
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical C–H functionalization reaction of N-heterocycles with alkyl iodides†
Yaseen Hussaina,
Ganga Sankara,
Magnus Rueping *b and
Rene M. Koenigs*ac
*b and
Rene M. Koenigs*ac
aInstitute of Organic Chemistry, RWTH Aachen University, Landoltweg 1, D-52074 Aachen, Germany. E-mail: rene.koenigs@rwth-aachen.de
bKAUST Catalysis Center (KCC), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: magnus.rueping@kaust.edu.sa
cDepartment of Chemistry, University of Bayreuth, Universitätsstraße 30, 95447 Bayreuth, Germany. E-mail: rene.koenigs@uni-bayreuth.de
First published on 29th April 2025
Abstract
Herein, we report on an electrochemical protocol for the C–H alkylation of N-heterocycles with easily accessible alkyl halides. A wide range of azauracil derivatives including bioactive tethered azauracil, pyrazinone and quinoxalinone were well accommodated and delivered the alkylated products in good to excellent yield.
The direct functionalization of C–H bonds is one of the most straight-forward approaches to increase molecular complexity and to introduce new functional groups onto an existing molecular framework. In this context, C–H functionalization reactions of N-heterocycles are of particular interest owing to the widespread use of such building blocks in modern drugs and agrochemicals.1 Current methods for the direct functionalization of N-heterocycles often proceed through the use of highly reactive intermediates, such as low-valent carbene or radical intermediates for the construction of new C–C bonds.2–8 Such radical C–H functionalization reactions have recently attracted significant interest and today a range of different approaches to access the pivotal alkyl radical intermediate have been described.2 Common approaches involve the utilization of suitable radical precursors, such as N-hydroxyphthalimide esters,3 carboxylic acids,4 or alkyl halides.5 Besides these substrates, aliphatic aldehyde,6 boronic acids,7 or alkyl hydrazines8 have also been documented for radical functionalization of N-heterocycles (Scheme 1a). Although these molecules have been utilized for alkylation reaction, the development of more sustainable and mild protocols by using more easily accessible radical precursor feedstocks are highly desirable.
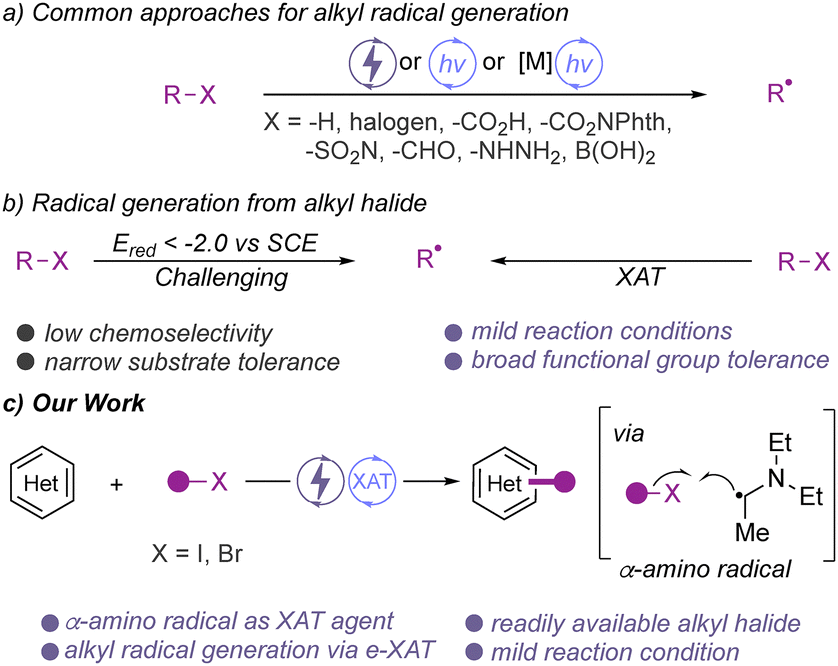 | ||
| Scheme 1 Approaches for the radical C–H functionalization of heterocycles with alkyl halides. | ||
