
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic generation of hypochlorous acid on plasmonic Au/AgCl catalysts prepared by microwave-assisted reduction†
Yasuhiro
Shiraishi
 *ab,
Yoko
Hiroaki
a,
Satoshi
Ichikawa
c,
Shunsuke
Tanaka
d and
Takayuki
Hirai
a
*ab,
Yoko
Hiroaki
a,
Satoshi
Ichikawa
c,
Shunsuke
Tanaka
d and
Takayuki
Hirai
a
aResearch Center for Solar Energy Chemistry and Division of Chemical Engineering, Graduate School of Engineering Science, Osaka University, Toyonaka 560-8531, Japan. E-mail: shiraishi.yasuhiro.es@osaka-u.ac.jp
bInnovative Catalysts Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), Osaka University, Suita 565-0871, Japan
cResearch Center for Ultra-High Voltage Electron Microscopy, Osaka University, Ibaraki 567-0047, Japan
dDepartment of Chemical, Energy, and Environmental Engineering, Kansai University, Suita 564-8680, Japan
First published on 9th May 2025
Abstract
Plasmonic Au particles loaded on AgCl powders (Au/AgCl) prepared by microwave-assisted reduction, when photoirradiated under visible light in aerated chloride solutions, efficiently generate hypochlorous acid owing to the large Au–AgCl periphery sites by the formation of small Au particles.
Hypochlorous acid (HClO) is a strong, water-soluble oxidant that is commonly used for disinfection, bleaching, and sterilization.1 HClO is industrially produced from Cl2 gas manufactured by the electrolysis of Cl− solutions,2 which involves the oxidation of Cl− at the anode (eqn (1)) and the reduction of O2 (eqn (2)) at the cathode.3 The total electrolysis can be expressed as shown in eqn (3).4 The dissolution of Cl2 gas in water generates HClO by disproportionation (eqn (4)). The electrolysis (eqn (3)) is an up-hill reaction with a large Gibbs free energy gain and requires a huge amount of electrical energy.5 Therefore, a clean and sustainable method is necessary for HClO production.
| 2Cl− → Cl2 + 2e− (+1.36 V vs. NHE) | (1) |
| O2 + 4H+ + 4e− ⇌ 2H2O (+1.23 V vs. NHE) | (2) |
| 2Cl− + 1/2O2 + 2H+ → Cl2 + H2O (ΔG° = +105 kJ mol−1 at pH 7.0) | (3) |
| Cl2 + H2O ⇌ HClO + H+ + Cl− (K = 3.94 × 10−4 M2 at 298 K) | (4) |
Semiconductor photocatalysis can generate HClO in Cl− solutions with O2 at ambient temperature (eqn (3)). The photogenerated valence band holes (hVB+) oxidize Cl− to form Cl2 (eqn (1)), which generates HClO via disproportionation (eqn (4)), while the conduction band electrons (eCB−) reduce O2 (eqn (2)). However, early reported photocatalysts produced low-HClO-concentration solutions (<3 ppm).6–9 This is because (i) the oxidation of bulk Cl− by hVB+ is difficult to promote due to its deep oxidation potential (eqn (1)); and (ii) these catalysts are mainly photoexcited by UV light (λ < 400 nm), which readily promotes photolysis of HClO into O2 and Cl− (eqn (5)).4 The design of photocatalysts that efficiently oxidize Cl− under visible light is therefore necessary.
