
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalyzed iodonium ion promoted activation of conventional thioglycosides for stereoselective 1,2-cis furanosylations†
Surya Pratap Singh,
Umesh Chaudhary,
Adrienne Daróczi and
Indrajeet Sharma *
*
Department of Chemistry and Biochemistry, University of Oklahoma, 101 Stephenson Parkway, Norman, OK-73019, USA. E-mail: isharma@ou.edu; Web: https://indrajeetsharma.com
First published on 30th May 2025
Abstract
Iron-catalyzed activation of conventional thioglycosides using the iodonium ion enabled the selective synthesis of 1,2-cis furanosides. This strategy accommodates diverse furanosyl donors (D-ribose, D-arabinose, L-arabinose) and various O-nucleophiles (1°, 2°, 3°). Additionally, 1,2-cis hexaribofuranoside was synthesized, and the influence of the C2 functional group on selectivity was investigated.
The synthesis of furanosides has garnered significant attention due to their essential role in biological systems and their presence as core structures in approved drugs, deoxyribonucleic acid (DNA) and ribonucleic acid (RNA).1 Stereochemically, furanosides exist in two distinct forms: (1) 1,2-cis and (2) 1,2-trans.2 While the synthesis of 1,2-trans furanosides is well-established through anchimeric assistance, constructing 1,2-cis furanosides remains a formidable challenge for synthetic chemists.3 Several catalytic approaches4 have been developed, employing chiral bis-thiourea,5 phenanthroline,6 gold,7 and copper catalysis.8 Although these strategies are elegant, they pose significant limitations, such as the laborious synthesis of chiral thiourea catalysts, multiple steps to access furanoside donors, and reliance on precious metals (Scheme 1a). A carefully designed and efficient strategy is required to address these challenges, which employs (1) an inexpensive, non-toxic catalyst with a high turnover number and (2) a furanosyl donor that can be easily accessible and remains benchtop stable for extended periods.
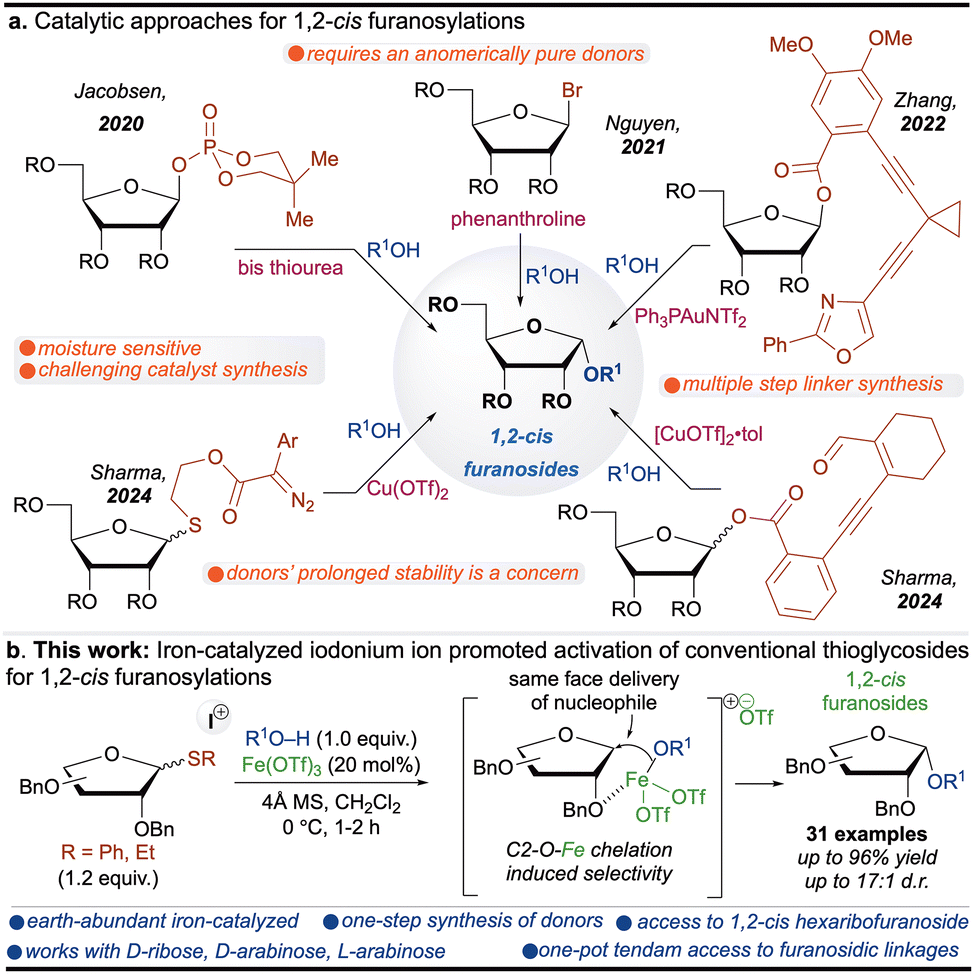 | ||
| Scheme 1 (a) Catalytic approaches for 1,2-cis furanosylations. (b) Our work. | ||
Iron (Fe), the second most abundant metal in Earth's crust (∼5%),9 is essential in biological systems, where it functions as a key component of various enzymatic processes.10 Its abundance, low cost, non-toxicity, and environmentally benign nature make it an attractive alternative to toxic, rare, and expensive coinage and platinum group metals (PGMs).11 Given these advantages, iron holds great potential as a catalyst for the stereoselective synthesis of 1,2-cis furanosides in a cascade manner. However, its use in such transformations remains underexplored, primarily due to its tendency to rapidly cycle through multiple oxidation states during chemical reactions.12 Identifying suitable additives to mitigate these limitations, along with easily accessible benchtop stable glycosyl donors, could enhance the efficiency of iron-catalyzed glycosylation.
