
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimerization-enhanced aggregation of chlorophyll into helical supramolecular polymers†
Ryo
Kudo‡
a,
Ryuichi
Kawai‡
a,
Sho
Takamiya‡
a,
Sougata
Datta
 b,
Hiroki
Hanayama
c,
Nobuyuki
Hara
de,
Hitoshi
Tamiaki
d and
Shiki
Yagai
*bc
b,
Hiroki
Hanayama
c,
Nobuyuki
Hara
de,
Hitoshi
Tamiaki
d and
Shiki
Yagai
*bc
aDivision of Advanced Science and Engineering, Graduate School of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
bInstitute for Advanced Academic Research (IAAR), Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan. E-mail: yagai@faculty.chiba-u.jp
cDepartment of Applied Chemistry and Biotechnology, Graduate School of Engineering, Chiba University, 1-33 Yayoi-cho, Inage-ku, Chiba 263-8522, Japan
dGraduate School of Life Sciences, Ritsumeikan University, Kusatsu, Shiga 525-8577, Japan
eDepartment of Chemistry, College of Humanities & Sciences, Nihon University, Setagaya-ku, Tokyo 156-8550, Japan
First published on 8th May 2025
Abstract
We designed a chlorophyll dyad that folds like a closed pair of scissors. Despite possessing identical interaction sites to its monad counterpart, the folded dyad exhibited markedly enhanced aggregation behavior, forming micrometer-scale helical nanofibers. We propose that this enhanced aggregation propensity upon dimerization arises from conformational fixation through folding.
The extraordinary self-organization properties of proteins originate from their intricately folded tertiary structures. Through precise intrachain interactions, one-dimensional polypeptide chains undergo a remarkable transformation into supramolecular monomeric units whose shapes, polarities, and interaction sites are exquisitely and three-dimensionally organized.1 This mechanism of (macro)molecular preorganization prior to self-assembly, elegantly exemplified by biomacromolecules, offers profound inspiration for the rational design of synthetic systems aimed at constructing tailored nano- to mesoscale architectures.2–5
In pursuit of this principle, we have investigated “scissor-shaped” homo-dyads of functional molecules that undergo folding before self-assembly.6 These dyads, consisting of π-conjugated molecules linked by xylene spacers bearing long alkyl chains, fold via intramolecular hydrogen bonding between amide groups and π–π stacking interactions. This folding confers two distinctive features. First, the resulting wedge-shaped geometry facilitates self-assembly into specific nanostructures not only helically extended nanofibers7,8 but also nanotoroids9 and nanotubes.10–12 Second, the folded dyads exhibit supramolecular chirality; specifically, introducing chiral side chains induces either left- or right-handed scissor-shaped conformations, and this conformational chirality is transferred to chiral toroidal and tubular nanostructures13 and one-dimensional supramolecular polymers.14
In this study, we investigated a previously unexplored aspect: the intrinsic self-assembly propensity of the folded dyads. If these scissor-shaped dyads were to assemble in an open conformation,15,16 their four π-conjugated surfaces would theoretically result in a higher aggregation propensity compared to the corresponding monad reference. However, as they actually adopt a folded conformation, only two π-conjugated surfaces remain accessible for intermolecular association, a situation equivalent to that of the monad reference. Additionally, intramolecular hydrogen bonding effectively sequesters one pair of N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O sites, thereby restricting intermolecular interactions to a single N–H and CO pair—once again paralleling the monad reference. Despite these structural similarities, only the dyad exhibits markedly enhanced self-assembly behavior.13
O sites, thereby restricting intermolecular interactions to a single N–H and CO pair—once again paralleling the monad reference. Despite these structural similarities, only the dyad exhibits markedly enhanced self-assembly behavior.13
To elucidate the role of folding in self-assembly, we synthesized a chlorophyll-based scissor-shaped dyad, Chld and compared its self-assembly behavior with its monad counterpart, Chlm (Fig. 1). Chlorophyll derivatives exhibit rich changes in their electronic absorption spectra upon π-stacking, an effect particularly pronounced in their Qy absorption bands.17 Furthermore, owing to the intrinsic asymmetric centers at the 17 and 18 positions, the chlorin scaffold induces strong supramolecular chirality, enabling investigation of their aggregation by circular dichroism (CD) spectroscopy.18 Our results reveal that while Chlm fails to form any well-defined aggregates, Chld assembles into extended, left-handed helical fibers (Fig. 1a). Remarkably, the superior self-assembling propensity of the dyads persists even when the chlorin core is replaced with a porphyrin scaffold, underscoring the generality of folding-induced supramolecular polymerization.