Among the radical precursors, alkyl halides dominate as one of the most readily accessible substrates that are commercially available or can be easily prepared from the corresponding alcohols. However, due to the low reduction potential (Ered < −2 V versus SCE) of alkyl halides, their application as radical precursors in chemical transformation poses a challenge in terms of compatibility with reacting partners.9 This challenge could be overcome by employing halogen-atom transfer (XAT)10 agents under mild reaction conditions, and the generation of alkyl radicals via SET11 has been reported (Scheme 1b). In contrast, very recently, photoinduced palladium catalysis has been explored to activate alkyl halides for various alkylation reactions.12 In this context, our group has demonstrated the radical C–H alkylation of N/O heterocycles using photoinduced metal catalysis.13 Similarly, electrochemistry has recently opened a window to generate alkyl radicals from alkyl halides through an electrochemical halogen-atom transfer (e-XAT) strategy without compromising its efficiency and substrate compatibility.14,15 To the best of our knowledge, this strategy has not been applied for the alkylation of heterocycles (Scheme 1c). Due to the mild and sustainable nature, we anticipated that the e-XAT strategy could be applied for the alkylation of N-heterocycles.
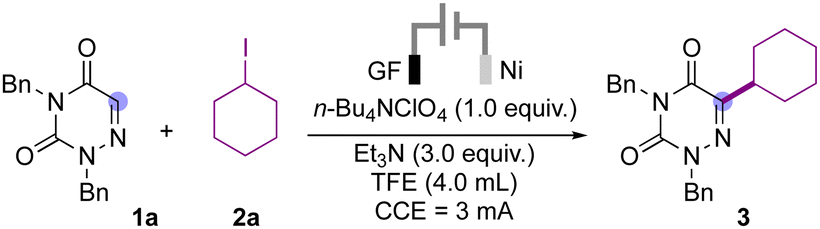
In the initial experiment, we performed the reaction of azauracil derivative 1a with cyclohexyl iodide (2a) and triethyl amine as an XAT agent under electrochemical conditions using graphite as the electrodes in acetonitrile solvent (Table 1, entry 1). To our delight we obtained the desired alkylation product 3 in 13% yield. With this positive result we exchanged the graphite cathode with a nickel electrode and the product yield was increased to 33% (Table 1, entry 2). We further evaluated various solvents using Ni-foam as the cathode and observed that trifluoroethanol proved to be the best solvent for this transformation to give the desired product in a yield of 86% (Table 1, entry 2). When the reaction was carried out in DMF, only trace amounts of the desired product were observed. Further screening of the electrolytes (Table 1, entries 3–5) for this transformation was carried out and revealed that n-tetrabutylammonium perchlorate (TBAClO4) was superior to other electrolytes to afford the desired product in 90% yield (Table 1, entry 3). When using Hunig's base as an XAT agent, a reduced yield of the product was observed (Table 1, entry 6). Similarly, lowering the equivalents of triethyl amine provided a lower yield of the product (Table 1, entry 7). A sharp decrease in the yield of product 3 was observed on reducing the equivalents of alkyl iodide 2a (Table 1, entry 8). Further increasing the current to 6 mA improved the yield and the product was isolated in 93% yield (Table 1, entry 10). However, at 9 mA current, the product was obtained in a lower yield (Table 1, entry 10).
