
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArtificial amidase with modifiable active sites and designable substrate selectivity for aryl amide hydrolysis†
Mohan Lakavathu and
Yan Zhao *
*
Department of Chemistry, Iowa State University, Ames, Iowa 50011-3111, USA. E-mail: zhaoy@iastate.edu; Tel: +1-515-294-5845
First published on 19th May 2025
Abstract
Hydrolases are used by cells to process key biomolecules including peptides and esters. Previous synthetic mimics of proteases generally only hydrolyze highly active ester derivatives. We report a synthetic catalyst with an acid/base dyad in its active site that hydrolyzes aryl amides under near physiological conditions. The aspartic protease mimic achieves substrate selectivity by its imprinted active site, which is tunable through different template molecules used during molecular imprinting. It can be designed to maintain or override the intrinsic activities of aryl amides in a predictable manner.
Hydrolases are vital for cells to process key biomolecules including peptides, lipids, nucleic acids, and carbohydrates. Spontaneous hydrolyses of amide, phosphate ester, or glycosidic bonds take hundreds to tens of millions of years.1 Chemists can speed up these reactions by activating the electrophile (e.g., protonation of carbonyl), the nucleophile (e.g., deprotonation of water), and/or the leaving group (e.g., by protonation of a glycosidic oxygen). Enzymes employ the same fundamental strategies to speed up their catalytic reactions,2 but they do so inside their active sites instead of changing the entire reaction media as chemists do. This allows enzymes to target specific substrates (or specific sites on a substrate) in the presence of similar or more reactive functional groups in the same solution—a selectivity difficult to realize with synthetic catalysts.
Aspartic proteases employ a pair of carboxylic acids for catalytic amide hydrolysis.3 One of the two acids is deprotonated and the resulting carboxylate/carboxylic acid dyad is able to activate the electrophilic substrate, nucleophilic water, and the amine leaving group cooperatively.4