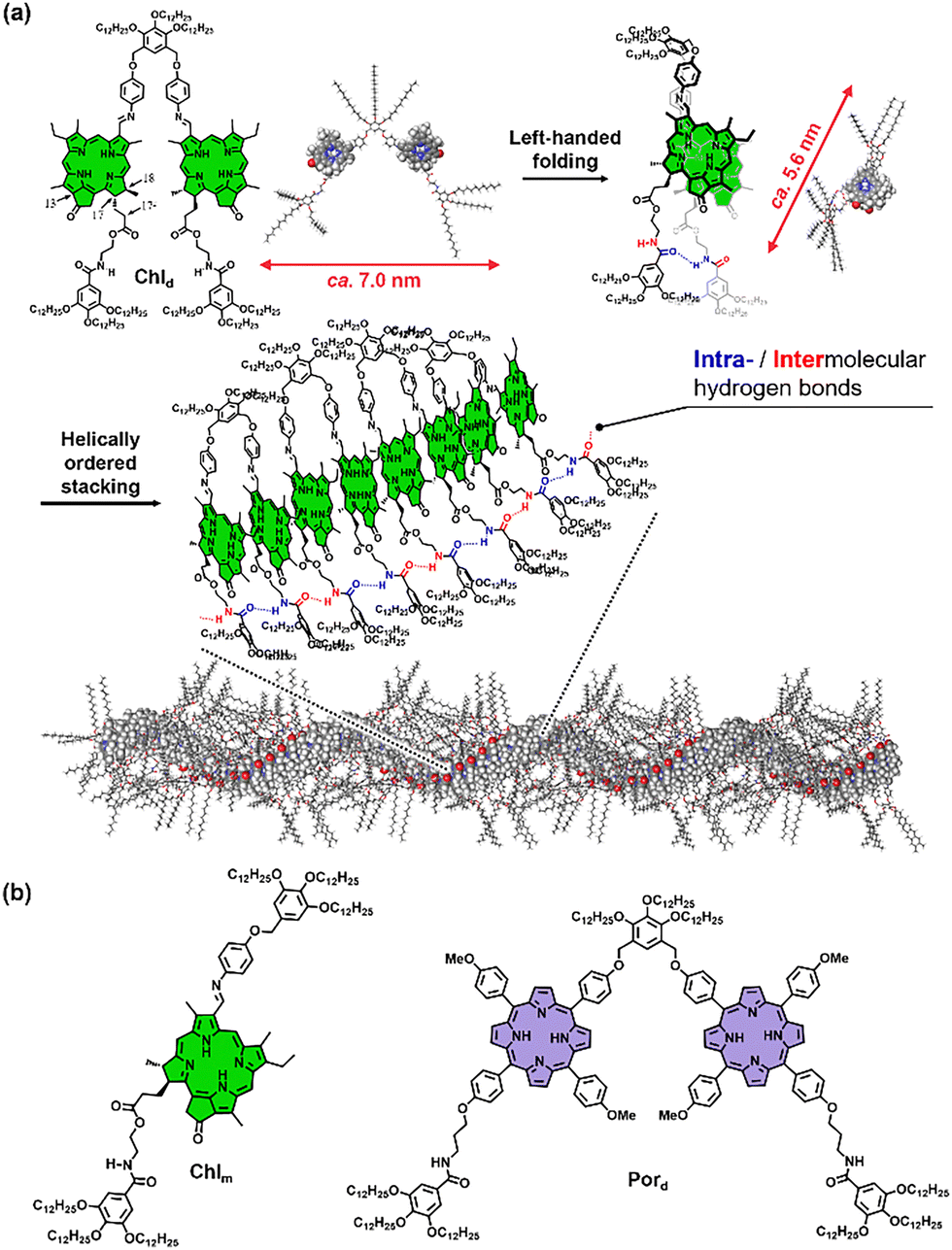 | ||
| Fig. 1 (a) Molecular structure of Chld and schematic representation of its self-assembly process to form helical nanofibers. (b) Molecular structures of reference compounds Chlm and Pord. | ||
Chld and Chlm were synthesized and characterized by 1H and 13C NMR spectroscopy as well as mass spectrometry (Scheme S1, ESI†). The self-assembly behavior of these molecules in methylcyclohexane (MCH) was investigated using variable-temperature UV/Vis absorption and CD spectroscopy. At 100 °C, both Chld and Chlm (c = 100 μM) exhibited almost identical absorption spectra, showing Soret and Qy bands at 414 and 691 nm, respectively (Fig. 2a and b, red lines). This observation indicates the absence of intramolecular electronic interaction between the two chlorin chromophores of Chld in the monomeric state. Upon cooling the solutions to 20 °C at a rate of 1 °C min−1, the Qy band of Chld exhibited a bathochromic shift and split into two distinct absorption bands (J-bands) with maxima at 708 and 728 nm (Fig. 2a, blue line). Such spectral changes are indicative of the formation of two distinct J-type (slipped) stacking modes between the chlorin chromophores. In contrast, no such spectral changes were observed for Chlm under identical conditions (Fig. 2b).