|
|||
|---|---|---|---|
| Entry | Deviation from the reaction conditions | Time (h) | Yield [%] of 3 |
| a Reaction conditions: 1a (0.2 mmol), 2a (2.0 equiv.), n-Bu4NClO4 (1.0 equiv.) and Et3N (3.0 equiv.) in TFE (4.0 mL) at rt under CCE of 3 mA. TFE (trifluoroethanol). TBA (tetrabutylammonium).b The yield in parentheses is with bromocyclohexane. | |||
| 1 | TBAI, graphite electrodes and MeCN as solvent | 30 | 13 |
| 2 | TBAI as electrolyte, and MeCN/CH2Cl2/THF/HFIP/TFE/MeOH/DMF as solvent | 17–30 | 33/61/31/77/86/18/traces |
| 3 | None | 30 | 90 |
| 4 | TBAPF6 instead of TBAClO4 | 50 | 13 |
| 5 | TBABr instead of TBAClO4 | 30 | 69 |
| 6 | DIPEA instead of Et3N | 42 | 53 |
| 7 | Et3N (1.0 equiv.) was used | 42 | 82 |
| 8 | 2a (1.0 equiv.) was used | 42 | 69 |
| 9 | CCE at 6.0 mA | 22 | 93(76)b |
| 10 | CCE at 9.0 mA | 17 | 86 |
After obtaining the optimized reaction conditions, we proceeded with the evaluation of the substrate scope by varying the alkyl halide component (Scheme 2). Both cyclic and acyclic alkyl bromide or – iodide were well accommodated in this transformation to provide the products 3–11 in moderate to good yield. The ring size of the cycloalkane shows a moderate effect on the yield as the cyclobutyl bromide provided the product 4 in lower yield compared to product 3 obtained from cyclohexyl bromide. Similarly, iodocycloalkanes provided the desired product in higher yield than the bromo analogue probably due to the lower bond dissociation energy of alkyl iodides. Cyclopentyl iodide gave the desired product 5 in 63% yield and cycloheptyl bromide also