
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Short-bite PSP-type ligands: coordination chemistry and ligand rearrangement reactions†
Franziska Flecken a,
Toni Grellb and
Schirin Hanf*a
a,
Toni Grellb and
Schirin Hanf*a
aInstitute for Inorganic Chemistry, Karlsruhe Institute of Technology, Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: schirin.hanf@kit.edu
bDipartimento di Chimica, Università degli Studi di Milano, Via Camillo Golgi 19, 20131 Milano, Italy
First published on 20th May 2025
Abstract
Sulphur-containing short-bite diphosphine ligands are widely overlooked despite their analogy to well-explored PCP-based ligands. This work investigates the impact of the monoatomic sulphur-based ligand backbone in nickel complexes [NiHal2(PSP)], PSP = Ph2PSPPh2, Hal = Br (1), I (2) as counterparts of related [NiHal2(PCP)] complexes. The highly flexible PSP ligand backbone has shown a postive effect in Kumada–Tamao–Corriu coupling reactions of sterically demanding substrates.
Although bidentate PCP-type (R2P–(CH2)n–PR2) ligands with carbon-based backbones are widely recognized for their importance, the incorporation of a heteroatom, such as sulphur, into the ligand backbone has been largely overlooked. This might be due to the fact that the simplest PSP-type ligand, namely Ph2P–S–PPh2, exhibits an interesting tautomeric equilibrium, which is thought to complicate the synthetic access. Under ambient conditions, the equilibrium favours the PPS tautomer (Ph2P(
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)–PPh2), but PSP-based compounds can be stabilised by electron-withdrawing groups or transition metals, forming coordination-stabilised tautomers, as demonstrated by Weigand's group and ours.1,2
S)–PPh2), but PSP-based compounds can be stabilised by electron-withdrawing groups or transition metals, forming coordination-stabilised tautomers, as demonstrated by Weigand's group and ours.1,2
Despite the limited exploration of PSP-type ligands, a few PPS- and PSP-based transition metal complexes have been reported. However, chelate-type metal complexes remain rare due to the significant ring strain within the resulting four-membered M–P–S–P ring. Only two reports describe the isolation of PSP-based chelate Mo(0)3 and Ru(II)4 complexes. In contrast, more often the formation of dinuclear complexes, including Ni(0),5 Cu(I),2 W(0),6 Ag(I)1 and Mn(I),7 in which the PSP-type ligand adopts a bridging coordination mode, has been reported. Furthermore, mononuclear complexes of Fe(0)1 and Cr(0),8 in which a PPS ligand coordinates via its P atom, have been isolated (Fig. 1).
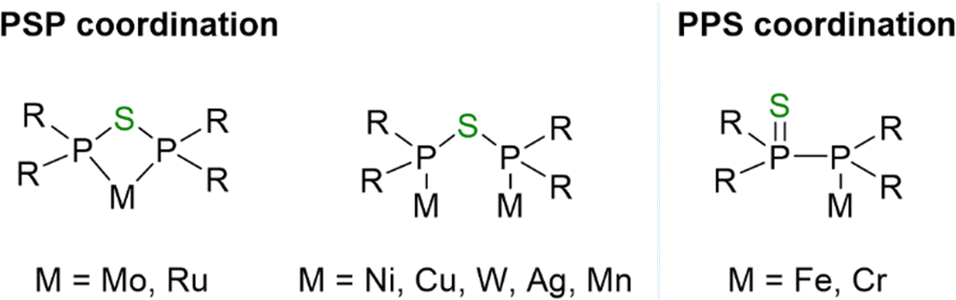 | ||
| Fig. 1 Different coordination modes of the PSP (left) and PPS (right) tautomer with R = Ph for Mo(0), Cu(I), Ru(II); Cy for Ru(II); tBu for Ag(I); CH3 for Mn(I); CF3 for Ni(0); 3,4-Me-C4H5P for W(0).1–7 Chelate compounds are only known for Mo(0)3 and Ru(II).4 | ||
Only recently we have explored the impact of the backbone of short-bite ligands in transition metal complexes. In this context, we have reported PXP-stabilised trinuclear Cu complexes of the form [Cu3(μ3-Hal)2(μ-PXP)3]PF6 (PXP = Ph2P–O–PPh2, Ph2P–S–PPh2; Hal = Cl, Br, I), which are formed via the in situ formation of the PXP-type ligand from the reaction of [Cu(MeCN)4]PF6 with K(X)PPh2 (X = O or S) and HalPPh2. The ligand backbone significantly impacts both molecular structure and photo-physics. Replacing oxygen (Ph2P–O–PPh2) with sulphur (Ph2P–S–PPh2) can activate or deactivate photo-emission.2