| 2HClO → O2 + 2H+ + 2Cl− | (5) |
Recently, we found that visible light irradiation of Au particles loaded on semiconductor AgCl powders, synthesized by a deposition–precipitation (DP) method [Au(DP)/AgCl], efficiently generated HClO in Cl− solutions under air flow.4 As shown in Scheme 1A(a), visible light activation of the localized surface plasmon resonance (LSPR) of Au particles produces the hot electrons (ehot−) and hot holes (hhot+). As shown in (b), the ehot− is injected into the AgCl CB and reduces O2 (eqn (2)). The hhot+ oxidizes the lattice Cl− of AgCl (ClL−) in the periphery of Au particles and generates Cl2, which is disproportionated to afford HClO (eqn (4)). As shown in Scheme 1B, the ClL− oxidation occurs at ca. +0.5 V [vs. reversible hydrogen electrode (RHE)], which is more negative than that for bulk Cl− oxidation (1.36 V), owing to the strong Ag–Cl orbital hybridization. This results in efficient HClO generation under visible light. As shown in Scheme 1A(c), the eliminated ClL− is compensated from the solution. The ClL− oxidation/compensation cycle on the Au(DP)/AgCl catalysts generated 38 ppm of HClO, which was 10-fold higher than that generated by earlier reported systems.6–9
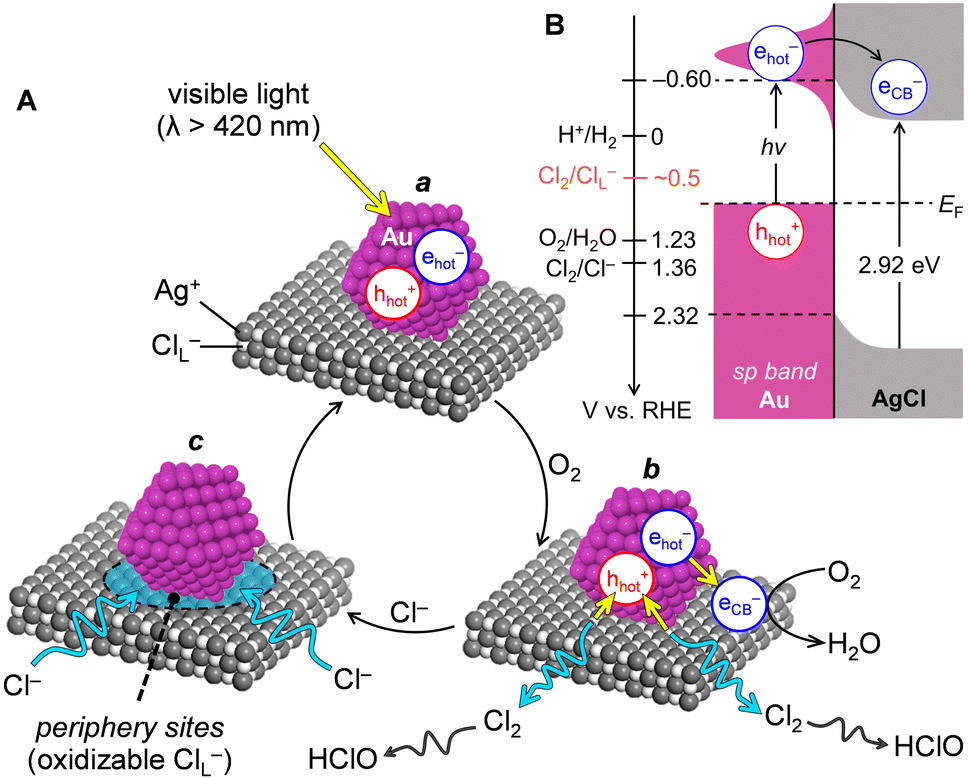 | ||
| Scheme 1 Proposed mechanism for (A) HClO generation on plasmonic Au/AgCl catalysts in Cl− solutions with O2, which includes (a) LSPR activation, (b) oxidation/reduction, and (c) compensation of ClL−. (B) Band structures at the Au–AgCl interface. | ||
The next challenge is the enhancement of catalytic activity. The rate-determining step is the ClL− oxidation on the peripheral sites at the Au–AgCl interface (Scheme 1A(c)). We hypothesized that creating a large number of small Au particles on AgCl would increase the number of peripheral sites, which may increase the number of ClL− that can be oxidized by ehot− photogenerated on the Au particles. The DP method used for deposition of Au particles in our previous work4 involves the adsorption of Au precursors (HAuCl4·4H2O) onto AgCl and subsequent calcination at a high temperature (673 K).10 In this case, Au(DP)/AgCl with 2.7 wt% Au showed the highest activity. However, higher Au loadings created larger Au particles owing to their migration and coalescence.11 This decreases the number of peripheral ClL− and showed lower activity.4 Therefore, a method to create a large number of small Au particles is necessary.