In this work, we set our goal to employ an acid/base dyad to duplicate the function of aspartic protease inside a synthetically constructed active site. Many efforts have been made to prepare protease-mimicking synthetic catalysts, but they generally only hydrolyze highly activated esters.5–19 Limited success has been reported in creating artificial enzymes to hydrolyze amides, i.e., artificial amidase.20–22 Natural enzymes, for example, use their active sites to distinguish closely related substrates and are even able to overturn intrinsic reactivities of the substrates. These features are yet to be realized in the synthetic mimics of protease.
To create our artificial amidase, we first designed template molecule 1a, whose structure is color-coded to highlight the different purposes of the substructures (Scheme 1): the red moiety is the space holder for the substrate and the blue part indicates the to-be-installed catalytic dyad; and the black part allows the template to be polymerized into a cross-linked polymer network.
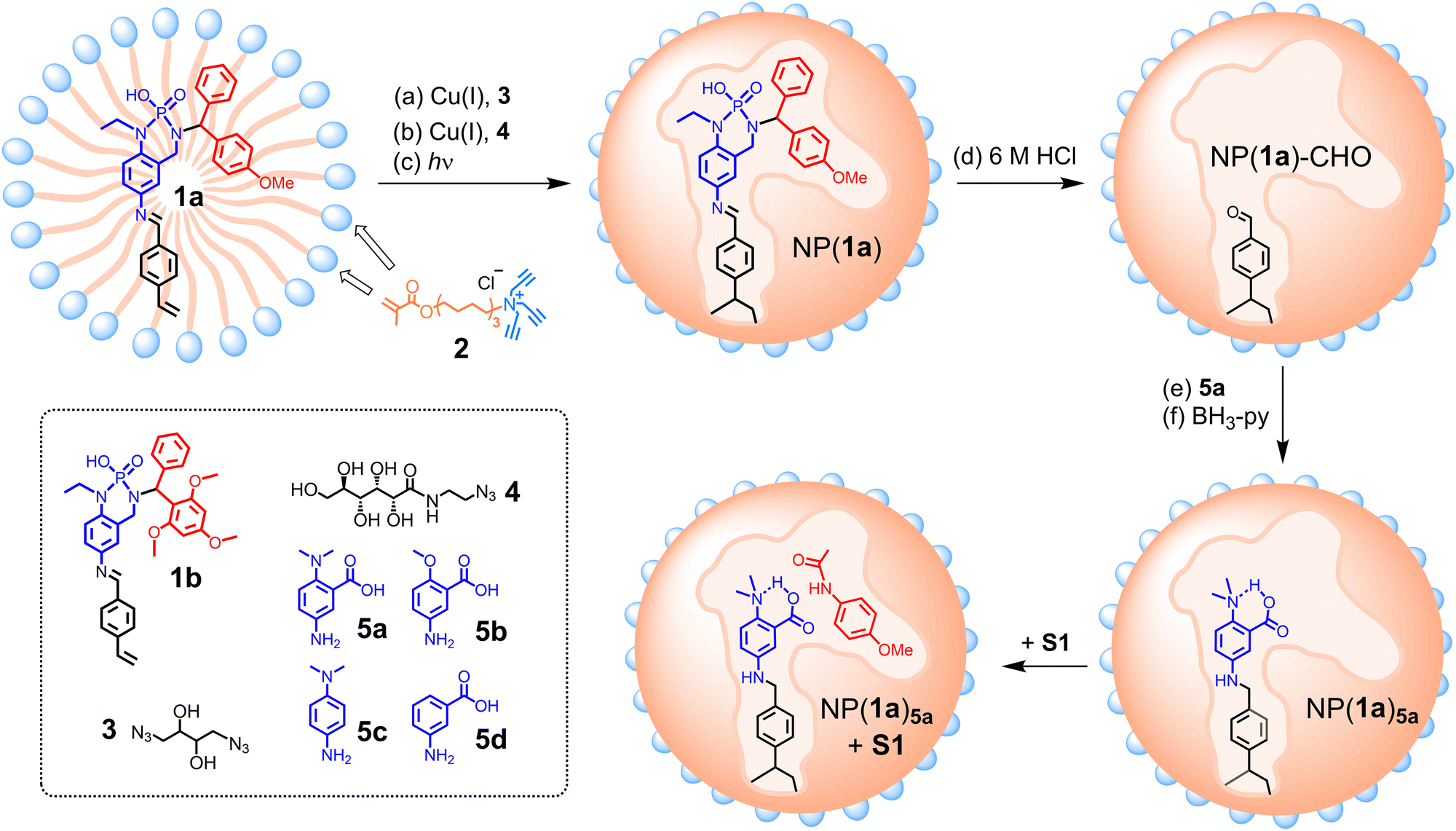 | ||
| Scheme 1 Preparation of molecularly imprinted nanoparticles NP(1a)5a as artificial amidase for the hydrolysis of S1. Surface ligands (i.e., clicked 4) on the micelle surface are omitted for clarity. | ||
To create enzyme-like water-soluble nanoparticles, we performed molecular imprinting23–32 of 1a in the micelle of cross-linkable surfactant 2. The mixed micelle, containing divinyl benzene (DVB, a free radical cross-linker) and 2,2-dimethoxy-2-phenylacetophenone (DMPA, a photo initiator), is first cross-linked on the surface by diazide 3 via the highly efficient click reaction (Scheme 1, step a). A second round of click reaction with monoazide 4 introduces a layer of hydrophilic ligands on the micelle surface (step b). In step c, UV-induced photopolymerization co-polymerizes the template with polymerizable surfactant 2 and DVB, solidifying the micelle core to afford nanoparticles, NP(1a).
In the post-modification, the imine bond of the polymerized template inside NP(1a) is first hydrolyzed in 6 M HCl to afford an aldehyde group in NP(1a)-CHO (step d), which is functionalized through reductive amination with 5a (steps e and f). The resulting NP(1a)5a, i.e., the imprinted nanoparticle prepared with 1a as the template and post-modified with 5a in the reductive amination, contains an acid/base pair in the active site, with a nearby substrate-binding site similar in size and shape to the red-colored moiety of the template. As shown in Scheme 1, this site is designed to accommodate 4-methoxyphenyl acetanilide (S1), with the carbonyl close to the catalytic dyad.