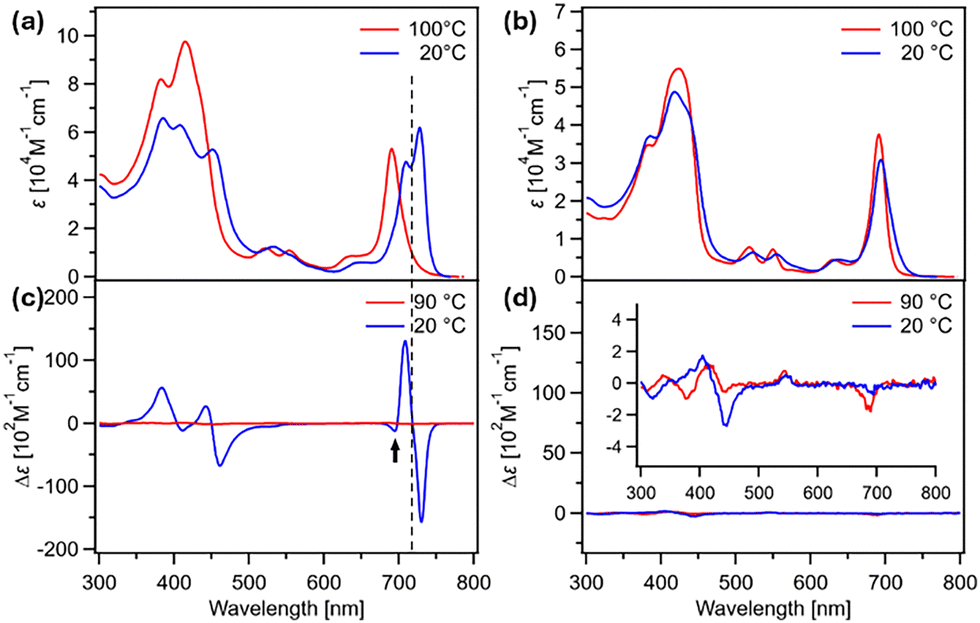 | ||
| Fig. 2 (a) and (b) Changes in the UV/Vis absorption and (c) and (d) CD spectra of (a) and (c) Chld (100 μM) and (b) and (d) Chlm (300 μM) upon cooling from 100 to 20 °C at a rate of 1 °C min−1 in MCH. | ||
The temperature-dependent UV/Vis absorption spectra of Chld (c = 100 μM) were analyzed by plotting changes in the absorbance at the J-band maxima of 708 and 728 nm as a function of temperature (Fig. S1, ESI†). Identical temperature responses were observed at both wavelengths, yielding non-sigmoidal curves. This result indicates that the two J-bands originate from the same aggregate species. Such temperature-dependent aggregation behavior is characteristic of a nucleation–elongation (cooperative) supramolecular polymerization mechanism. During the cooling process, the elongation temperature (Te) was observed at approximately 57 °C (Fig. S1b and c; blue lines, ESI†). In contrast, the subsequent heating measurement exhibited a similar non-sigmoidal curve but with a substantially elevated Te at 82 °C (Fig. S1b and c; red lines, ESI†). Such pronounced thermal hysteresis indicates that the aggregation of Chld during the cooling process is under kinetically controlled. Therefore, we analyzed the heating curves recorded at various concentrations using the nucleation–elongation model19 and obtained thermodynamic parameters from a van’t Hoff analysis. The standard enthalpy (ΔH°) and entropy (ΔS°) for Chld were determined to be −194 kJ mol−1 and −472 J mol−1 K−1, respectively (Fig. S2, ESI†). The obtained ΔS° value is significantly larger compared to those reported for analogous compounds.19–22 This result strongly suggests that the self-assembly process of Chld involves an intramolecular folding process. In stark contrast, the supramolecular polymerization of Chlm, unable to involve a folding process, did not exhibit the features of a cooperative mechanism, as indicated by the heating and cooling curves (Fig. S3, ESI†).