Inspired by the significant influence of the ligand backbone, this study explores sulphur-containing PSP-type ligands, as counterparts to the well-known PCP ligand sets, and their coordination chemistry towards nickel. In analogy to the formation of trinuclear PSP-stabilised Cu(I) complexes, initial studies for the isolation of Ni(II) compounds concentrated on the in situ ligand formation via the reaction of [Ni(MeCN)4](BF4)2 with KPS (K(S)PPh2) and HalPPh2 (Hal = Cl, Br, I). From these reactions the desired complexes [NiHal2(PSP)] [Hal = Br (1), I (2)] were isolated for the first time. Alternatively, both complexes were successfully synthesised via the direct reaction of the PPS tautomer with nickel bromide and iodide in acetonitrile. This contrasts with the reported synthesis of Cu(I)-based compounds, where the in situ ligand formation was required to prevent ligand decomposition reactions.2 The decomposition observed in the presence of Cu(I) may result from the simultaneous coordination of the PPS/PSP ligand through both P and S atoms to the soft Cu(I) centre. The differing reactivity observed with Cu(I) and Ni(II) highlights the crucial impact of the metal precursor's nature and the hardness or softness of the metal ion in governing the reactivity of the PPS/PSP ligand.
Violet crystals of 1 and 2 could be grown from a saturated DCM solution layered with n-heptane. Single-crystal X-ray diffraction analysis reveals that both compounds adopt closely related solid-state structures, featuring nearly planar four-membered Ni–P–S–P chelate rings (Fig. 2). Due to the larger atomic radius of sulphur compared to carbon within PCP-type ligands, the P–S bonds are significantly longer than P–C bonds, resulting in greater flexibility of the P–S–P fragment. This increased flexibility leads to notably acute P–S–P angles of 80.81(3)° for 1 and 81.51(15)° for 2, in contrast to the wider P–C–P angle of 91.94° observed in the PCP-type ligand of [NiBr2(dppm)] (91.94°), dppm = bis(diphenyl-phosphino)methane).9 The acute bond angle is essential for forming PSP-based chelate complexes, whereas in many cases, the high ring strain prevents chelation, favouring either monodentate coordination or bridging coordination of two separate metal centres.3,10
 | ||
| Fig. 2 Molecular structures of [NiHal2(PSP)] 1 (Hal = Br) and 2 (Hal = I). Selected bond lengths (Å) and angles (°) for 1: P–S 2.1146(7)–2.1183(7) P–Ni 2.1347(6)–2.1425(6) Ni–Br 2.3271(4)–2.3370(4) P–C 1.799(2)–1.812(2) Br–Ni–Br 97.234(13) Br–Ni–P 91.613(19)–171.07(2) Ni–P–S 99.43(3)–99.80(3) P–S–P 80.81(3) P–Ni–P 79.80(2) and for 2: P–S 2.117(3) P–Ni 2.137(3) Ni–I 2.5251(7) P–C 1.814(6)–1.816(7) I–Ni–I 100.57(8) I–Ni–P 89.93(7)–167.36(6) Ni–P–S 98.93(10) P–S–P 81.51(15) P–Ni–P 80.62(13). | ||
A comparison of the electronic donor properties can be made by analysing the P–Ni bond lengths. Complexes 1 and 2 exhibit shorter P–Ni distances than related PCP-type nickel complexes, such as [NiBr2(dppe)] (2.141(1), 2.156(1) Å)11 and [NiBr2(dppm)] (2.1423(16) Å).9 This trend suggests stronger metal–ligand interactions in PSP-type ligands, likely due to their enhanced ligand π-backbonding capability compared to PCP-type analogues, as shown by the calculated Löwdin charges (Table S10, ESI†).