NP(1a)5a indeed is able to perform catalytic hydrolysis of aryl amides (S1 and S2), but not less reactive amides such as S3 and S4 (Fig. 1a). Note that, once taken out of the active site, 5a is completely inactive toward either 4-nitrophenyl acetate (an activated ester) or 4-nitrophenyl acetanilide (S2) (Fig. S3–S6, ESI†). Substrate S2 has a better leaving group than S1, but only affords ca. 1/3 of the hydrolytic product in the NP(1a)5a-catalyzed reaction (Fig. 1a). The overturn of intrinsic reactivity is a strong indicator for successful molecular imprinting. After all, template 1a with its para-methoxyphenyl group is designed to bind S1 that contains the same substructure. Molecularly imprinted micelles have been shown to have outstanding abilities to distinguish small changes in their guests during binding, including the addition,33 removal,33 and shift34 of a single methyl (or methylene) group. Covalent imprinting, as in our case, is also known to have high fidelity in the imprinting process.35,36
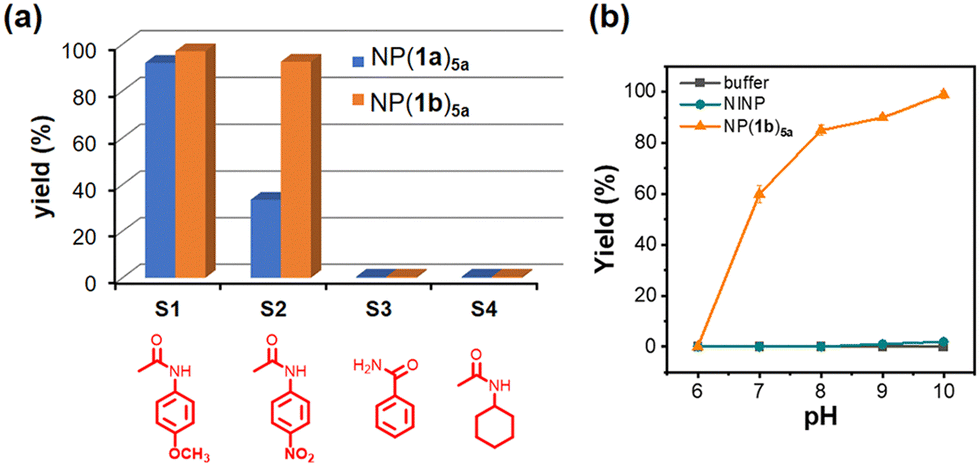 | ||
| Fig. 1 (a) Yields of amide hydrolyses by NP(1a)5a and NP(1b)5a after 20 h at 80 °C in pH 8 buffer. [substrate] = 50 μM. [catalyst] = 10 μM. (b) The pH profile in the hydrolysis of S2 by NP(1b)5a. | ||
Since the imprinted active site strongly influences the substrate selectivity, we prepared NP(1b)5a using template 1b that is expected to afford a larger and more accommodating substrate-binding site. Both S1 and S2 become equally reactive with this catalyst under the experimental conditions (20 h at 80 °C in pH 8 buffer) while S3 and S4 stay unreactive (Fig. 1a). It seems that the larger imprinted pocket can now reasonably accommodate the para-nitro-substituted substrate, and the higher intrinsic reactivity allows S2 to catch up with S1 (see below for additional discussion).
Fig. 1b shows the pH profile for the hydrolysis of S2 by NP(1b)5a. A large change in catalytic activity happens over pH 6–8 while a further increase of pH brings a less significant effect. The same figure indicates that the aryl amide has negligible reactivity in the presence of the nonimprinted nanoparticles (NINPs, similarly prepared nanoparticles in the absence of the template) or in the buffer. To further confirm the importance of the imprinted pocket and the catalytic dyad, we studied the hydrolysis of S2 by several other control catalysts, i.e., NP(1b)-CHO or NP(1b) with an aldehyde group in the imprinted site after the imine hydrolysis, and NP(1b)5b–d (i.e., the nanoparticles obtained through reductive amination of NP(1b)-CHO using compounds 5b–d). The fact that none of these nanoparticles display significant activities (Fig. S19, ESI†) indicates that both the amine and the carboxylic acid in NP(1b)5a are critical to the observed activity.