Thioglycosides are particularly robust and versatile glycosyl donors, valued for their prolonged stability, which makes them ideal for the automated synthesis of complex oligosaccharides.13 Notably, they can be synthesized in a single step from commercially available materials.14 However, their activation traditionally requires potent stoichiometric activating agents, presenting a challenge for catalytic transformations.15 Interestingly, sulfur's unique reactivity with halonium ions under metal-catalyzed conditions enables the catalytic activation of thioglycosides.16
Here, we propose a synergistic approach leveraging iron catalysis, halonium ion activation, and conventional thioglycosides to achieve challenging furanosidic linkages. This strategy enables a one-pot cascade activation, where thioglycosides are first activated, forming an oxocarbenium ion. Subsequent C2-O-iron coordination directs the approach of the glycosyl acceptor from the cis face, as demonstrated in our previous research.8 Utilizing these stable thioglycosides, this methodology provides access to unique and synthetically challenging 1,2-cis hexaribofuranosides (Table 1).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 2 | 3 (equiv.) | MLn | t (°C) | Time (h) | 4, yieldb | 4, dr(α/β) |
| a All the reactions were performed with acceptor (0.025 mmol, 1.0 equiv.), donor (0.03 mmol, 1.2 equiv.), additive (0.025 mmol, 1.0 equiv.), and metal catalyst (0.005 mmol, 0.2 equiv.), molecular sieves MS (100 wt%), and CH2Cl2 (0.05 M).b Yields were calculated after analyzing 1H-NMR using 1,3,5-trimethoxybenzene as internal standard.c Isolated yield. | |||||||
| 1. | 2a | 3a (1.5) | Fe(OTf)3 | rt | 1 | 28% | 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2. | 2a | 3b (1.5) | Fe(OTf)3 | rt | 1 | 46% | 3:1 |
| 3. | 2a | 3c (1.5) | Fe(OTf)3 | rt | 1 | 63% | 5:1 |
| 4. | 2a | 3c (1.5) | Fe(OTf)3 | 0 | 1.5 | 81% | 6:1 |
| 5. | 2a | 3c (1.5) | Fe(OTf)3 | −10 | 2 | 79% | 6:1 |
| 6. | 2b | 3c (1.5) | Fe(OTf)3 | 0 | 1.5 | 73% | 5:1 |
| 7. | 2c | 3c (1.5) | Fe(OTf)3 | 0 | 1.5 | 86% | 5:1 |
| 8. | 2a | 3c (1.0) | Fe(OTf)3 | 0 | 1.5 | 94(91%)c | 6:1 |
| 9. | 2a | 3c (1.0) | — | 0 | 17 | 61% | 3:1 |
| 10. | 2a | — | Fe(OTf)3 | 0 | 24 | n.r. | n.d. |
| 11. | 2a | 3c (1.0) | Fe(OTf)2 | 0 | 2 | 83% | 5:1 |
| 12. | 2a | 3c (1.0) | Cu(OTf)2 | 0 | 2 | 62% | 5:1 |
| 13. | 2a | 3c (1.0) | Zn(OTf)2 | 0 | 2 | 72% | 5:1 |
| 14. | 2a | 3c (1.0) | Ag(OTf) | 0 | 2 | 59% | 2:1 |