The quasi square planar coordination of the nickel(II) centres is indicated by their structural index parameters12 of 0.13 (1) and 0.18 (2) (more information can be found in the ESI,† Table S2). This is further confirmed by the deviation from the best plane, which is defined by the two P atoms of the PSP ligand and the two halide atoms (0.008 Å (1) and 0.000 Å (2), (Table S2, ESI†), and explains the diamagnetic nature of the compounds, which is supported by the observation of sharp signals in the corresponding NMR spectra (31P{1H} NMR: −17.5 ppm (1); −14.5 ppm (2), Fig. 3 and Fig. S2, S7, ESI†). The order of the chemical shifts is somehow unexpected, since the bromide should lead to a downfield shift of the 31P NMR signal of 1 compared to 2. Similar observations were made by Fergusson and Heveldt for other square planar complexes of the type [MHal2(PR3)2] (M = Pd, Pt; Hal = Cl, Br, I), which they attributed to either a polarisation effect or a M → Hal π-back-bonding in the order of I > Br > Cl.13 Compared to the dppm analogue14 of 1, which shows a singlet at −22.5 ppm in the 31P{1H} NMR spectrum, the phosphorus signal of 1 is slightly downfield shifted, which indicates a higher deshielding of the P atoms in the sulphur-based PSP ligand.
 | ||
| Fig. 3 UV/Vis absorption spectra and photographs of the complexes 1 and 2 in DCM solutions (c = 1 × 10−4 mol L−1, left). Broad maxima in the visible range at 510 nm (1) and 564 nm (2) are causing the intense red-violet (1) and dark-violet (2) colours. 31P{1H} NMR spectra of 1 and 2 recorded in CD2Cl2 (right). | ||
UV/Vis absorption spectra have been recorded for the two colourful complexes (Fig. 3). 1 in DCM solution shows a broad absorption band at around 510 nm (ε = 2391 dm3 mol−1 cm−1) in the visible region, while for 2 this absorption band is shifted to lower energies with a maximum at around 564 nm (ε = 2297 dm3 mol−1 cm−1), due to the larger ligand field splitting induced by the bromo ligand. The respective transitions are responsible for the red-violet (1) and dark violet (2) colours of the compounds and can mainly be ascribed to Ni–L(PSP) charge transfer transitions, with a small contribution of the iodo ligands in the case of 2. This was confirmed by TD-DFT calculations and by the corresponding difference densities (Tables S11 and S12, ESI†).
Attempts to isolate the chloride analogue [NiCl2(PSP)] via the direct reaction of NiCl2 and PPS, via the conversion of [Ni(MeCN)4](BF4)2 with PPS or KPS and ClPPh2, as well as via ligand exchange reactions using PPS and [NiCl2(dppe)] or [NiCl2(PPh3)2] failed. Instead, several signals with a strong downfield shift are displayed in the 31P{1H} NMR spectra, which point towards the formation of new P-containing compounds. This is confirmed by the isolation and characterisation of [Ni2(μ2-Ph2P)(μ2-Ph2PS)(Ph2PSS)2] (3) via single-crystal X-ray diffraction (Fig. 4). Further, reactions of [Ni(COD)2] (COD = cycloocta-1,5-diene), as alternative nickel source, with PPS or KPS and ClPPh2 were attempted. In these cases, either the trinuclear nickel complex 4, [Ni3(μ2-Ph2P)2(μ2-Ph2PS)2(Ph2PS)2], or the dinuclear nickel complex 5, [Ni2(μ2-Ph2P)(μ2-Ph2PS)(Ph2PS)(Ph2PSS)], were isolated, in which the nickel(II) centres are coordinated by Ph2PSS−, Ph2P− and Ph2PS− ligands (Fig. 4).