NP(1b)5a exhibits enzyme-like Michaelis–Menten kinetics in its catalytic hydrolysis of S2 under near physiological conditions (Fig. 2a). With a Michaelis constant (Km) of 94 ± 11 μM and the catalytic turnover (kcat) of 0.0065 min−1, the catalytic efficiency (kcat/Km) is 69 M−1 min−1. Natural enzymes tend to have high turnovers but moderate substrate binding. In contrast, our catalysis is dominated by substrate binding, likely because the template bears too close a resemblance to the substrate instead of the transition state.
 | ||
| Fig. 2 (a) Michaelis–Menten plot of the hydrolysis of S2 by NP(1b)5a in a 25 mM HEPES buffer at 40 °C and pH 7.4. [NP(1b)5a] = 8 μM. (b) Hammett σ–ρ correlation in the hydrolysis of para-substituted-phenyl acetates catalyzed by NP(1b)5a. | ||
We probed the catalytic mechanism of NP(1b)5a by two methods, employing more reactive p-substituted phenyl acetates as commonly done in both natural37,38 and artificial enzymes.5–19 Hammett plots reveal the amount of negative charge developed on the phenyl oxygen in aryl ester hydrolysis. Anionic oxygen-based nucleophiles tend to give a reaction constant (ρ) of 1–1.2.39–41 In contrast, nucleophilic attack of the ester by a neutral nitrogen affords a ρ of 2–3, while a general-base-catalyzed water attack has a ρ of 0.5–0.7. The ρ value of NP(1b)5a for aryl ester hydrolysis is 1.33 (Fig. 2b), consistent with an anionic nucleophile in the active site, likely the carboxylate after intramolecular proton transfer to the ortho amine (Scheme 1).
The second mechanistic investigation involves the solvent kinetic isotope effect (KIE): reactions with a cleavage of the O–H bond in the rate-determining step such as a general-base catalysis has a primary KIE of kH2O/kD2O = 2–3.41–43 In our case, a KIE value of 1.06 is obtained for the hydrolysis of p-nitrophenyl acetate by NP(1b)5a (Table S1, ESI†), ruling out such mechanisms. Apparently, the proton transfer from the carboxylic acid to the neighboring amine occurred prior to the rate-determining step, allowing the carboxylate to carry out the nucleophilic attack as an anionic oxygen nucleophile, also supported by the Hammett plot as discussed above.
It should be mentioned that nucleophilic attack on the carbonyl is just one of the many steps in the amide hydrolysis, which involves a much poorer leaving group than the aryl esters. Departure of the leaving group and hydrolysis of the acylated catalyst need to occur before the catalyst is ready for another round of catalysis. The pH profile shown in Fig. 1b should be a composite effect, as all these steps are pH-dependent.
Why is the same dyad highly active inside the molecularly imprinted site but completely inactive in solution? A key reason is probably proximity, since the substrate is bound by the imprinted site, with its carbonyl right next to the catalytic groups. Desolvation likely is also highly important. The carboxylate can hydrogen-bond easily with solvent molecules in water and become less nucleophilic. This explains why 5a is inactive in methanol or water. Once the dyad resides in a hydrophobic pocket, not only does the lack of nearby protic solvent molecules increase the nucleophilicity of the carboxylate, but the ortho amine after turning into the ammonium ion (i.e., the conjugate acid) can also stabilize the anionic tetrahedral intermediate. Electrostatic interactions are especially strong in a nonpolar microenvironment and have been proposed to be a major contributor to enzyme catalysis.44
Fig. 1a shows that NP(1b)5a with its larger substrate-binding site is less selective than NP(1a)5a. It nonetheless is able to differentiate the substitution pattern on the phenyl group of aryl amides. As shown in Fig. 3, it tolerates substrates with various para groups on the phenyl ring (S1, S2, S5, and S6) and also ortho-nitrophenyl acetamide (S7). Consistent with the 2,4-6-trimethoxy substitution on its template (1b), despite its broad substrate selectivity, NP(1b)5a completely excludes meta-nitrophenyl acetamide (S8).
 | ||
| Fig. 3 The yields of aryl amide hydrolysis catalyzed by NP(1b)5a after 24 h in HEPES buffer (pH 7.4) at 40 °C. | ||
S1 is more reactive than S2 with NP(1a)5a but the two become similarly reactive in the presence of NP(1b)5a (Fig. 1a). Under a milder condition (at 40 °C and pH 7.4), S2 overtakes S1. It seems that when competing factors are at play (e.g., intrinsic reactivity versus templating effect), the final selectivity becomes a tradeoff and can vary under different reaction conditions.