To optimize the reaction conditions, we initially selected phenyl 2,3,5-tri-O-benzyl-1-thio-D-ribofuranoside (2a) as the model furanosyl donor, methyl 6-OH-2,3,4-tri-O-benzyl-α-D-glucopyranoside (1a) as the glycosyl acceptor, and N-chlorosuccinimide (NCS, 3a) as the halonium ion source. Based on our previous studies highlighting the role of triflate ions in glycosylation chemistry,17 we chose Fe(OTf)3 as the iron catalyst, considering its efficiency in promoting glycosylation reactions. Upon activation of donor 2a, we obtained the desired furanoside in low yield with moderate selectivity (α:β = 2:1, entry 1). Introducing N-bromosuccinimide (NBS, 3b) as an additive improved both the yield and selectivity (entry 2). A further enhancement was observed when N-iodosuccinimide (NIS, 3c) was employed, significantly increasing both yield and selectivity (α:β = 5:1) under ambient conditions (entry 3). To refine the reaction parameters, we tested lower temperatures (entries 4 and 5, see Table S-1 in ESI†), finding that 0 °C was optimal, delivering a higher yield with improved selectivity (α:β = 6:1). Next, we screened alternative thioglycoside donors, including tolyl 2,3,5-tri-O-benzyl-1-thio-D-ribofuranoside (2b, entry 6) and ethyl 2,3,5-tri-O-benzyl-1-thio-D-ribofuranoside (2c, entry 7). Reducing the additive amount to 1 equivalent under the optimized conditions further improved the yield (entry 8). To probe the role of Fe(OTf)3, we conducted the reaction without the catalyst (entry 9). As expected, the yield declined significantly, and the reaction proceeded sluggishly with reduced selectivity (α:β = 3:1), underscoring the catalyst's importance. Additionally, when the reaction was performed without an additive (entry 10), no product formation was observed even after 24 hours, highlighting the essential role of the iodonium ion in initiating the reaction. These findings demonstrate the synergistic effect between the iodonium ion and the iron catalyst in driving the glycosylation process. To further evaluate the catalytic system, we screened other earth-abundant metal triflate salts, including Fe(OTf)2 (entry 11), Cu(OTf)2 (entry 12), and Zn(OTf)2 (entry 13). Among these, Fe(OTf)3 emerged as the most effective catalyst. Screening AgOTf (entry 14) resulted in a marked decrease in yield and diastereoselectivity. We also screened triflic acid (TfOH) as a catalyst, with and without molecular sieves, and it produced lower yields and a mixture of diastereomers (see Table S-1, entries 1 and 2 in ESI†). In summary, thioglycoside donors 2a and 2c were identified as the most efficient, with 2a showing superior performance. The optimized conditions were found to be Fe(OTf)3 as the catalyst, NIS (3c) as the most suitable additive, and a reaction temperature of 0 °C.