 | ||
| Fig. 4 Molecular structures of multinuclear Ni(II) complexes, resulting from the conversion of NiCl2 and PPS (3), [Ni(COD)2] and PPS (4) and [Ni(COD)2] and KPS and ClPPh2 (5). | ||
Interestingly, during the reaction with [Ni(COD)2] an oxidation of Ni(0) to Ni(II) occurred, which is induced by the rearrangement of the parent PPS ligand and the subsequent formation of Ph2PSS−, Ph2P− and Ph2PS−. Ogawa reported similar rearrangements of PPS, which involved, initiated by UV radiation or radical starters,15–18 a homolytic P–P bond cleavage yielding one Ph2P(S)˙ and one Ph2P˙ radical, that could recombine to give new P-containing compounds. Density functional theory (DFT) calculations proved the localisation of the HOMO mostly at the P(S)–P unit rather than at the phenyl rings, which is the driving force in terms of reactivity.15 We assume, that the coordination of the PSP ligand to nickel facilitates similar rearrangement reactions in the presence of certain Ni(II) or Ni(0) precursors, resulting in the formation of 3–5. Small amounts of 3–5 appear as by-products in the synthesis of 1 and 2, but with NiCl2 or [Ni(COD)2], 3–5 form as main products. Hereby, the absence or presence of light did not show an impact on the product formation and the UV radiation of complexes 1 and 2 in solution did not initiate the formation of multinuclear Ni complexes (Fig. S11, ESI†). These findings again underline the high reactivity of the PPS/PSP ligand scaffold in the presence of selected transition metal precursors.
In the multinuclear complexes 3–5, the Ni(II) atoms are coordinated in a quasi-planar arrangement (Fig. 4). In all three complexes the nickel atoms are bridged by a combination of one Ph2P− ligand and one diametrically opposite Ph2PS− ligand, leading to five-membered Ni2P2S metallacycles. Based on the structural index parameters12 of 3–5, a nearly square planar coordination mode can be confirmed for all nickel(II) centres, which is less ideal for 4 and 5 in comparison to 3 (Table S4, ESI†). The comparison of the distances of the Ni(II) atoms to the best plane, defined by the four coordinating P and S atoms, indicates the highest deviation from an ideal planarity for 4.
The NMR spectra of compounds 3–5 agree with the molecular structures obtained from single-crystal X-ray diffraction. However, a higher paramagnetic character can be witnessed for 4, as indicated by the occurrence of very broad signals in the 31P{1H} NMR spectrum (see ESI†). This might be due to an enhanced dynamic behaviour in solution, which disrupts the square-planar geometry of 4 and enables the formation related of paramagnetic tetrahedral complexes. Compounds 3 and 5 exhibit 31P{1H} NMR spectra with distinct signals, whereby 2JPP couplings > 30 Hz and 3JPP couplings < 10 Hz can be identified (Fig. S21 and S22, ESI†). The signal corresponding to the Ph2PSS− ligand is found in the region of 73 ppm [3: 72.7 ppm (Pd), 5: 73.0 ppm (Pa)]. The different coordination environments of the terminal and internal Ph2PS− ligands cause a drastic shift in their 31P{1H} NMR signals. Whereas the terminal Ph2PS− ligand is found at chemical shifts of 31.1 ppm (5, Pd), signals at around 90 ppm can be related to internal Ph2PS− ligands [3: 92.0 ppm (Pb), 5: 90.1 ppm (Pb)]. Signals at chemical shifts around 60 ppm can be attributed to the Ph2P− ligand [3: 60.4 ppm (Pc), 5: 56.3 ppm (Pc)].