This work shows that a simple acid/base catalytic dyad inside a substrate-tailored imprinted pocket can hydrolyze aryl amides under near physiological conditions (pH 7.4 at 40 °C) but the same dyad is completely unreactive in solution. A highlight of the system is the readily tunable substrate-binding site, via different template molecules (1a and 1b). Tunable reactivity is then achieved, a feature commonly seen in natural enzymes but rare in synthetic systems.45,46 As a result, aryl amides with less intrinsic reactivity can be made more reactive and substitution patterns on the substrate are easily distinguished in a predictable manner.
We thank NSF (CHE-2246635) for financial support.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Wolfenden, Annu. Rev. Biochem., 2011, 80, 645–667 CrossRef CAS PubMed.
- E. Erez, D. Fass and E. Bibi, Nature, 2009, 459, 371–378 CrossRef CAS PubMed.
- B. M. Dunn, Chem. Rev., 2002, 102, 4431–4458 CrossRef CAS PubMed.
- D. B. Northrop, Acc. Chem. Res., 2001, 34, 790–797 CrossRef CAS PubMed.
- D. J. Cram, H. E. Katz and I. B. Dicker, J. Am. Chem. Soc., 1984, 106, 4987–5000 Search PubMed.
- V. T. D'Souza and M. L. Bender, Acc. Chem. Res., 1987, 20, 146–152 CrossRef.
- K. Skorey, V. Somayaji and R. Brown, J. Am. Chem. Soc., 1989, 111, 1445–1452 CrossRef CAS.
- T. Ema, D. Tanida, T. Matsukawa and T. Sakai, Chem. Commun., 2008, 957–959 RSC.
- H. B. Albada and R. M. J. Liskamp, J. Comb. Chem., 2008, 10, 814–824 CrossRef CAS PubMed.
- M. Kheirabadi, N. Çelebi-Ölçüm, M. F. L. Parker, Q. Zhao, G. Kiss, K. N. Houk and C. E. Schafmeister, J. Am. Chem. Soc., 2012, 134, 18345–18353 CrossRef CAS PubMed.
- M. J. MacDonald, L. D. Lavis, D. Hilvert and S. H. Gellman, Org. Lett., 2016, 18, 3518–3521 CrossRef CAS PubMed.
- J. J. Garrido-González, M. M. Iglesias Aparicio, M. M. García, L. Simón, F. Sanz, J. R. Morán and Á. L. Fuentes de Arriba, ACS Catal., 2020, 10, 11162–11170 CrossRef.
- J. J. Garrido-González, E. Sánchez-Santos, A. Habib, Á. V. Cuevas Ferreras, F. Sanz, J. R. Morán and Á. L. Fuentes de Arriba, Angew. Chem., Int. Ed., 2022, 61, e202206072 CrossRef PubMed.
- P. Pengo, S. Polizzi, L. Pasquato and P. Scrimin, J. Am. Chem. Soc., 2005, 127, 1616–1617 CrossRef CAS PubMed.
- M. D. Nothling, A. Ganesan, K. Condic-Jurkic, E. Pressly, A. Davalos, M. R. Gotrik, Z. Xiao, E. Khoshdel, C. J. Hawker, M. L. O'Mara, M. L. Coote and L. A. Connal, Chem, 2017, 2, 732–745 CAS.
- M. D. Nothling, Z. Xiao, N. S. Hill, M. T. Blyth, A. Bhaskaran, M.-A. Sani, A. Espinosa-Gomez, K. Ngov, J. White, T. Buscher, F. Separovic, M. L. O’Mara, M. L. Coote and L. A. Connal, Sci. Adv., 2020, 6, eaaz0404 CrossRef CAS PubMed.
- I. Bose and Y. Zhao, ACS Catal., 2021, 11, 3938–3942 CrossRef CAS PubMed.
- N. Liu, S.-B. Li, Y.-Z. Zheng, S.-Y. Xu and J.-S. Shen, ACS Omega, 2023, 8, 2491–2500 CrossRef CAS PubMed.
- Z. Wang, Y. Lu, J. Yang, W. Xiao, T. Chen, C. Yi and Z. Xu, Langmuir, 2023, 39, 5929–5935 CrossRef CAS PubMed.