In the CD measurements, both compounds were nearly CD-silent at 90 °C, exhibiting significantly limited Cotton effects. Upon cooling to 20 °C, the Chld solution displayed a bisignate CD couplet with a zero-crossing point at 719 nm, featuring a negative peak on the longer wavelength side and a positive peak on the shorter wavelength side (negative/positive Cotton effect, Fig. 2c). The zero-crossing point does not coincide with either of the Qy absorption maxima. Given that a CD signal becomes weakly negative around 690 nm (shown by the arrow), the observed CD spectrum is likely a superposition of two excitonic couplings:23,24 strong negative/positive Cotton effects arising from the long-wavelength J-band, where excitonic coupling is stronger, and weaker positive/negative Cotton effects from the short-wavelength J-band, where excitonic coupling is comparatively weaker. According to the exciton chirality rule,25,26 the dominant (stronger) Cotton effect suggests the predominant involvement of a left-handed helical stacking arrangement of the chlorin units. In contrast, Chlm remained nearly CD-silent even at 20 °C, consistent with the UV/Vis absorption result (Fig. 2d).
Atomic force microscopy (AFM) provided unambiguous evidence for the distinct self-assembly properties of Chld and Chlm. Solutions of each compound in MCH were first cooled to induce self-assembly, subsequently spin-coated onto highly oriented pyrolytic graphite (HOPG) substrates, and then imaged by AFM. For Chld, a well-developed network of fibrous aggregates was observed (Fig. 3a and Fig. S4, S5, ESI†). High-resolution imaging revealed that the fibers were bundled together during self-assembly (Fig. 3b), and the thinnest fibers within these bundles had widths of approximately 5.2 nm (Fig. 3d). This value is significantly smaller than the estimated length (ca. 7 nm) of Chld in an extended conformation but closely matches its length in a folded conformation with ca. 5.6 nm (Fig. 1a, upper), suggesting that the molecules adopt the folded conformation within the fibers. Furthermore, periodic grooves inclined at an angle of 36° relative to the short axis of the fibers were observed along the long axis of the fibers (Fig. S6a, ESI†). The thickness (height) measured at the groove positions was approximately 3.0 nm, while that at the inter-groove regions was measured to be around 3.5 nm (Fig. S6b, ESI†). These features strongly support a model in which Chld assembles into helically stacked fibers (Fig. 1a, lower). The helices exhibited an exclusively left-handed twist, with a helical pitch of approximately 5.1 nm, as determined by height profiling along the fiber long axis (Fig. 3e). This handedness is consistent with the supramolecular chirality inferred from CD spectroscopy. In sharp contrast, Chlm exhibited only ill-defined aggregates even at concentrations as high as 300 μM, lacking any discernible fibrous morphology (Fig. 3c and Fig. S4c, d, ESI†). These results suggest that dimerization of chlorophyll molecules plays a pivotal role in significantly enhancing their propensity for supramolecular polymerization.