Beyond the structural characterisation of PSP-containing nickel complexes, such compounds can be employed in Kumada–Tamao–Corriu coupling reactions as alternative to Pd catalysts. This investigation is driven by the fact, that Ni-based complexes of the type [NiHal2(PCP)], including PCP-type ligands, are leading catalyst examples for such reactions, whereby a drastic impact of the ligand backbone has been reported.19–21 To explore the impact of the sulphur incorporation into the ligand backbone on the catalytic activity and selectivity, complexes 1 and 2 were applied as homogenous catalysts in coupling reactions. To the best of our knowledge the application of PSP-type ligands in catalysis has not yet been described in the literature, probably due to possible catalyst poisoning effects induced by sulphur. However, a tautomeric shift from the PSP to the PPS tautomer during the catalytic cycle could generate vacant coordination sites, which can facilitate substrate binding. This tautomeric behaviour would present new opportunities for designing hemilabile ligands (Scheme 1).
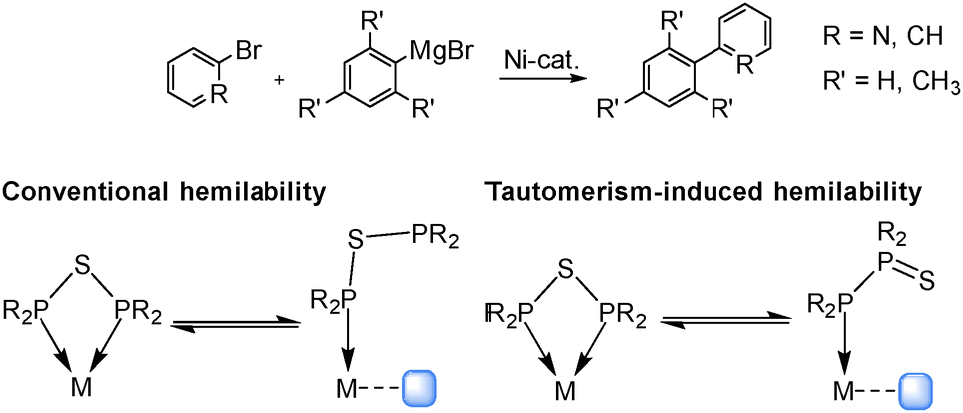 | ||
| Scheme 1 Application of PSP-stabilised Ni complexes in Kumada coupling reactions and the proposed tautomerism-induced hemilability, leading to a reduced steric demand. The empty coordination site is marked in blue. | ||
To gain first insights, C(sp2)–C(sp2) coupling reactions were conducted in THF, toluene and benzene using 1, 2 and [NiBr2(dppe)] as reference catalyst, since it has been widely applied as catalyst for Kumada coupling reactions (Table S7, ESI†).10 Therefore, coupling reactions of bromobenzene and -pyridine with PhMgBr or MesMgBr (Mes = mesityl) were conducted, and the successful conversions of the aryl halides prove that PSP-based complexes can be applied as efficient catalysts for Kumada coupling reactions. Hereby no indication of any catalyst poisoning, induced by the sulphur ligand backbone, could be observed. These results agree with a study of Li and coworkers, who applied secondary phosphine sulphide-based Ni complexes for the coupling of aryl chlorides with aryl Grignard reagents.22
Interestingly, the choice of solvent drastically influences the catalytic performance across all three systems (Table S7, ESI†). For [NiBr2(dppe)], the bromobenzene conversion with MesMgBr reaches 74% in THF but is negligible in benzene or toluene. In contrast, the PSP-based catalysts 1 and 2 show higher activity in toluene than in benzene or THF.