Microwave (MW)-assisted reduction is a simple method for the deposition of metal particles. This facilitates the deposition of several kinds of metal particles (Au,12 Pt,13 Pd14) onto several inorganic/organic supports by a relatively short time (∼30 min) MW irradiation of a mixture containing metal precursors, supports, and reducing reagents such as ethylene glycol at low temperatures (373–453 K). In the present work, we deposited Au particles on AgCl by the MW method using ethylene glycol as a reducing reagent. The obtained Au(MW)/AgCl powders contained a large number of small Au particles. This increases the number of peripheral ClL− and exhibits enhanced activity.
AgCl powder was prepared by a precipitation method using AgNO3 and KCl in water.15 The AgCl powder was stirred in ethylene glycol with different amounts of HAuCl4·4H2O. MWs were irradiated onto the mixture for 17 min at 433 K under magnetic stirring. The resultant product was recovered by centrifugation, washed with water, and dried in vacuo, yielding Au(MW)x/AgCl powders, where x denotes the amount of Au loaded, which is determined by X-ray fluorescence (XRF) analysis [x (wt%) = Au/AgCl × 100]. The powder X-ray diffraction (XRD) patterns of Au(MW)x/AgCl (Fig. S1, ESI†) showed a peak at 38° assigned to the {111} diffraction peak of metallic Au (JCPDS 04-0784) as is the case for Au(DP)x/AgCl,4 indicating the formation of Au0. They also showed peaks assigned to cubic AgCl (JCPDS 31-1238) as is the case for pristine AgCl and Au(DP)x/AgCl, indicating that the AgCl structure was maintained even after the MW irradiation.
Photoreactions were performed in 550 mM NaCl solutions whose Cl− concentration was similar to that of seawater.4 A NaCl solution (50 mL) containing catalyst (0.1 g) was photoirradiated under visible light (λ > 420 nm) using a Xe lamp under air flow (1.0 L min−1) with magnetic stirring at 303 K, where the emission spectrum of the lamp is shown in Fig. S2 (ESI†). Fig. 1 (blue bars) shows the amount of HClO generated on the respective catalysts during 24 h of photoirradiation. In the case of Au(DP)x/AgCl, Au(DP)2.7/AgCl showed the highest activity, and further Au loading decreased the activity.4 In contrast, in the case of Au(MW)x/AgCl, Au(MW)4.9/AgCl and Au(MW)6.1/AgCl showed the highest activity with the amount of HClO generated being about twice those generated on Au(DP)2.7/AgCl. In addition, Au(MW)2.7/AgCl also showed higher activity than Au(DP)2.7/AgCl. This indicates that the MW method produces more active Au/AgCl catalysts than the DP method.
 | ||
| Fig. 1 Amount of HClO generated on the respective catalysts during 24 h of photoirradiation in (red) water and (blue) 550 mM NaCl solution. Reaction conditions: solution (50 mL), catalyst (0.1 g), air flow (1.0 L min−1), λ > 420 nm (Xe lamp), and temperature (303 K). | ||
The action spectrum for HClO generation on Au(MW)4.9/AgCl (Fig. S3, ESI†) agrees with the Au LSPR band, as is the case for Au(DP)2.7/AgCl,4 confirming that the Au LSPR activation by visible light triggers the reaction (Scheme 1A(a)). The apparent quantum yields (ΦAQY) of Au(MW)4.9/AgCl are higher than those of Au(DP)2.7/AgCl at the entire wavelengths, suggesting that the hhot+ and ehot− generated on Au(MW)4.9/AgCl are consumed more efficiently. Cyclic voltammetry (CV) was performed using the catalyst-loaded fluorinated tin oxide (FTO) electrodes to confirm the ClL− oxidation/compensation cycle (Scheme 1A(b) and (c)). The CV of Au(MW)4.9/AgCl measured in NaCl solution under O2 (Fig. S4, ESI†) showed the anodic current for ClL− oxidation at >0.6 V (vs. RHE) and the cathodic current for O2 reduction at <0.2 V. These currents appeared reversibly, as was the case for Au(DP)2.7/AgCl.4 In contrast, the absence of Cl− in solution showed a weak current for ClL− oxidation, which disappeared after the second cycles. The results suggest that the catalyst photoexcited in Cl− solution generates HClO via the Au LSPR activation by visible light and ClL− oxidation/compensation cycle (Scheme 1A). The electrochemical impedance spectroscopy (EIS) Nyquist plots of Au(MW)4.9/AgCl− and Au(DP)2.7/AgCl− loaded FTO measured in NaCl solution in the dark (Fig. S5, ESI†) showed similar charge transfer resistance (RCT). Visible light irradiation decreased the RCT of both catalysts with Au(MW)4.9/AgCl showing lower RCT. This indicates that the hhot+ photogenerated on Au(MW)4.9/AgCl is consumed more efficiently by oxidation of ClL−: the efficient consumption of hhot+ by ClL− results in higher photocatalytic activity of Au(MW)/AgCl (Fig. 1). It is noted that Au(MW)4.9/AgCl, when reused for photoreactions, showed activity similar to that of the fresh catalyst (Fig. S6, ESI†), suggesting that the catalyst is reusable without significant loss of activity.