With the optimized reaction conditions in hand, we explored the substrate scope for 1,2-cis (α)-furanosylation using D-ribosyl donor 2a and a diverse range of glycosyl acceptors. We began by evaluating the deactivated glucosyl acceptor, methyl 6-OH-2,3,4-tri-O-benzoyl-α-D-glucopyranoside (1b), which reacted efficiently to afford the desired furanoside 5 with excellent selectivity. Next, we investigated mannose acceptors 1c and 1d, representing both activating and deactivating systems. Despite literature reports suggesting poor reactivity due to the axial C2 stereochemistry,5,6 our methodology proved highly effective, delivering the corresponding furanosides 6 and 7 in high yields with excellent 1,2-cis selectivity. Given the efficient reactivity of primary nucleophiles with donor 2a, we extended our study to secondary nucleophiles, which are typically less reactive due to their lower nucleophilicity. Using methyl 4-OH-2,3,6-tri-O-benzoyl-α-D-glucopyranoside (1e) as a model secondary acceptor, we successfully obtained furanoside 8 in high yield with excellent selectivity, overcoming its previously reported low reactivity. To further expand the scope, we tested isopropanol and the bioactive natural product L-menthol. Both exhibited high reactivity and selectivity, yielding furanosides 9 and 10, respectively. Finally, we examined the tertiary alcohol 1-adamantanol, which coupled efficiently with the ribosyl donor to afford furanoside 11 in excellent yield and selectivity. Late-stage modification of bioactive and pharmaceutically relevant molecules remains a major challenge in synthetic chemistry, making such transformations highly valuable.18 In this regard, we successfully achieved late-stage 1,2-cis furanosylation of biologically important molecules, including cholesterol and oleanolic acid, yielding the desired compounds (12, 13) with high efficiency and selectivity.19 Having successfully established 1,2-cis (α)-furanosylation, we next focused on achieving 1,2-cis (β)-selectivity using L- and D-arabinose thioglycoside donors (2d, 2e). We began by examining the activated glucosyl acceptor (1a) and the deactivated glucosyl acceptor (1b), both of which coupled efficiently with the L-arabinose donor 2d to furnish disaccharides 14 and 15 in good yields with high selectivity. Similarly, the deactivated mannose acceptor (1d) performed well, yielding furanoside 16 with excellent selectivity. For D-arabinose, both activating and deactivating glycosyl acceptors, including glucose- and mannose-derived acceptors (1a–1d, 1f), reacted efficiently, affording the desired furanosides (17–21) with high 1,2-cis selectivity. Expanding the scope beyond sugar-based acceptors, the secondary nucleophile L-menthol once again demonstrated high reactivity, producing furanosides 22 in excellent yield and selectivity. Additionally, the tertiary alcohol 1-adamantanol coupled successfully with the arabinose donor, delivering furanoside 23 with remarkable efficiency. Oligosaccharides are vital biomolecules with crucial functions in biological systems, yet their synthesis remains a significant challenge. Constructing 1,2-cis oligosaccharides is notoriously difficult due to steric hindrance from the C2 position, and efficient strategies for assembling furanose-based oligosaccharides are limited.20 To overcome this hurdle, we utilized our newly designed donor 2f to achieve the iterative synthesis of a hexaribosaccharide. Remarkably, our method enabled the construction of the target hexaribosaccharide 32 with high 1,2-cis selectivity over nine steps, yielding the final product in good yield. This success highlights the effectiveness of our approach in streamlining the synthesis of complex 1,2-cis oligosaccharides (Scheme 2).