underwent a smooth transformation to provide product 6 in 62% yield. Also, acyclic primary and secondary alkyl bromides and iodides were well suited for this transformation and provided the products 7–11 in good yield. However, neopentyl bromide only gave the product 8 in 14% yield, which may be related to undesired side reactions of putative alkyl radical intermediates. Additionally, Boc-protected 4-iodopiperidyl was also tested under a slightly modified reaction condition to provide the product 12 in good yield. Limitations lie within the use of tertiary alkyl halides, which proved incompatible with the reaction conditions.
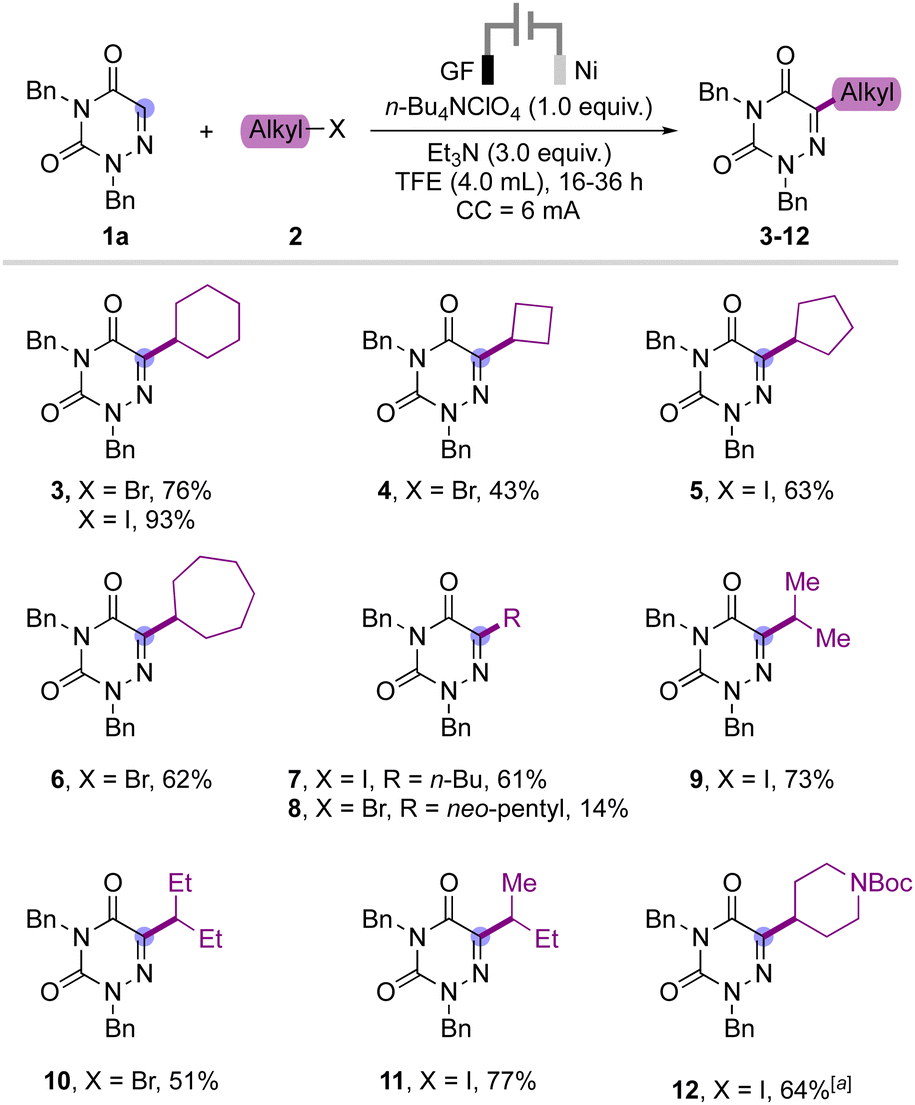 | ||
Scheme 2 Substrate scope with various alkyl halides. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) The reaction was carried out at 3 mA for 72 h. The reaction was carried out at 3 mA for 72 h. | ||
We then proceeded with the evaluation of the scope by changing the substituents on the heterocycle 1 (Scheme 3). The reaction of mono substituted azauracil also worked smoothly to deliver the product 13 in 68% yield. Furthermore, we varied the substitution on the N-benzyl azauracil such as propargyl, allyl, alkyl, cinnamyl, cyclopropyl and acetate that were well tolerated under the standard reaction conditions to provide the corresponding products 14–19 in moderate to excellent yield. In the case of propargyl and allyl bearing azauracil, the desired products 14 and 15 were isolated in moderate yields. In addition, quinoxaline and the pyrazine heterocycle were also well accommodated to deliver the