The higher photocatalytic activity of Au(MW)/AgCl originates from the formation of smaller Au particles. Fig. 2a shows the results of scanning transmission electron microscopy (STEM) observation of Au(MW)4.9/AgCl. The energy-dispersive X-ray spectroscopy (EDS) elemental maps indicate that Au particles are dispersed on the AgCl surface, as is the case for Au(MW)1.3/AgCl (Fig. S7, ESI†), Au(DP)2.7/AgCl (Fig. S8, ESI†), and Au(DP)4.5/AgCl (Fig. S9, ESI†). The size distributions of the Au particles (Fig. 2b) show that Au(DP)4.5/AgCl contains Au particles with an average diameter of 50 nm, which is larger than that of Au(DP)2.7/AgCl (41 nm). This indicates that, in the DP method, higher Au loadings create larger Au particles due to the migration and coalescence of the particles by high temperature calcination (673 K).4,11 In contrast, Au(MW)4.9/AgCl (Fig. 2c) contains smaller Au particles (36 nm), which are similar to those of the lower-Au-loading Au(MW)1.3/AgCl catalyst (38 nm). This indicates that the MW method creates smaller Au particles even with higher Au loadings. This may be because rapid heating by MW irradiation creates numerous nuclei16 and low temperature heating (433 K) suppresses their migration and coalescence.
 | ||
| Fig. 2 (a) STEM-EDS results of Au(MW)4.9/AgCl. Size distributions of Au particles on (b) Au(DP)x/AgCl and (c) Au(MW)x/AgCl catalysts. | ||
The smaller Au particles created by the MW method increase the number of peripheral sites (Scheme 1A(c)), which efficiently promotes the oxidation of ClL− by hhot+ photogenerated on Au particles, enhancing photocatalytic activity. The number of peripheral ClL− can roughly be determined by the photoreaction in pure water without Cl−. In this case, ClL− of AgCl oxidized by hhot+ is not compensated from the solution; therefore, the amount of HClO generated reflects the number of peripheral ClL− that can be oxidized by hhot+ on Au particles. As shown in Fig. 1 (red bars), the amount of HClO generated on Au(MW)x/AgCl is much larger than that generated on Au(DP)2.7/AgCl and increases with Au loading. These results agree with the photocatalytic activity (blue bars) and the EIS Nyquist plots (Fig. S5, ESI†). This suggests that the MW method produces small Au particles on the AgCl surface and creates a larger number of peripheral ClL− that are active for the consumption of the hhot+ photogenerated, thereby enhancing photocatalytic activity.
The amount of HClO generated on Au(MW)x/AgCl (blue bars in Fig. 1) is saturated with ≥4.9 wt% Au, although the number of peripheral ClL− increases with ≥4.9 wt% Au (red bars). This is because of the decrease in the LSPR band of the Au particles. The diffuse-reflectance (DR) UV-vis spectra of Au(MW)x/AgCl (Fig. 3) indicate that the intensity of the LSPR band decreases with Au loading, together with a red shift of the band. This is ascribed to the decrease in the electron density of Au particles.11 X-ray photoelectron spectroscopy (XPS) analysis (Fig. S10, ESI†) was used to clarify the electronic states of Au, Ag, and Cl. The Ag 3d (Fig. S11, ESI†) and Cl 2p (Fig. S12, ESI†) spectra of Au(DP)2.7/AgCl and Au(MW)x/AgCl are similar, indicating that the Ag and Cl components of these catalysts have similar electronic states. The Au 4f spectra of the catalysts (Fig. S13, ESI†) show Au0 components.17 The increase in the Au loading shifts the peaks to higher binding energy, confirming the decrease in the electron density of the Au particles. The Au loading leads to a transfer of AgCl eCB− to the Au particles to balance their Fermi levels, thereby increasing the electron density of the Au particles.18 However, loading of excess amounts of Au may lead to a charge dispersion on the Au particles.19 This decreases the electron density of the Au particles and weakens the LSPR band (Fig. 3). The weak LSPR band suppresses the activation by visible light and decreases the photocatalytic activity, although the number of peripheral ClL− increases with Au loadings (red bars in Fig. 1). The results indicate that loading an appropriate amount of Au particles is necessary for high photocatalytic activity.