 | ||
| Fig. 3 (a)–(c) AFM images of aggregates spin-coated immediately after cooling MCH solutions of (a) and (b) Chld (100 μM) and (c) Chlm (300 μM) from 100 to 20 °C at a rate of 1 °C min−1. Inset in (b) shows the magnified view of the area surrounded by a dashed square in (b); the white arrows in the inset of (b) show the left-handed helicity of a nanofiber. (d) and (e) AFM height analysis of Chld nanofibers along the (d) red and (e) yellow arrows in (b). | ||
To investigate the role of the amide group in the self-assembly of Chld, FT-IR spectroscopy was performed on its fibrous aggregates in MCH and compared with the spectrum of its monomeric state in THF (Fig. 4).27 The spectrum of the MCH solution showed significant shifts in the N–H and CO stretching vibrations of the amide groups compared to the THF solution, namely from 3575/3505 to 3300 cm−1, and from 1655 to 1633 cm−1, respectively (Fig. 4, upper). In contrast, the two CO stretching vibration bands of the 172-ester and 13-keto groups showed only small shifts (1738/1702 → 1740/1693 cm−1), suggesting that these groups are not significantly involved in intermolecular interactions. In contrast, Chlm exhibited considerably smaller shifts (3575/3505 → ≈3350 cm−1) in the amide N–H stretching vibrations. The amide CO stretching vibration displayed a shift to higher wavenumber (1665 → 1677 cm−1), which can be attributed to solvent effects (Fig. 4, lower). These findings indicate a weaker involvement of the amide group of Chlm in directional hydrogen bonding. Furthermore, considerable broadening was observed for both the ester- and keto-derived CO stretching bands of Chlm, implying the presence of multiple, less specific hydrogen-bonding interactions. These results suggest that while Chld forms highly ordered aggregates through well-defined hydrogen bonds between amide groups, Chlm tends to form less ordered assemblies stabilized by a broader range of hydrogen bonding motifs.
 | ||
| Fig. 4 FT-IR spectra of Chld (c = 200 μM, upper) and Chlm (c = 1.2 mM, lower) in THF (dashed lines) and MCH (solid lines). | ||
The findings described above clearly demonstrate that the dimerization of chlorophyll molecules significantly enhances the propensity for supramolecular polymerization. Our combined spectroscopic and AFM analyses strongly suggest that Chld initially undergoes left-handed intramolecular folding prior to aggregation. This folding event is likely crucial in facilitating both intra- and intermolecular chiral J-type stacking interactions among chlorin chromophores. Moreover, FT-IR spectral analysis indicates that the two amide groups in Chld participate in hydrogen-bonding interactions, likely involving distinct intra- and intermolecular interactions (Fig. 1a).
However, the above finding poses a fundamental question: if folding indeed occurs within Chld, the π-surfaces and amide hydrogen bonding sites available for one-dimensional stacking should theoretically be equivalent to those in the Chlm. Thus, why is it that only Chld successfully aggregates into one-dimensional supramolecular polymers? We propose that this remarkable difference arises specifically due to the intramolecular folding effect. Folding effectively rigidifies the molecular conformation, particularly stabilizing the otherwise flexible side chains bearing amide groups. This structural preorganization significantly promotes efficient intermolecular associations. Although the initial entropy loss associated with supramolecular polymerization via intramolecular folding is considerable, the subsequent nucleation and elongation steps become kinetically more favorable for Chld relative to Chlm, due to the preorganized, folded conformation of the former.
To further validate whether this dimerization approach can be generalized to structurally analogous porphyrin derivatives, we synthesized Pord and Porm (Fig. 1b and Scheme S2, ESI†) and compared their self-assembly behaviors. Temperature-dependent absorption spectroscopy revealed a notable hypochromic shift of the Soret band upon cooling, exclusively for Pord, strongly indicative of aggregate formation (Fig. S7, ESI†). Consistent with these spectroscopic observations, AFM imaging showed that while Porm produced only amorphous aggregates (Fig. S8, ESI†), Pord formed well-defined cylindrical fibers (Fig. S9, ESI†). Although the pronounced aggregation propensity of Pord fibers resulted in limited dispersibility, notably, AFM observations revealed a gentle twisting of the fiber but did not reveal any distinct helical morphology. This finding suggests that the inherent chiral centers at the 17 and 18 positions in the chlorin ring are essential for the emergence of uniform helical morphology in Chld fibers.
In conclusion, this study demonstrates that dimerization of chlorophyll and porphyrin molecules designed to adopt folded conformations effectively promotes their self-assembly behavior. Particularly in the case of chlorophyll derivatives, molecular chirality originating from the intrinsic asymmetry of the chlorin framework is transmitted to the supramolecular level, thereby enabling the formation of helically biased supramolecular polymers. We are currently extending this strategy to the zinc complex of these molecules, and preliminary results indicate that introducing additional interaction sites leads to even more complex self-assembly behavior. These findings will be reported in due course.
This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI grant no. 22H02203, 22H00331, 23K23470 and 23H04873 in a Grant-in-Aid for Transformative Research Areas “Materials Science of Meso-Hierarchy.” This work was the result of using research equipment shared in MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting construction of core facilities) Grant Number JPMXS0440200025. The authors thank the Materials Analysis Division, Institute of Science Tokyo for MALDI-TOF-MS analysis.