These activity differences stem from the distinct solution behaviour of the complexes. [NiBr2(dppe)] dissolves well in THF without ligand dissociation but poorly in benzene/toluene (Fig. S25, ESI†), explaining its low activity in non-coordinating solvents. Conversely, 1 dissolves readily in benzene but it predominately precipitates in toluene (Fig. S26, ESI†). Nevertheless, the catalytically active concentration is sufficient at the low catalyst loadings employed. In THF, 1 forms a dark red-violet solution, with NMR signals indicating the partial PPS ligand liberation, which can create open coordination sites. For 2, THF solutions show no free PPS ligand but suggest the formation of new Ni species (e.g., monodentate PSP- or PPS-bound complexes, Fig. S27, ESI†). This dynamic behaviour of complexes 1 and 2 arises from the strain in the four-membered Ni–P–S–P ring and the underlying PSP/PPS tautomerism. When comparing the selectivity of 1 and 2 with [NiBr2(dppe)], a drop in selectivity occurs, due to an enhanced formation of homocoupling products of the Grignard reagents. This might be due to the higher dynamic behaviour of the PSP ligand, which is accompanied by a more challenging reaction control. However, a significant advantage of the PSP-based nickel complexes over [NiBr2(dppe)] emerges in the coupling of sterically demanding Grignard reagents, such as MesMgBr. Whereas with 2 82% conversion of 2-bromopyridine and MesMgBr can be achieved in toluene, only 53% conversion were observed using the reference catalyst. The dynamic coordination sphere and reduced steric hindrance in PSP complexes enhance the catalytic activity, likely via a tautomerism-induced hemilability.
To further investigate this effect, reactions of PhMgBr and MesMgBr with 2-bromonaphthalene were performed, highlighting the significant influence of sulphur in the ligand backbone (Table S8, ESI†). While no notable backbone effect is observed with PhMgBr, the sulphur backbone substantially enhances the catalytic activity in the case of MesMgBr compared to the dppe counterpart ([NiBr2(PSP)]: 78% vs. [NiBr2(dppe)]: 8%). NMR studies of the reaction between 2 and MesMgBr further confirm the dynamic behaviour of the PSP ligand and suggest the formation of PPS-coordinated nickel complexes and a tautomerism-induced hemilability in addition to the conventionally observed hemilability of bidentate ligands (Fig. S28, ESI†). In the context of new drug developments, research focuses on establishing novel pathways of C(sp2)–C(sp3) couplings.23,24 As part of this study, the coupling reactions of bromobenzene or -pyridine with CyMgBr (Cy = cyclohexyl) were investigated and demonstrate the substrate scope (Table S9, ESI†).
In conclusion, we report the synthesis of PSP-coordinated nickel halide complexes, namely [NiHal2(PSP)], PSP = Ph2PSPPh2, Hal = Br (1), I (2), as counterparts of the well-studied PCP-type ligands. The incorporation of sulphur into the ligand backbone drastically influences the compounds’ molecular structure, the spectroscopic (UV/Vis, IR and NMR) properties as well as their catalytic performance in coupling reactions. Interestingly, the reactions of NiCl2 or [Ni(COD)2] with PPS or the precursors KPS and ClPPh2 lead to P–P bond cleavage and the formation of multinuclear nickel complexes, containing Ph2PS−, Ph2P− and Ph2PSS− ligands.
The authors wish to thank the Stiftung der Deutschen Wirtschaft for a doctoral scholarship for F. F. We wish to thank Prof. Dieter Fenske for his help with single-crystal XRD and Prof. Peter Roesky for support. Further we thank Sven Becker, Nhu Phong Nguyen and Lorena Damsch for their experimental help.
Data availability
Further information is available within the ESI,† and as open file formats on Zenodo (https://doi.org/10.5281/zenodo.13234535). A pre-print was published on ChemRxiv (https://doi.org/10.26434/chemrxiv-2024-fdgcn).Conflicts of interest
The authors declare no competing financial interest.Notes and references
- S. Yogendra, S. S. Chitnis, F. Hennersdorf, M. Bodensteiner, R. Fischer, N. Burford and J. J. Weigand, Inorg. Chem., 2016, 55, 1854–1860 CrossRef CAS PubMed.