- Y.-M. Wong, Y. Hoshino, K. Sudesh, Y. Miura and K. Numata, Biomacromolecules, 2015, 16, 411–421 CrossRef CAS PubMed.
- M. Divya, T. Benny, P. Christy, E. P. Aparna and K. S. Devaky, Bioorg. Chem., 2017, 74, 91–103 CrossRef CAS PubMed.
- T. Matsushita, H. Yamochi, S. Omiya, T. Koyama, K. Hatano and K. Matsuoka, Bioorg. Med. Chem., 2023, 92, 117422 CrossRef CAS PubMed.
- G. Wulff and J. Liu, Acc. Chem. Res., 2012, 45, 239–247 Search PubMed.
- X. Shen, C. Huang, S. Shinde, K. K. Jagadeesan, S. Ekström, E. Fritz and B. Sellergren, ACS Appl. Mater. Interfaces, 2016, 8, 30484–30491 CrossRef CAS PubMed.
- Y. Yuan, Y. Yang, M. Faheem, X. Zou, X. Ma, Z. Wang, Q. Meng, L. Wang, S. Zhao and G. Zhu, Adv. Mater., 2018, 30, 1800069 CrossRef PubMed.
- J. Pan, W. Chen, Y. Ma and G. Pan, Chem. Soc. Rev., 2018, 47, 5574–5587 RSC.
- S. Li, P. A. Lieberzeit, S. Piletsky and A. P. F. Turner, Smart polymer catalysts and tunable catalysis, Elsevier, Amsterdam, Netherlands; Cambridge, MA, 2019 Search PubMed.
- H. Zhang, Adv. Mater., 2020, 32, 1806328 CrossRef CAS PubMed.
- K. Haupt, P. X. Medina Rangel and B. T. S. Bui, Chem. Rev., 2020, 120, 9554–9582 Search PubMed.
- S. Muratsugu, S. Shirai and M. Tada, Tetrahedron Lett., 2020, 61, 151603 CrossRef CAS.
- J. Li, M. Zhu, M. Wang, W. Qi, R. Su and Z. He, Soft Matter, 2020, 16, 7033–7039 RSC.
- W. Wei, V. K. Thakur, Y. J. Chew and S. Li, Mater. Today Chem., 2020, 17, 100286 Search PubMed.
- K. Chen and Y. Zhao, Org. Biomol. Chem., 2019, 17, 8611–8617 RSC.
- J. K. Awino, R. W. Gunasekara and Y. Zhao, J. Am. Chem. Soc., 2017, 139, 2188–2191 CrossRef CAS PubMed.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812–1832 CrossRef CAS.
- G. Wulff, Chem. Rev., 2002, 102, 1–28 CrossRef CAS PubMed.
- J. A. Verpoorte, S. Mehta and J. T. Edsall, J. Biol. Chem., 1967, 242, 4221–4229 CrossRef CAS PubMed.
- F. J. Kezdy and M. L. Bender, Biochemistry, 1962, 1, 1097–1106 CrossRef CAS PubMed.
- T. C. Bruice and S. J. Benkovic, J. Am. Chem. Soc., 1964, 86, 418–426 CrossRef CAS.
- J. F. Kirsch, W. Clewell and A. Simon, J. Org. Chem., 1968, 33, 127–132 CrossRef CAS.
- R. A. Gibbs, P. A. Benkovic, K. D. Janda, R. A. Lerner and S. J. Benkovic, J. Am. Chem. Soc., 1992, 114, 3528–3534 CrossRef CAS.
- M. L. Bender, E. J. Pollock and M. C. Neveu, J. Am. Chem. Soc., 1962, 84, 595–599 CrossRef CAS.
- B. M. Anderson, E. H. Cordes and W. P. Jencks, J. Biol. Chem., 1961, 236, 455–463 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. B. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- M. D. Arifuzzaman, I. Bose, F. Bahrami and Y. Zhao, Chem. Catal., 2022, 2, 2049–2065 CrossRef CAS PubMed.
- F. Bahrami and Y. Zhao, J. Org. Chem., 2024, 89, 5148–5152 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, characterization data, additional data, and NMR spectra. See DOI: https://doi.org/10.1039/d5cc01868d |
| This journal is © The Royal Society of Chemistry 2025 |