 | ||
| Fig. 3 Diffuse-reflectance UV-vis spectra of Au(MW)x/AgCl. | ||
Active Au(MW)/AgCl catalysts can be synthesized at relatively low temperatures (≥413 K). The catalysts synthesized by MW irradiation at ≥413 K showed similar Au loadings and activities (Fig. S14, ESI†). In contrast, lower temperature synthesis created lower-Au-loading catalysts with lower activity: they showed weaker LSPR band (Fig. S15, ESI†) and Au{111} diffraction (Fig. S16, ESI†) because the reduction of Au3+ to Au0 was suppressed at lower temperatures.20 Also note that a relatively short time MW irradiation (≥7 min) produces active catalysts. As shown in Fig. S17 (ESI†), the catalysts synthesized by different MW irradiation times (7–25 min) showed similar Au loadings and activities, where they showed similar LSPR absorption (Fig. S18, ESI†) and Au (111) diffraction (Fig. S19, ESI†). These results indicate that active Au(MW)/AgCl catalysts can be synthesized by short time MW irradiation (≥7 min) at relatively low temperatures (≥413 K).
The solar-to-chemical conversion (SCC) performance of Au(MW)4.9/AgCl was evaluated under irradiation with AM1.5G simulated sunlight21 over the wavelength range of 420–2500 nm (Fig. S2, ESI†). Fig. 4 shows the change in the amount of HClO generated and the SCC efficiency with time. Au(MW)4.9/AgCl (square) generates more HClO than than of Au(DP)2.7/AgCl (circle). The SCC efficiency for HClO generation on Au(MW)4.9/AgCl (∼0.05%) is higher than that on Au(DP)2.7/AgCl (∼0.03%)4 and remains almost constant during the photoirradiation, indicating stable and efficient HClO generation on Au(MW)4.9/AgCl. Note that the irradiation of UV region light (300–400 nm) is ineffective: as shown in Fig. S20 (ESI†), irradiation with entire wavelength light (λ > 300 nm) decreases the HClO amount and SCC efficiency because UV light promotes photolysis of HClO generated (eqn (5)). The results indicate that visible light irradiation is effective for HClO generation. The visible light irradiation of Au(MW)4.9/AgCl for 24 h generated 63 ppm of HClO, which was higher than that obtained on Au(DP)2.7/AgCl (38 ppm).4 This HClO concentration (63 ppm) exceeds the concentration that is recommended by the WHO for the disinfection of drinking water (>3 ppm)22 and the concentration that was reported effective for the inactivation of several types of bacteria and viruses such as SARS-CoV-2 (60 ppm).23 Furthermore, as shown in Fig. S21 (ESI†), the high oxidation capability of the HClO solution (63 ppm) obtained by Au(MW)4.9/AgCl is confirmed by the bleaching tests using a black mold and a piece of denim pants.
 | ||
| Fig. 4 Change in the amount of HClO generated and the SCC efficiency with time under simulated sunlight irradiation of the respective catalysts (λ > 420 nm). Conditions: 550 mM NaCl (50 mL), catalyst (0.1 g), air flow (1.0 L min−1), and temperature (303 K). | ||
In summary, we demonstrated that the MW method rapidly (≥7 min) created a large number of small Au particles (∼40 nm) on AgCl powders under mild temperature (≥413 K) conditions. Visible light irradiation of the obtained Au(MW)/AgCl catalysts in aerated Cl− solutions generated high-HClO-concentration solutions via the ClL− oxidation/compensation cycles owing to the enlarged Au–AgCl periphery site that efficiently promotes ClL− oxidation. The results based on the creation of small Au particles by MW irradiation may contribute to the clean and sustainable production of HClO using solar energy.