 | ||
| Scheme 2 The substrate scope of our methodology. (a) acceptor (1.0 equiv.), donor (1.2 equiv.), NIS (1.0 equiv.), Fe(OTf)3 (20 mol%), 4 Å MS (100 wt%), and CH2Cl2(0.05 M)., (b) TBAF in THF (1 mol L−1, 1.5 equiv.), THF (2.0 M). NIS, N-iodosuccinimide; TBAF, tetrabutylammonium fluoride. | ||
To investigate the influence of C2-functional groups on stereoselectivity (Scheme 3a), we examined donors with OMe (2g, coordinating but less bulky), OTIPS (2h, coordinating and bulky), and N3 (2i, weakly coordinating and linear). The consistent selectivity outcomes observed in the resulting furanosides (33–35) further reinforce the role of chelation control in selectivity induction. In our previous reports, we discovered C2-copper chelated stereoselectivity induction.8
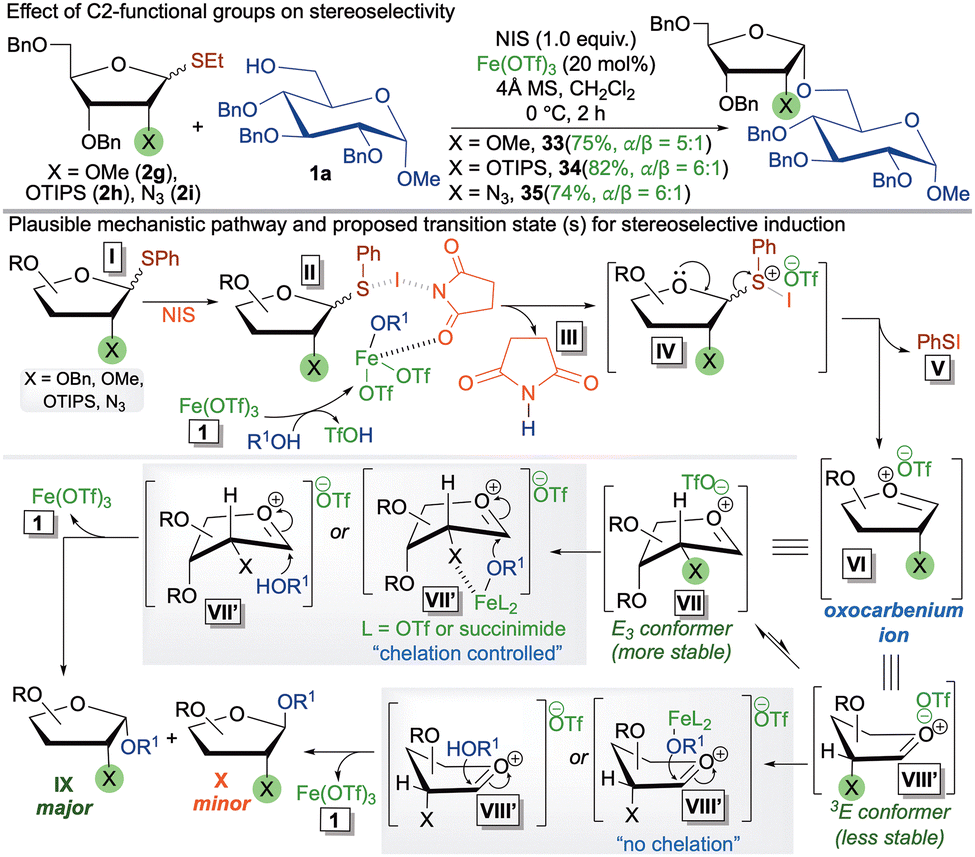 | ||
| Scheme 3 Mechanistic studies and plausible reaction pathways for 1,2-cis selectivity. | ||
These mechanistic studies provide valuable insight into the reaction pathways involved. Initially, Fe(OTf)3 undergoes ligand exchange with the glycosyl acceptor, while the thioglycoside donor I gets activated through chelation with NIS, forming complex II. This intermediate then dissociates to generate intermediate IV, with the concurrent release of succinimide III. The intermediate IV subsequently transforms into the reactive oxocarbenium ion VI, a key intermediate in glycosylation chemistry, along with the formation of phenylsulfenyl iodide V. This oxocarbenium ion adopts two envelope conformations: E3 (VII, more stable) and 3E (VIII, less stable).8 The nucleophilic attack occurs from the inner face of each conformer (see Fig. S-6 in ESI†), facilitated by the iron-chelation with C2–heteroatom in E3 conformation, furnishing the major 1,2-cis furanoside IX and the 3E conformation leading to the minor 1,2-trans furanoside X.
