alters SEI composition, resulting in the incorporation of CaO, CaBr2, organic compounds, and polymeric structures. This SEI composition enhances Ca2+ transport and stabilises Ca plating/stripping, reducing overvoltage. The suppression of CaCO3 formation is ascribed to CIP formation induced by Br−, which suppresses solvent decomposition during desolvation and promotes the formation of SEI structures that are more conducive to Ca2+ conduction.
To assess the viability of the developed electrolyte, an initial investigation was performed using a Ca–S battery (Fig. 4a), known for its notably high theoretical specific capacity.24 A brief evaluation of oxidative stability revealed that all electrolytes were anodically stable above 3.3 V (Fig. S8, ESI†), which was sufficient for operation with sulfur cathodes. The initial discharge and charge capacities were determined as 536 and 512 mA h g−1, respectively, demonstrating a reversible electrochemical behaviour (Fig. 4b). These values were lower than those achieved for Ca(CB11H12)2 in DME/THF (805 and 705 mA h g−1, respectively),5 possibly because the higher solubility of reaction intermediates in G2 led to active material loss and reduced capacity during cycling (Fig. S9, ESI†).
 | ||
| Fig. 4 (a) Schematic of a prototype Ca metal battery using an electrolyte of 0.4 M Ca(CB11H12)2 with 0.05 M CaBr2 in G2. (b) Voltage profile during the first cycle. (c) S 2p spectra of S/C electrodes in pristine (top) and discharged (bottom) states. | ||
To investigate the conversion processes in the S/C cathode, we characterised pristine and discharged electrodes by XPS. The S 2p spectrum of the pristine cathode showed a spin–orbit doublet characteristic of elemental sulphur at 164.0 and 165.2 eV (Fig. 4c, top). After discharge to 0.5 V, the spectrum featured three doublets attributable to residual sulphur, CaSn, and CaS (Fig. 4c, bottom).24,25 The predominance of CaS indicated the effective conversion of sulphur to sulphides, validating the active participation of the sulphur cathode. This finding suggests that the capacity loss in G2 stems from CaSn dissolution, not unreacted sulphur, and implies that the further optimisation of the solvent composition to suppress polysulfide dissolution could enhance the electrochemical performance.5 Compared with DME/THF,5 G2 facilitated the dissolution of CaSn intermediates, leading to a decreased surface coverage by CaS and CaSn and relatively enhanced visibility of these oxidised species in the XPS spectra.
In summary, this study revealed that the incorporation of CaBr2 into Ca(CB11H12)2-based electrolytes in G2 substantially enhanced Ca plating/stripping efficiency by modifying both solvation structure and SEI composition. The Br− anions promoted CIP formation, suppressing solvent decomposition and minimising CaCO3 formation, which hindered Ca2+ mobility. This improvement was reflected in the enhanced long-term electrochemical stability and reduced overvoltage. Furthermore, the optimised electrolyte enabled the successful operation of a Ca–S battery, demonstrating its potential for the development of high-energy-density systems. These findings highlight the role of anion engineering in advancing practical Ca metal batteries.
This work was supported by a JSPS KAKENHI Grant-in-Aid for Scientific Research B (No. 22H01803) and JST FOREST Program (Grant Number JPMJFR236F). Additional support was provided by Sumitomo Metal Mining Co., Ltd., the Murata Science and Education Foundation (No. M24AN132), and the Iketani Science and Technology Foundation (No. 0361179-A).
Data availability
All relevant experimental data within the article will be provided by the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Notes and references
- X. L. Huang, X. Li, M. Yang, Y. Yang, J. Qian, L. Yao, K. Zhu, H. K. Liu and Y. X. Wang, Chem. Commun., 2025, 61, 2156–2172 RSC.
- A. M. Melemed, A. Khurram and B. M. Gallant, Batteries Supercaps, 2020, 3, 570–580 CrossRef CAS PubMed.
- Z. Li, O. Fuhr, M. Fichtner and Z. Zhao-Karger, Energy Environ. Sci., 2019, 12, 3496–3501 RSC.
- A. Shyamsunder, E. Blanc, A. Assoud and F. Nazar, ACS Energy Lett., 2019, 4, 2271–2276 CrossRef CAS.
- K. Kisu, S. Kim, T. Shinohara, K. Zhao, A. Zuttel and S. Orimo, Sci. Rep., 2021, 11, 7563 CrossRef CAS PubMed.