This study was supported by JSPS KAKENHI Grant Number (21H01707 and 23K17346).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- W. A. Rutala and D. J. Weber, Clin. Miclobiol. Rev., 1997, 10, 597–610 CrossRef CAS PubMed.
- R. K. B. Karlsson and A. Cornell, Chem. Rev., 2016, 116, 2982–3028 CrossRef CAS PubMed.
- I. Moussallem, J. Jörissen, U. Kunz, S. Pinnow and T. Turek, J. Appl. Electrochem., 2008, 38, 1177–1194 CrossRef CAS.
- Y. Shiraishi, Y. Shimabukuro, K. Shima, S. Ichikawa, S. Tanaka and T. Hirai, JACS Au, 2023, 3, 1403–1412 CrossRef CAS PubMed.
- K. Sayama, ACS Energy Lett., 2018, 3, 1093–1101 CrossRef CAS.
- L. Huang, R. Li, R. Chong, G. Liu, J. Han and C. Li, Catal. Sci. Technol., 2014, 4, 2913–2918 RSC.
- B. Reichman and C. E. Byvik, J. Phys. Chem., 1981, 85, 2255–2258 CrossRef CAS.
- R. Pang, Y. Miseki, S. Okunaka and K. Sayama, ACS Sustainable Chem. Eng., 2020, 8, 8629–8637 CrossRef CAS.
- R. Pang, Y. Miseki and K. Sayama, Catal. Sci. Technol., 2022, 12, 2935–2942 RSC.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- D. Tsukamoto, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2012, 134, 6309–6315 CrossRef CAS PubMed.
- H. Li, F. Qin, Z. Yang, X. Cui, J. Wang and L. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef CAS PubMed.
- E. A. Anumol, P. Kundu, P. A. Deshpande, G. Madras and N. Ravishankar, ACS Nano, 2011, 5(10), 8049–8061 CrossRef CAS PubMed.
- A. M. R. Galletti, C. Antonetti, A. M. Venezia and G. Giambastiani, Appl. Catal., A, 2010, 386, 124–131 CrossRef CAS.
- S. Liu, Q. Zhang, H. Li, Y. Yang, X. Tian and A. Whiting, Chem. – Eur. J., 2015, 21, 9671–9675 CrossRef CAS PubMed.
- M. Tsuji, M. Hashimoto, Y. Nishizawa, M. Kubokawa and T. Tsuji, Chem. – Eur. J., 2005, 11, 440–452 CrossRef CAS PubMed.
- M. P. Casaletto, A. Longo, A. Martorana, A. Prestianni and A. M. Venezia, Surf. Interface Anal., 2006, 38, 215–218 CrossRef CAS.
- S. Liu, J. Chai, S. Sun, L. Zhang, J. Yang, X. Fu, J. Hai, Y. H. Jing and B. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 46451–46463 CrossRef CAS PubMed.
- Y. Lykhach, S. M. Kozlov, T. Skála, A. Tovt, V. Stetsovych, N. Tsud, F. Dvořák, V. Johánek, A. Neitzel, J. Mysliveček, S. Fabris, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2016, 15, 284–288 CrossRef CAS PubMed.
- Y. J. Zhu and X. L. Hu, Chem. Lett., 2003, 32, 1140–1141 CrossRef CAS.
- I. Gordon, F. C. Krebs, X. Mathew, C. M. Lampert, A. Rougier, G. P. Smestad and A. Subrahmanyam, Sol. Energy Mater. Sol. Cells, 2015, 133, A1–A6 CrossRef CAS.
- Guidelines for drinking-water quality: incorporating the first and second addenda, WHO, 4th edn, 2022, https://www.who.int/publications/i/item/9789240045064.
- N. Hatanaka, M. Yasugi, T. Sato, M. Mukamoto and S. Yamasaki, J. Appl. Microbiol., 2022, 132, 1496–1502 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, data (Fig. S1–S21), and references. See DOI: https://doi.org/10.1039/d5cc01929j |
| This journal is © The Royal Society of Chemistry 2025 |