In conclusion, we have developed iron-catalyzed activation of conventional thioglycosides for 1,2-cis furanosylation. In this work, we have successfully reported the synthesis of a challenging 1,2-cis hexaribosaccharide. Installation of different chelating functional groups at the C2 position probed the iron chelation-induced cis selectivity.
SPS and IS conceptualized the idea. SPS performed the substrate scope and synthesis of hexaribosaccharide. UC performed the C2 functional groups studies. AD helped purify a few substrates. SPS and IS completed the writing of this manuscript, and all authors approved the final version.
We sincerely thank the National Science Foundation (NSF, CHE-1753187) for their generous support of this work. We sincerely thank Dr Novrus G. Akhmedov and Dr Steven Foster from the Research Support Services at the University of Oklahoma for their assistance with NMR and mass spectral analyses, respectively.
Data availability
The study's data are available in the published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Imamura and T. Lowary, Trends Glycosci. Glycotechnol., 2011, 23, 134–152 CrossRef CAS; (b) B. Lindberg, in Adv. Carbohydr. Chem. Biochem., ed., R. S. Tipson and D. Horton, Academic Press, 1990, vol. 48, pp. 279–318 Search PubMed; (c) D. C. Crick, S. Mahapatra and P. J. Brennan, Glycobiology, 2001, 11, 107R–118R CrossRef CAS PubMed; (d) T. L. Lowary, Acc. Chem. Res., 2016, 49, 1379–1388 CrossRef CAS PubMed.
- (a) P. Peltier, R. Euzen, R. Daniellou, C. Nugier-Chauvin and V. Ferrières, Carbohydr. Res., 2008, 343, 1897–1923 CrossRef CAS; (b) A. Varki, Glycobiology, 1993, 3, 97–130 CrossRef CAS PubMed; (c) S. A. Thadke, B. Mishra and S. Hotha, J. Org. Chem., 2014, 79, 7358–7371 CrossRef CAS.
- Y. Xu, H.-C. Bin, F. Su and J.-S. Yang, Tetrahedron Lett., 2017, 58, 1548–1552 CrossRef CAS.
- M. Islam, G. Gayatri and S. Hotha, J. Org. Chem., 2015, 80, 7937–7945 CrossRef CAS.
- A. B. Mayfield, J. B. Metternich, A. H. Trotta and E. N. Jacobsen, J. Am. Chem. Soc., 2020, 142, 4061–4069 CrossRef CAS PubMed.
- H. Xu, R. N. Schaugaard, J. Li, H. B. Schlegel and H. M. Nguyen, J. Am. Chem. Soc., 2022, 144, 7441–7456 CrossRef CAS.
- X. Ma, Y. Zhang, X. Zhu and L. Zhang, CCS Chem., 2022, 4, 3677–3685 CrossRef CAS PubMed.
- (a) B. Ghosh, A. Alber, C. W. Lander, Y. Shao, K. M. Nicholas and I. Sharma, ACS Catal., 2024, 14, 1037–1049 CrossRef CAS; (b) B. Ghosh, A. Alber, C. W. Lander, Y. Shao, K. M. Nicholas and I. Sharma, Org. Lett., 2024, 26, 9436–9441 CrossRef.
- (a) R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef; (b) C. Bolm, Nat. Chem., 2009, 1, 420 CrossRef PubMed.
- (a) E. W. Hunsaker and K. J. Franz, Inorg. Chem., 2019, 58, 13528–13545 CrossRef; (b) N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, Chem. Rev., 2021, 121, 14682–14905 CrossRef PubMed; (c) K. Jomova, M. Makova, S. Y. Alomar, S. H. Alwasel, E. Nepovimova, K. Kuca, C. J. Rhodes and M. Valko, Chem. – Biol. Interact., 2022, 367, 110173 CrossRef.