Data availability
The data that support the findings of this work have been included in the main text and ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- B. J. G. E. Pieters, M. B. Van Eldijk, R. J. M. Nolte and J. Mecinović, Chem. Soc. Rev., 2016, 45, 24 RSC.
- S. De, B. Chi, T. Granier, T. Qi, V. Maurizot and I. Huc, Nat. Chem., 2018, 10, 51 CrossRef CAS PubMed.
- Y. Vyborna, S. Altunbas, M. Vybornyi and R. Häner, Chem. Commun., 2017, 53, 12128 RSC.
- X. Hu, J. O. Lindner and F. Würthner, J. Am. Chem. Soc., 2020, 142, 3321 CrossRef CAS PubMed.
- K. Jalani, A. D. Das, R. Sasmal, S. S. Agasti and S. J. George, Nat. Commun., 2020, 11, 3967 CrossRef CAS PubMed.
- K. Tashiro, T. Saito, H. Arima, N. Suda, B. Vedhanarayanan and S. Yagai, Chem. Rec., 2022, 22, e202100252 CrossRef CAS PubMed.
- M. Yamauchi, T. Ohba, T. Karatsu and S. Yagai, Nat. Commun., 2015, 6, 8936 CrossRef CAS PubMed.
- N. Suda, H. Arima, T. Saito, T. Aizawa and S. Yagai, Chem. Lett., 2022, 51, 700 CrossRef CAS.
- J. S. Valera, H. Arima, C. Naranjo, T. Saito, N. Suda, R. Gómez, S. Yagai and L. Sánchez, Angew. Chem., Int. Ed., 2022, 134, e202114290 CrossRef.
- S. Yagai, M. Yamauchi, A. Kobayashi, T. Karatsu, A. Kitamura, T. Ohba and Y. Kikkawa, J. Am. Chem. Soc., 2012, 134, 18205 CrossRef CAS PubMed.
- T. Saito and S. Yagai, Org. Biomol. Chem., 2020, 18, 3996 RSC.
- T. Saito and S. Yagai, Eur. J. Org. Chem., 2020, 2475 CrossRef CAS.
- T. Saito, T. Kajitani and S. Yagai, J. Am. Chem. Soc., 2023, 145, 443 CrossRef CAS PubMed.
- T. Saito, D. Inoue, Y. Kitamoto, H. Hanayama, T. Fujita, Y. Watanabe, M. Suda, T. Hirose, T. Kajitani and S. Yagai, Nat. Nanotechnol., 2025 DOI:10.1038/s41565-025-01882-8.
- S. Matsubara and H. Tamiaki, Chem. Lett., 2021, 50, 99 Search PubMed.
- T. Ishii, S. Matsubara and H. Tamiaki, Chem. Commun., 2023, 59, 1967 RSC.
- S. Matsubara, S. Shoji and H. Tamiaki, Chem. Commun., 2024, 60, 12513 RSC.
- N. Hara and H. Tamiaki, Bull. Chem. Soc. Jpn., 2024, 97, uoae032 CrossRef CAS.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, T. F. A. De Greef, P. Van Der Schoot, A. P. H. J. Schenning and E. W. Meijer, Chem. – Eur. J., 2010, 16, 362 CrossRef CAS PubMed.
- S. Ogi, K. Sugiyasu, S. Manna, S. Samitsu and M. Takeuchi, Nat. Chem., 2014, 6, 188 CrossRef CAS PubMed.
- S. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300 CrossRef CAS PubMed.
- M. F. J. Mabesoone, A. J. Markvoort, M. Banno, T. Yamaguchi, F. Helmich, Y. Naito, E. Yashima, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2018, 140, 7810 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752 CrossRef CAS PubMed.
- N. Harada and K. Nakanishi, Acc. Chem. Res., 1972, 5, 257 CrossRef CAS.
- G. Pescitelli, Chirality, 2022, 34, 333 CrossRef CAS PubMed.
- S. Shoji, T. Ogawa, T. Hashishin and H. Tamiaki, ChemPhysChem, 2018, 19, 913 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General information, synthesis, structural characterization data, NMR spectra, photophysical results, and morphological images. See DOI: https://doi.org/10.1039/d5cc02060c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |