
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of red-shifted 1,2-azaborinine-based molecular solar thermals†
Adrian J.
Müller
 a,
Johannes
Markhart
a,
Holger F.
Bettinger
b and
Andreas
Dreuw
*a
a,
Johannes
Markhart
a,
Holger F.
Bettinger
b and
Andreas
Dreuw
*a
aInterdisciplinary Center for Scientific Computing, Ruprecht-Karls University, Im Neuenheimer Feld 205, 69120 Heidelberg, Germany. E-mail: adrian.mueller@iwr.uni-heidelberg.de; dreuw@uni-heidelberg.de
bInstitute of Organic Chemistry, University of Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany
First published on 5th May 2025
Abstract
1,2-Azaborinines are a promising molecular class for application as molecular solar thermal (MOST) systems offering a solution for solar energy storage. Our computational screening of 4,5-substituted derivatives using density functional theory targeting excitation and storage energies revealed push–pull systems with significantly red-shifted absorption bands, enhancing their spectral overlap with the solar spectrum. Among these, the 4-cyano-5-hydroxy derivative, with a 1.1 eV red-shift of the first excited state compared to the unsubstituted molecule, was investigated in detail and was shown to retain the favourable properties of the parent system. It is thus particularly well suited as a MOST system.
Molecular solar thermal (MOST) systems1 are promising solar fuels for sustainable energy storage. MOST systems are a particular subgroup of photoswitches that store solar energy in the form of chemical energy via a reversible intramolecular conversion upon photoreaction forming a higher-energy, long-lived metastable isomer. The stored energy can then be released on demand in the form of heat. Harvest, conversion, and release are handled by a single molecule making this approach very comprehensive. Various key criteria have been formulated to characterise suitable systems:2–4 (1) non-negligible spectral overlap with the solar spectrum is mandatory and the two isomers must be spectrally separated. Optimally, the metastable isomer absorbs at shorter wavelengths and has no overlap with the solar spectrum to avoid competing photoreactions. (2) A high quantum yield of MOST photoconversion. (3) A sufficiently long lifetime of the metastable state matching the desired application. (4) A high energy storage density achieved by maximising the storage energy ES, while keeping the molecular weight low. (5) The energy release should be triggerable by locally controllable external stimuli, e.g. by catalysis, electrochemistry, or heat. (6) No degradation after numerous switching cycles. (7) The system should be sustainable and non-toxic. It is clear that optimising all these aspects simultaneously is a challenging or even impossible task. For example, introducing substituents to tune the photophysical properties typically leads to a decreased storage energy density.5
1,2-Dihydro-1,2-azaborinine 1 is a molecule with great potential for MOST applications with a predicted storage energy density of 3.1 MJ kg−1 (ES = 59.3 kcal mol−1).6,7 The planar form of 1 resembles benzene where one CC unit is replaced with a BN unit and hence it is also denoted as BNB (BN-benzene). Although it is isoelectronic to benzene, it has a distinct photochemical behaviour, which was studied extensively.6,8–15 The Dewar form 1-BND was the only observed photoproduct upon irradiation with 253.7 nm in a neon matrix and complete conversion was achieved within 105 min (Fig. 1).8 Quantum chemical calculations revealed that after photoexcitation to the first excited state S1, the compound relaxes to the ground state via a conical intersection (CI) to form an energetically shallow prefulvene-like intermediate 1-PF.9,10,16 From there, facile thermal conversion occurs to either 1-BNB or 1-BNDvia the transition states 1-TS1 or 1-TS2, respectively.7,9 A major step towards MOST applicability was achieved when substituents in the 1,2-position were introduced.12 For example, the 1-tert-butyldimethylsilyl-2-mesityl derivative 1a exhibits a red-shifted absorption spectrum, undergoes photoisomerisation with a quantum yield of 46%, and even its Dewar isomer could be isolated at room temperature under inert conditions and was stable for weeks.12 Computations of 1 and a 1a analogue showed that the CI is significantly lower in energy driving the photoisomerisation and the 1a analogue shows no PF intermediate hence explaining the dramatically increased quantum yield compared to 1.13 Furthermore, active back conversion catalysts are so far: Wilkinson's catalyst,12 Ag salts,17 and Au(111) surfaces.18 Nonetheless, the published azaborinine derivatives so far show only minor spectral overlap with the solar spectrum.
 | ||
| Fig. 1 Photoisomerisation and back conversion of 1,2-dihydro-1,2-azaborinine. | ||
Inspired by these previous works, we started to rationally design derivatives with lower first excitation energies employing a (time dependent) density functional theory (TD)DFT approach which has been compared to high-level ab initio methods (see ESI† for details). Substituents altering the electronic structure and especially substituents with +M (mesomeric) and −M effects, known as push and pull substituents, respectively, are considered effective for tuning the photophysics of chromophores.3,5 First, substitution patterns were motivated by inspecting the highest occupied and lowest unoccupied molecular orbital, HOMO and LUMO (see Fig. 2 and ref. 9 Fig. S1), respectively, because they often dictate nucleo- and electrophilicity and comprise 92% of the first excitation, which is a π–π* transition. The positions with the biggest lobes suggest the largest impact. Interestingly, the 3- and 6-position are ambiguous as they have large lobes in both MOs. On the contrary, the 5- and 4-position appear ideal since they possess lobes only in the HOMO or LUMO, respectively. To support our rational design pattern, an initial vertical excitation energy screening of substituted BNB compounds was performed at their equilibrium geometry. Each of the six distinct substitution sites was subsequently substituted with either a representative +M (methoxy) or −M (nitrile) substituent yielding twelve different structures. From each, we assessed the first excitation energy. The largest and second largest red-shift for CN was observed in positions 4 and 3, respectively, while it was position 5 and 3, respectively, for OMe (ESI†).
 | ||
| Fig. 2 Visualisation of the main orbital contributions of the first excited state of 1-BNB at its equilibrium geometry from TDDFT calculations. Depicted with an isovalue of 0.100. | ||
Subsequently, various 4,5-substituted derivatives were screened to investigate the impact of substitution. A total of 49 compounds, including 1, were systematically generated by combining two unique sets of substituents: [H, Me, OH, OMe, NH2, NMe2] at the 5-position and [F, Cl, CF3, CN, CHO, COOH, NO2, dicyanovinyl (DCV)] at the 4-position. The first set primarily features +I (inductive) or +M effects, whereas the second set predominantly exhibits −I and/or −M effects. This approach ensured a comprehensive exploration of electronic influences across the molecular framework. The 49 compounds were then classified based on the presence of push and/or pull substituents as no mesomeric effects (noM, 7 compounds), push-only (12), pull-only (10), and push–pull (20).
The first excitation energies of the benzene (BNB) and Dewar (BND) form of all compounds are visualised in Fig. 3A as well as kernel density estimations of each class (Fig. 3B and C) highlighting compounds 1 and 4-cyano-5-hydroxy-1,2-dihydro-1,2-azaborinine 2 (vide infra). In general, introducing push or pull substituents systematically red-shifts S1 (BNB). However, push-only substitution appears to impact the first excitation energies less than pull-only substitution. On the other hand, push-only systems ensure spectral separation better than pull systems. The pull-only systems separate into two groups: one ensuring spectral separation between 4.4 to 4.8 eV (COOH/CN combined with H/Me), while other pull-only substitutions cause a tremendous red-shift of S1 for BNB, which is even larger for BND diminishing spectral separation. This is a consequence of the nature of the respective strong pull substituents and the structure of the BNB and BND isomers. In the Dewar forms, the substituents have more space facilitating planarisation. For example for the 4-NO2-5-Me compound, the nitro group cannot planarise in the BNB form due to steric hindrance, while it becomes almost coplanar to the 4,5-CC bond in the BND form extending the π-system leading to a larger red-shift in the BND form. This also holds for the DCV group. Substitution with formyl results in the largest red-shift since formyl can planarise even in the BNB form. Furthermore, the push–pull subset, despite its broad distribution, is well separated from the noM subset, which is illustrated in Fig. 3B. This readily shows that any combination of the chosen push and pull substituents results in a significant red-shift of the first excited state compared to 1. In addition, this emphasises the superiority of mesomeric over inductive effects for tuning the first excitation energy. Moreover, the strengths of the push and pull subsets can be leveraged, yielding the most promising candidates with a significant red-shift of the excitation energy while maintaining – or even enhancing – the spectral separation of the BNB and BND isomers. The corresponding oscillator strengths can be found in the ESI.†
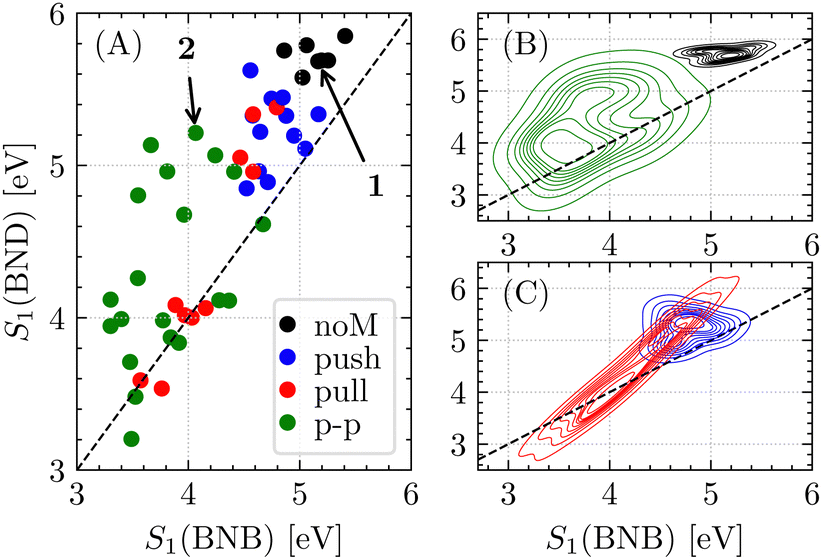 | ||
| Fig. 3 (A) First excitation energies of the benzene and Dewar form of the screened compounds (TDDFT). Compounds 1 and 2 are highlighted. (B) and (C) show kernel density estimate plots for the classified compounds: noM (black), push–pull (green), push-only (blue), and pull-only (red). | ||
Aside from tuning the photophysics, we also investigated how the storage energy ES varies throughout the set of compounds (see Fig. 4). Compounds of the noM class are all close to the value of unsubstituted 1 (ES = 59.6 kcal mol−1). The pull-only and push-only systems show in general a lowered storage energy, while most push-only systems are lower compared to the pull-only systems. The largest subset of push–pull systems, however, shows the biggest variance and the lowest ES values on average.
 | ||
| Fig. 4 Violin plots of ES of the screened compounds (DFT). The width is proportional to the number of compounds in each class. Boxplots are overlaid in the centre to highlight key summary statistics. | ||
From all screened compounds, 2, combining a cyano with a hydroxy group, was chosen for further investigations due to its favourable properties summarised in Table 1 and because it is structurally the simplest and lightest push–pull system. Compared to 1, 2 shows a significant red-shift of 1.10 eV, and a slightly decreased oscillator strength and storage energy, but a largely improved spectral separation. A thorough investigation of the S1 state of 2-BNB showed that the substitution pattern did not change its character, which is still a π–π* transition with a contribution of 98% of the HOMO–LUMO transition (Fig. 5, left). Indeed, the orbitals as well as the S1 state changed as expected and intended. The lobes on the BNB core retain their shape while the HOMO and LUMO are extended by π lobes originating from atomic π and π* orbitals of the hydroxy (push) and cyano (pull) substituents, respectively. The HOMO and LUMO are then delocalised over the whole chromophore. In contrast, since the BND form is not planar, the π system cannot delocalise as strongly and hence the BND form generally absorbs at shorter wavelengths, which is very pronounced in 2 (Fig. 5, right). The significant red-shift is apparent in Fig. 6, comparing the simulated UV/Vis spectra of compounds 1 and 2 in their BNB form as well as illustrating the spectral separation of 2-BNB and 2-BND. Obviously, not only the first excited state is red-shifted. Five states of 2-BNB lie energetically below 6.5 eV (190 nm) while only two states are present for 1-BNB. With compound 2, we could demonstrate how the absorption of monocyclic 1,2-azaborinines can be shifted to wavelengths >300 nm. Given that the employed method commonly overestimates vertical excitation energies and due to the absence of solvation effects (simulated solvent effect in ESI,† Fig. S3), the experimental absorption band can be expected at even longer wavelengths resulting in a significantly improved spectral overlap with the solar spectrum making 2 a very promising candidate for MOST applications.
| Property | 1 | 2 |
|---|---|---|
| S 1(BNB)/eV | 5.17 (240 nm) | 4.07 (305 nm) |
| f osc,S1(BNB) | 0.17 | 0.13 |
| Spectral separation ΔS1/eV | 0.52 | 1.15 |
| E S/kcal mol−1 | 59.6 | 52.2 |
 | ||
| Fig. 5 Visualisation of the main orbital contributions of the first excited state of 2-BNB (left) and 2-BND (right) at the equilibrium geometry from TDDFT calculations. Depicted with an isovalue of 0.100. | ||
 | ||
| Fig. 6 Simulated TDDFT UV/Vis spectra of (A) 1-BNB and 2-BNB and (B) of 2-BNB and 2-BND (full width at half maximum = 0.1 eV). | ||
For the thermal back reaction as well as the photoreaction after relaxation along the conical intersection, it is important to investigate how the ground state potential energy surface is altered upon substitution. Inspired by the reaction pathway of 1,9,10,16 we could construct a plausible analogous pathway for 2 with a PF intermediate. The 2-PF structure is directly connected to 2-BNB and 2-BNDvia2-TS1 and 2-TS2, respectively (ESI,† Fig. S1). The pathways of the two compounds are shown in Fig. 7. For the thermal back reaction, the first barrier of 2 equals ΔE2-TS2,2-BND = 20.3 kcal mol−l and is 1.7 kcal mol−1 higher than the equivalent for 1 directly extending the lifetime of the storage state but slightly diminishing the effective amount of stored energy. The subsequent barrier equals ΔE2-TS1-PF = 9.1 kcal mol−l. Thus, the rate-determining step is the conversion of BND to PF for both compounds. However, in practice the energy release should occur on demand using external stimuli following a different coordinate with ideally lower barriers to ensure controllability and to maximise the released energy. Hence, the longer natural half-life is generally favourable for MOST applications. Moreover, if we assume analogous photoconversion pathways, the higher barrier ΔE2-TS1,2-PF of 2 is very advantageous: Upon formation of intermediate 2-PF after irradiation, formation of 2-BND is kinetically clearly more favourable than reforming 2-BNB which should significantly improve the quantum yield. A similar improvement was observed for 1-silyl-2-chloro-azaborinine.13
 | ||
| Fig. 7 Thermal back reaction pathway for compounds 1 and 2 and the equilibrium geometry of 2-PF (DFT). | ||
To further deepen our understanding of the impact of this substitution pattern on the switching behaviour, we used a spin–flip TDDFT approach to find the minimum energy crossing point of the S1/S0 conical intersection (CI), which is the minimum in the multidimensional seam space, offering a simple picture of the photoreaction. A spin–flip approach is required to obtain correct CI topologies. The distance between the carbons in the 3- and 6-positions serves as the most intuitive parameter to assess structural proximity. Although it provides a robust approximation of the reaction coordinate, it alone does not fully encompass the complexity of the entire process. The distances along the proposed pathway of 2 are given in Table 2, showing that the optimised CI is structurally close to 2-TS1 and similar to previously found CIs (≈2.56 Å, see ref. 13 Fig. S2) strongly indicating switchability. Importantly, the CI was found to be only 1 meV higher in energy than the Franck–Condon point of 2-BNB making it energetically easily accessible, especially at room temperature.
This study focused on the rational computational design of azaborinine derivatives to improve their suitability as MOST systems, primarily by improving spectral overlap with the solar spectrum. It revealed that push–pull systems featuring an acceptor in 4- and a donor in the 5-position provide great tunability and a significant red-shift in the first excited state. However, storage energies slightly decrease for most 4,5-substituted compounds. In particular, 4-cyano-5-hydroxy-1,2-dihydro-1,2-azaborinine 2 exhibits favourable properties as a MOST compound. Comparison to unsubstituted 1 showed analogue S1 states as well as thermal back reaction pathways with a PF intermediate. At the cost of a slightly decreased storage energy, the storage state has a longer thermal lifetime and the pathway indicates higher quantum yields. In conclusion, 2 is a highly promising candidate for MOST applications with significantly improved spectral properties and we hope to inspire synthetic chemists and spectroscopists to challenge our computational prediction.
| Structure | BNB | CI | TS1 | PF | TS2 | BND |
|---|---|---|---|---|---|---|
| CC distance/Å | 2.82 | 2.59 | 2.56 | 2.35 | 2.12 | 1.56 |
The authors acknowledge support by the state of Baden-Wuerttemberg through bwHPC and the Landesgraduiertenfurderung (LGF), the German Research Foundation (DFG) through grant no. INST 40/575-1 FUGG (JUSTUS 2 cluster) and FOR 5499, and the Dr. K.-H. Eberle-Stiftung. We thank Manuel Eder for preliminary computations.
Data availability
The data supporting this article are included in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Giménez-Gómez, L. Magson, C. Merino-Robledillo, S. Hernáez-Troya, N. Sanosa, D. Sampedro and I. Funes-Ardoiz, React. Chem. Eng., 2024, 9, 1629–1640 RSC.
- Z.-i Yoshida, J. Photochem., 1985, 29, 27–40 CrossRef CAS.
- J. Orrego-Hernández, A. Dreos and K. Moth-Poulsen, Acc. Chem. Res., 2020, 53, 1478–1487 CrossRef PubMed.
- Z. Wang, P. Erhart, T. Li, Z.-Y. Zhang, D. Sampedro, Z. Hu, H. A. Wegner, O. Brummel, J. Libuda, M. B. Nielsen and K. Moth-Poulsen, Joule, 2021, 5, 3116–3136 CrossRef CAS.
- M. Quant, A. Lennartson, A. Dreos, M. Kuisma, P. Erhart, K. Börjesson and K. Moth-Poulsen, Chem. – Eur. J., 2016, 22, 13265–13274 CrossRef CAS PubMed.
- A. J. V. Marwitz, M. H. Matus, L. N. Zakharov, D. A. Dixon and S.-Y. Liu, Angew. Chem., Int. Ed., 2009, 48, 973–977 CrossRef CAS PubMed.
- H. F. Bettinger and O. Hauler, Beilstein J. Org. Chem., 2013, 9, 761–766 CrossRef CAS PubMed.
- S. A. Brough, A. N. Lamm, S.-Y. Liu and H. F. Bettinger, Angew. Chem., Int. Ed., 2012, 51, 10880–10883 CrossRef CAS PubMed.
- S. Jeong, E. Park, J. Kim and K. H. Kim, Phys. Chem. Chem. Phys., 2023, 25, 17230–17237 RSC.
- J. Kim, J. Moon and J. S. Lim, Chem. Phys. Chem., 2015, 16, 1670–1675 CrossRef CAS PubMed.
- K. Edel, R. F. Fink and H. F. Bettinger, J. Comput. Chem., 2016, 37, 110–116 CrossRef CAS PubMed.
- K. Edel, X. Yang, J. S. A. Ishibashi, A. N. Lamm, C. Maichle-Mössmer, Z. X. Giustra, S.-Y. Liu and H. F. Bettinger, Angew. Chem., Int. Ed., 2018, 57, 5296–5300 CrossRef CAS PubMed.
- E. M. Arpa, S. Stafström and B. Durbeej, Phys. Chem. Chem. Phys., 2024, 26, 11295–11305 RSC.
- T. Ozaki, S. K. Bentley, N. Rybansky, B. Li and S.-Y. Liu, J. Am. Chem. Soc., 2024, 146, 24748–24753 CrossRef CAS PubMed.
- S. Y. Toh, P. Djuricanin, T. Momose and J. Miyazaki, J. Phys. Chem. A, 2015, 119, 2683–2691 CrossRef CAS PubMed.
- M.-D. Su, Chem. – Eur. J., 2013, 19, 9663–9667 Search PubMed.
- R. C. Richter, S. M. Biebl, R. Einholz, J. Walz, C. Maichle-Mössmer, M. Ströbele, H. F. Bettinger and I. Fleischer, Angew. Chem., Int. Ed., 2024, 63, e202405818 CrossRef CAS PubMed.
- Z. Hussain, R. Einholz, S. M. Biebl, E. Franz, A. Müller, A. Dreuw, H. F. Bettinger, O. Brummel and J. Libuda, Top. Catal., 2025 DOI:10.1007/s11244-025-02089-w.
Footnote |
| † Electronic supplementary information (ESI) available: Computational details and results. See DOI: https://doi.org/10.1039/d5cc01963j |
| This journal is © The Royal Society of Chemistry 2025 |