corresponding products 20 and 21 in 76% and 40% yields, respectively. Furthermore, azauracil with N-bioactive tethered scaffolds such as isozepac, menthol, borneol and ciprofibrate were also tested under the optimized reaction conditions to afford the respective products 22–25 in moderate yields.
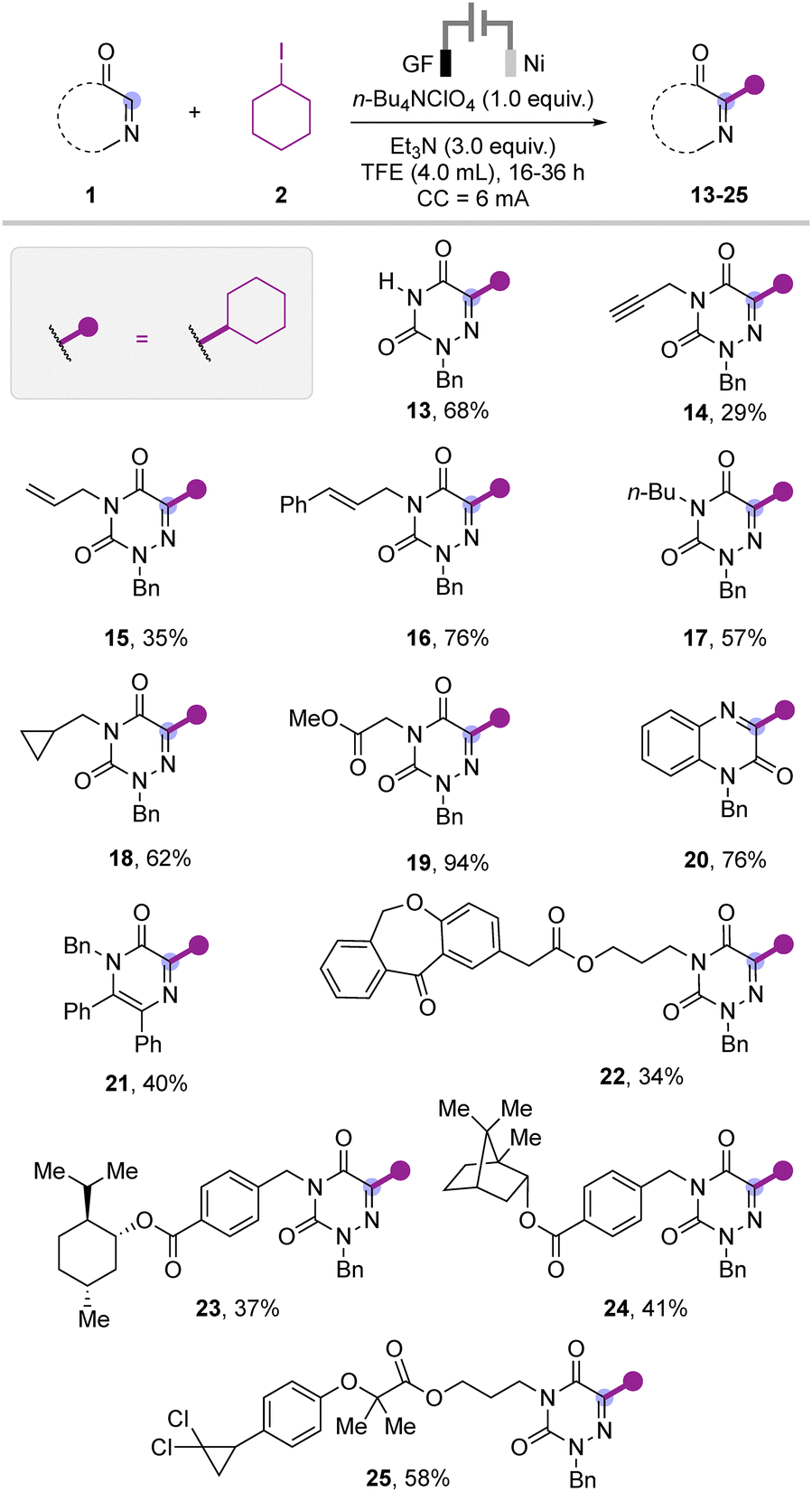 | ||
| Scheme 3 Substrate scope with N-substituted uracil and various heterocycles. | ||
To demonstrate the applicability of the developed protocol, the reaction was performed at 1 mmol scale and the product was obtained in 54% yield (Scheme 4a). Additionally, aza-Michael addition of 13 with maleimide 26 under electrochemical conditions provided the product 27 in 72% yield (Scheme 4b). Furthermore, the NH insertion reaction of 13 with phenyl diazoacetate 28 was also performed under photochemical conditions16 and the insertion product 29 was isolated in 67% yield (Scheme 4c).
 | ||
| Scheme 4 (a) Scale-up reaction, (b) post synthetic transformations, and (c) radical quenching experiment. | ||
From the previously reported literature,15 it is evident that triethyl amine acts as an XAT agent to generate the alkyl radical from the corresponding alkyl halide. This alkyl radical then adds onto the imine carbon, which upon subsequent oxidation delivers the alkylated product. To prove the radical nature of the electrochemical reaction, a radical quenching experiment was performed using TEMPO as a radical quencher, which resulted in complete suppression of the formation of product 3, suggesting the radical nature of the reaction (Scheme 4c).