- F. Flecken, A. Knapp, T. Grell, C. Dreßler and S. Hanf, Inorg. Chem., 2023, 62, 13038–13049 CrossRef CAS PubMed.
- F. A. Cotton, L. R. Falvello, M. Tomas, G. M. Gray and C. S. Krainhanzel, Inorg. Chim. Acta, 1984, 82, 129–139 CrossRef CAS.
- P. E. Sues, A. J. Lough and R. H. Morris, Chem. Commun., 2014, 50, 4707–4710 RSC.
- H. Einspahr and J. Donohue, Inorg. Chem., 1974, 13, 1839–1843 CrossRef CAS.
- M. P. Duffy, Y. Lin, L. Y. Ting and F. Mathey, New J. Chem., 2011, 35, 2001–2003 RSC.
- S. Hoehne, E. Lindner and J.-P. Gumz, Chem. Ber., 1978, 111, 3818–3822 CrossRef CAS.
- P. G. Jones, A. K. Fischer, M. Farkens and R. Schmutzler, Acta Crystallogr., Sect. E, 2002, 58, m478–m479 CrossRef CAS.
- D. Lomjanský, C. Rajnák, J. Titiš, J. Moncoľ, L. Smolko and R. Boča, Inorg. Chim. Acta, 2018, 483, 352–358 CrossRef.
- R. J. Puddephatt, Chem. Soc. Rev., 1983, 12, 99–127 RSC.
- J. A. Rahn, A. Delian and J. H. Nelson, Inorg. Chem., 1989, 28, 215–217 CrossRef CAS.
- A. Okuniewski, D. Rosiak, J. Chojnacki and B. Becker, Polyhedron, 2015, 90, 47–57 CrossRef CAS.
- J. E. Fergusson and P. F. Heveldt, Inorg. Chim. Acta, 1978, 31, 145–154 CrossRef CAS.
- J. A. S. Bomfim, F. P. d Souza, C. A. L. Filgueiras, A. G. d Sousa and M. T. P. Gambardella, Polyhedron, 2003, 22, 1567–1573 CrossRef CAS.
- Y. Sato, S.-i Kawaguchi, A. Nomoto and A. Ogawa, Chem. – Eur. J., 2018, 25, 2295–2302 CrossRef PubMed.
- Y. Yamamoto, K. Fujiwara and A. Ogawa, Organometallics, 2023, 42, 2590–2597 CrossRef CAS.
- Y. Yamamoto, R. Tanaka, M. Ota, M. Nishimura, C. C. Tran, S.-I. Kawaguchi, S. Kodama, A. Nomoto and A. Ogawa, J. Org. Chem., 2020, 85, 14708–14719 CrossRef CAS PubMed.
- Y. Yamamoto and A. Ogawa, Chem. – Asian J., 2023, 18, e202201269 CrossRef CAS PubMed.
- M. Kumada, Pure Appl. Chem., 1980, 52, 669–679 CrossRef CAS.
- K. Tamao, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 4374–4376 CrossRef CAS.
- K. Tamao, J. Organomet. Chem., 2002, 653, 23–26 CrossRef CAS.
- G. Y. Li and W. J. Marshall, Organometallics, 2002, 21, 590–591 CrossRef CAS.
- A. W. Dombrowski, N. J. Gesmundo, A. L. Aguirre, K. A. Sarris, J. M. Young, A. R. Bogdan, M. C. Martin, S. Gedeon and Y. Wang, ACS Med. Chem. Lett., 2020, 11, 597–604 CrossRef CAS PubMed.
- N. Palaychuk, T. J. DeLano, M. J. Boyd, J. Green and U. K. Bandarage, Org. Lett., 2016, 18, 6180–6183 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2375582 (1), 2375583 (2), 2375585 (3), 2375584 (4), and 2375581 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc01739d |
| This journal is © The Royal Society of Chemistry 2025 |