- K. Kisu, A. Dorai, K. Hatakeyama-Sato, T. Takano, S. Takagi, K. Oyaizu and S. Orimo, ACS Appl. Mater. Interfaces, 2025, 17, 1322–1331 CrossRef CAS PubMed.
- A. M. Melemed, D. A. Skiba and B. M. Gallant, J. Phys. Chem. C, 2022, 126, 892–902 CrossRef CAS PubMed.
- C. Li, R. D. Guha, A. Shyamsunder, K. A. Persson and L. F. Nazar, Energy Environ. Sci., 2024, 17, 190–201 RSC.
- J. Long, Y. Liu, Z. He, S. Tan, F. Xiong, H. Xu, W. Wang, G. Zhang, Z. Yang and Q. An, ACS Nano, 2024, 18, 15239–15248 CrossRef CAS PubMed.
- A. T. Landers, J. Self, S. A. McClary, K. J. Fritzsching, K. A. Persson, N. T. Hahn and K. R. Zavadil, J. Phys. Chem. C, 2023, 127, 23664–23674 CrossRef CAS.
- S. Yang, X. Wang, R. Li, Y. Zhou, H. Huang, M. Zhou, Y. Gao, W. Zhao, Y. Gao, Z. Pan and X. Yang, Energy Environ. Sci., 2025, 18, 1941–1951 RSC.
- Y. Yi, Y. Xing, H. Wang, Z. Zeng, Z. Sun, R. Li, H. Lin, Y. Ma, X. Pu, M. M. Li, K. Y. Park and Z. L. Xu, Angew. Chem., Int. Ed., 2024, 63, e202317177 CrossRef CAS PubMed.
- D. Chinnadurai, W. Y. Lieu, S. Kumar, G. Yang, Y. Li and Z. W. Seh, Nano Lett., 2023, 23, 1564–1572 CrossRef CAS PubMed.
- Z. Hou, R. Zhou, Z. Min, Z. Lu and B. Zhang, ACS Energy Lett., 2022, 8, 274–279 CrossRef.
- J. G. Connell, M. Zorko, G. Agarwal, M. Yang, C. Liao, R. S. Assary, D. Strmcnik and N. M. Markovic, ACS Appl. Mater. Interfaces, 2020, 12, 36137–36147 CrossRef CAS PubMed.
- Z. Hou, R. Zhou, K. Liu, J. Zhu and B. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202413416 CrossRef CAS PubMed.
- N. T. Hahn, D. M. Driscoll, Z. Yu, G. E. Sterbinsky, L. Cheng, M. Balasubramanian and K. R. Zavadil, ACS Appl. Energy Mater., 2020, 3, 8437–8447 CrossRef CAS.
- J. D. Forero-Saboya, E. Marchante, R. B. Araujo, D. Monti, P. Johansson and A. Ponrouch, J. Phys. Chem. C, 2019, 123, 29524–29532 CrossRef CAS PubMed.
- M. Shakourian-Fard, G. Kamath, S. M. Taimoory and J. F. Trant, J. Phys. Chem. C, 2019, 123, 15885–15896 CrossRef CAS.
- T. Hosaka and S. Komaba, Bull. Chem. Soc. Jpn., 2022, 95, 569–581 CrossRef CAS.
- N. R. Dhumal and S. P. Gejji, Chem. Phys., 2006, 323, 595–605 CrossRef CAS.
- S. A. McClary, D. M. Long, A. Sanz-Matias, P. G. Kotula, D. Prendergast, K. L. Jungjohann and K. R. Zavadil, ACS Energy Lett., 2022, 7, 2792–2800 CrossRef CAS.
- J. Forero-Saboya, C. Davoisne, R. Dedryvère, I. Yousef, P. Canepa and A. Ponrouch, Energy Environ. Sci., 2020, 13, 3423–3431 RSC.
- A. Scafuri, R. Berthelot, K. Pirnat, A. Vizintin, J. Bitenc, G. Aquilanti, D. Foix, R. Dedryvère, I. Arčon, R. Dominko and L. Stievano, Chem. Mater., 2020, 32, 8266–8275 CrossRef CAS.
- Z. Li, B. P. Vinayan, T. Diemant, R. J. Behm, M. Fichtner and Z. Zhao-Karger, Small, 2020, 16, e2001806 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Results of additional instrumental analyses. See DOI: https://doi.org/10.1039/d5cc01242b |
| This journal is © The Royal Society of Chemistry 2025 |