We consider that the reaction proceeds by initial oxidation of triethylamine under electrochemical conditions, followed by deprotonation to α-amino radical intermediate Int-A. Then, the radical intermediate abstracts a halogen atom from the alkyl halide to generate the alkyl radical Int-B and α-halo amine. Finally, the intermediate Int-B adds onto the imine carbon followed by deprotonation and subsequent oxidation to provide 3 (Scheme 5).
 | ||
| Scheme 5 Putative reaction mechanism. | ||
In conclusion, we have developed an electro-catalyzed alkylation of heterocycles with alkyl halide through an XAT strategy. A wide range of alkyl halides (Br, I) were tolerated to deliver the alkylated heterocycles. Furthermore, azauracil bearing various functionalities and bioactive molecules viz. isozepac, borneol, menthol etc. also delivered the corresponding alkylated products in an acceptable yield.
This publication is based upon work partially supported by King Abdullah University of Science and Technology (KAUST) under Award No. ORFS-CRG12-2024-6438.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no conflict to declare.Notes and references
- (a) N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS PubMed; (b) M. M. Heravi and V. Zadsirjana, RSC Adv., 2020, 10, 44247–44311 RSC.
- S. Crespi and M. Fagnoni, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed.
- For selected research articles, see: (a) W.-M. Cheng, R. Shang, M.-C. Fu and Y. Fu, Chem. – Eur. J., 2017, 23, 2537–2541 CrossRef CAS PubMed; (b) R. Dash, S. P. Panda, K. S. Bhati, S. Sharma and S. Murarka, Org. Lett., 2024, 26, 7227–7232 CrossRef CAS PubMed; (c) K. Niu, L. Song, Y. Hao, Y. Liu and Q. Wang, Chem. Commun., 2020, 56, 11673–11676 RSC; (d) M.-C. Fu, R. Shang, B. Zhao, B. Wang and Y. Fu, Science, 2019, 363, 1429–1434 CrossRef CAS PubMed; (e) L. Xu, X. Wang, D. Yang, X. Yang and D. Wang, Angew. Chem., Int. Ed., 2025, 64, e202416451 CrossRef CAS PubMed.
- For recent review, see: (a) L. Li, Y. Yao and N. Fu, Eur. J. Org. Chem., 2023, e202300166 CrossRef CAS; (b) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed ; For recent selected research articles, see: ; (c) D.-Y. Liu, X. Liu, Y. Gao, C.-Q. Wang, J.-S. Tian and T.-P. Loh, Org. Lett., 2020, 22, 8978–8983 CrossRef CAS PubMed; (d) T.-B. Zhang, X.-D. Guan, Y. Gao, S.-C. Lu and B.-L. Li, Org. Biomol. Chem., 2024, 22, 3439–3443 RSC; (e) X. Chen, X. Luo, X. Peng, J. Guo, J. Zai and P. Wang, Chem. – Eur. J., 2020, 26, 3226–3230 CrossRef CAS PubMed.
- (a) W. Deng, X. Li, Z. Li, Y. Wen, Z. Wang, Z. Lin, Y. Li, J. Hu and Y. Huang, Org. Lett., 2023, 25, 9237–9242 CrossRef CAS PubMed; (b) W. Zhang and S. Lin, J. Am. Chem. Soc., 2020, 142, 20661–20670 CrossRef CAS PubMed; S. Ye, T. Xiang, X. Li and J. Wu, Org. Chem. Front., 2019, 6, 2183–2199 Search PubMed.
- For selected articles, see: (a) H.-F. Liu, M.-X. He and H.-T. Tang, Org. Chem. Front., 2022, 9, 5955–5961 RSC; (b) M. Wang, J. Liu, Y. Zhang and P. Sun, Adv. Synth. Catal., 2022, 364, 2660–2665 CrossRef CAS; (c) X. Shi, Y. Cao, Y. Liu, K. Niu, H. Song, J. Zhang and Q. Wang, Org. Chem. Front., 2023, 10, 1296–1300 RSC.