- (a) P. Nuss and M. J. Eckelman, PLoS One, 2014, 9, e101298 CrossRef; (b) P. B. Kettler, Org. Process Res. Dev., 2003, 7, 342–354 CrossRef.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef.
- (a) S. Escopy and A. V. Demchenko, Chem. – Eur. J., 2022, 28, e202103747 CrossRef PubMed; (b) S. Kaeothip, S. J. Akins and A. V. Demchenko, Carbohydr. Res., 2010, 345, 2146–2150 CrossRef; (c) Z. Zhang, I. R. Ollmann, X.-S. Ye, R. Wischnat, T. Baasov and C.-H. Wong, J. Am. Chem. Soc., 1999, 121, 734–753 CrossRef.
- (a) Frank K. Griffin, Duncan E. Paterson, Paul V. Murphy and Richard J. K. Taylor, Eur. J. Org. Chem., 2002, 1305–1322 CrossRef; (b) S. Hiranuma, T. Kajimoto and C.-H. Wong, Tetrahedron Lett., 1994, 35, 5257–5260 CrossRef.
- (a) J. D. C. Codée, R. E. J. N. Litjens, L. J. van den Bos, H. S. Overkleeft and G. A. van der Marel, Chem. Soc. Rev., 2005, 34, 769–782 RSC; (b) M. Panza, S. G. Pistorio, K. J. Stine and A. V. Demchenko, Chem. Rev., 2018, 118, 8105–8150 CrossRef PubMed.
- H. H. Trinderup, T. L. P. Sandgaard, L. Juul-Madsen and H. H. Jensen, J. Org. Chem., 2022, 87, 4154–4167 CrossRef.
- (a) S. P. Singh, U. Chaudhary and I. Sharma, Molecules, 2024, 29, 5367 CrossRef; (b) S. Pratap Singh, B. Ghosh and I. Sharma, Adv. Synth. Catal., 2024, 366, 1847–1856 CrossRef CAS; (c) S. P. Singh, U. Chaudhary, A. Daróczi and I. Sharma, Nat. Commun., 2025, 16, 3651 CrossRef.
- (a) J. Batra and A. S. Rathore, Biotechnol. Prog., 2016, 32, 1091–1102 CrossRef CAS PubMed; (b) K. R. Feingold, in Endotext, ed., K. R. Feingold, B. Anawalt, M. R. Blackman, A. Boyce, G. Chrousos, E. Corpas, W. W. de Herder, K. Dhatariya, K. Dungan, J. Hofland, S. Kalra, G. Kaltsas, N. Kapoor, C. Koch, P. Kopp, M. Korbonits, C. S. Kovacs, W. Kuohung, B. Laferrère, M. Levy, E. A. McGee, R. McLachlan, R. Muzumdar, J. Purnell, R. Sahay, A. S. Shah, F. Singer, M. A. Sperling, C. A. Stratakis, D. L. Trence and D. P. Wilson, MDText.com, Inc., Copyright © 2000–2025, South Dartmouth (MA), 2000 Search PubMed.
- Y. Wang and K. Liu, Naunyn-Schmiedebergs Arch. Pharmakol., 2024, 397, 4537–4554 CrossRef CAS.
- (a) L. Zhao, Z. Ma, J. Yin, G. Shi and Z. Ding, Carbohydr. Polym., 2021, 258, 117695 CrossRef CAS; (b) Y.-Y. Zhang, M. Ghirardello, R. Williams, A. S. Diaz, J. Rojo, J. Voglmeir, J. Ramos-Soriano and M. C. Galan, JACS Au, 2024, 4, 4328–4333 CrossRef CAS PubMed; (c) K. Le Mai Hoang, A. Pardo-Vargas, Y. Zhu, Y. Yu, M. Loria, M. Delbianco and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 9079–9086 CrossRef CAS PubMed; (d) H. A. Taha, M. R. Richards and T. L. Lowary, Chem. Rev., 2013, 113, 1851–1876 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc01597a |
| This journal is © The Royal Society of Chemistry 2025 |