- L. Yao, D. Zhu, L. Wang, J. Liu, Y. Zhang and P. Li, Chin. Chem. Lett., 2021, 32, 4033–4037 CrossRef CAS.
- M. Tian, S. Liu, X. Bu, J. Yu and X. Yang, Chem. – Eur. J., 2020, 26, 369–373 CrossRef CAS PubMed.
- (a) C. A. Paddon, F. L. Bhatti, T. J. Donohoe and R. G. Compton, J. Phys. Org. Chem., 2007, 20, 115–121 CrossRef CAS; (b) D. Li, T.-K. Ma, R. J. Scott and J. D. Wilden, Chem. Sci., 2020, 11, 5333–5338 RSC.
- (a) X. Tian, J. Kaur, S. Yakubov and J. P. Barham, ChemSusChem, 2022, 15, e202200906 CrossRef CAS PubMed; (b) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8087 CrossRef CAS PubMed; (c) T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS PubMed; (d) Y. Jianga, Y. Yina and Z. Jiang, Chin. J. Org. Chem., 2024, 44, 1733–1759 CrossRef; (e) N. Sanosa, B. Peñín, D. Sampedro and I. F.-Ardoiz, Eur. J. Org. Chem., 2022, e202200420 CrossRef CAS; (f) T. Wan, Ł. W. Ciszewski, D. Ravelli and L. Capaldo, Org. Lett., 2024, 26, 5839–5843 CrossRef CAS PubMed; (g) P. Bellotti, H.-M. Huang, T. Faber, R. Laskar and F. Glorius, Chem. Sci., 2022, 13, 7855–7862 RSC; (h) X. Liu, Z. Guo, Y. Che, X. Liu, M. Yu, J. Lv, M. Li, H. Xing and P. Chen, J. Mater. Chem. A, 2023, 11, 2472–2481 RSC; (i) N. Ma, L. Guo, Z.-J. Shen, D. Qi, C. Yang and W. Xia, Org. Biomol. Chem., 2022, 20, 1731–1737 RSC; (j) S. Lu, Z. Hu, D. Wang and T. Xu, Angew. Chem., Int. Ed., 2015, 63, e202406064 CrossRef PubMed.
- For selected research articles on SET activation of alkyl halides, see: (a) J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 14726–14727 CrossRef CAS PubMed; (b) D. A. Everson, B. A. Jones and D. J. Weix, J. Am. Chem. Soc., 2012, 134, 6146–6159 CrossRef CAS PubMed; (c) H. Chen, X. Jia, Y. Yu, Q. Qian and H. Gong, Angew. Chem., Int. Ed., 2017, 56, 13103–13106 CrossRef CAS PubMed; (d) F. Zhou, J. Zhu, Y. Zhang and S. Zhu, Angew. Chem., Int. Ed., 2018, 57, 4058–4062 CrossRef CAS PubMed; (e) S. Kim, M. J. Goldfogel, M. M. Gilbert and D. J. Weix, J. Am. Chem. Soc., 2020, 142, 9902–9907 CrossRef CAS PubMed; S. Bera, R. Mao and X. Hu, Nat. Chem., 2021, 13, 270–277 Search PubMed; (f) D. Qian, S. Bera and X. Hu, J. Am. Chem. Soc., 2021, 143, 1959–1967 CrossRef CAS PubMed; (g) X. Wu, W. Hao, K.-Y. Ye, B. Jiang, G. Pombar, Z. Song and S. Lin, J. Am. Chem. Soc., 2018, 140, 14836–14843 CrossRef CAS PubMed; (h) A. Cai, W. Yan, C. Wang and W. Liu, Angew. Chem., Int. Ed., 2021, 60, 27070–27077 CrossRef CAS PubMed; (i) J. D. Nguyen, E. M. D’Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed; (j) H. Kim and C. Lee, Angew. Chem., Int. Ed., 2012, 51, 12303–12306 CrossRef CAS PubMed; (k) Y. Shen, J. Cornella, F. Juliá-Hernández and R. Martin, ACS Catal., 2017, 7, 409–412 CrossRef CAS; (l) D. Alpers, M. Gallhof, J. Witt, F. Hoffmann and M. A. Brasholz, Angew. Chem., Int. Ed., 2017, 56, 1402–1406 CrossRef CAS PubMed; (m) H.-Q. Do, S. Bachman, A. C. Bissember, J. C. Peters and G. C. Fu, J. Am. Chem. Soc., 2014, 136, 2162–2167 CrossRef CAS PubMed; (n) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef CAS PubMed; (o) J. Lan, W. Yu, K. You, M. Xu, B. Zhang, Y. Wang, T. Wang and J. Luo, Org. Lett., 2023, 25, 7434–7439 CrossRef CAS PubMed; (p) S. Wu, J. Huang, L. Kang, Y. Zhang and K. Yuan, Org. Lett., 2024, 26, 763–768 CrossRef CAS PubMed; (q) L. Lu, Y. Wang, W. Zhang, W. Zhang, K. A. See and S. Lin, J. Am. Chem. Soc., 2023, 145, 22298–22304 CrossRef CAS PubMed.
- S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Angew. Chem., Int. Ed., 2024, 63, e202311972 CrossRef CAS PubMed.
- (a) S. Senapati, S. K. Hota, L. Kloene, C. Empel, S. Murarka and R. M. Koenigs, Angew. Chem., Int. Ed., 2024, e202417107 Search PubMed; (b) S. Jana, C. Pei, S. B. Bahukhandi and R. M. Koenigs, Chem. Catal., 2021, 1, 467–479 CrossRef CAS; (c) H. Fang, C. Empel, I. Atodiresei and R. M. Koenigs, ACS Catal., 2023, 13, 6445–6451 CrossRef CAS; (d) C. Pei, C. Empel and R. M. Koenigs, Angew. Chem., Int. Ed., 2022, 61, e202201743 CrossRef CAS PubMed.
- For recent review articles on electrocatalysis, see: (a) H. R. Stephen and J. L. Röckl, ACS Org. Inorg. Au, 2024, 4, 571–578 CrossRef CAS PubMed; (b) T. Ali, H. Wang, W. Iqbal, T. Bashir, R. Shah and Y. Hu, Adv. Sci., 2023, 10, 2205077 CrossRef CAS PubMed; (c) L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC; (d) L. Ma, X. Gao, X. Liu, X. Gu, B. Li, B. Mao, Z. Sun, W. Gao, X. Jia and J. Chen, Chin. Chem. Lett., 2023, 34, 107735 CrossRef CAS; (e) N. Li, R. Sitdikov, A. P. Kale, J. Steverlynck, B. Li and M. Rueping, Beilstein J. Org. Chem., 2024, 20, 2500–2566 CrossRef CAS PubMed.
- (a) X. Sun and K. Zheng, Nat. Commun., 2023, 14, 6825 CrossRef CAS PubMed; (b) J. Wu, R. Purushothaman, F. Kallert, S. L. Homölle and L. Ackermann, ACS Catal., 2024, 14(15), 11532–11544 CrossRef CAS PubMed.
- (a) S. K. Hota and S. Murarka, Chem. – Asian J., 2024, 19, e202301027 CrossRef CAS PubMed; (b) C. Zhu, H. Chen, H. Yue and M. Rueping, Nat. Synth., 2023, 2, 1068–1081 CrossRef CAS; (c) G. S. Kumar, C. Zhu, R. Kancherla, P. S. Shinde and M. Rueping, ACS Catal., 2023, 13, 8813–8820 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5cc01836f |
| This journal is © The Royal Society of Chemistry 2025 |