
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unprecedented bis(silylene)-supported silylone–metal complexes with Si0→CuI, Si0→NiI, and Si0→NiII dative bonds†
Changkai Shana,
Chenshu Daib,
Shenglai Yao *a,
Jun Zhuc and
Matthias Driess*a
*a,
Jun Zhuc and
Matthias Driess*a
aDepartment of chemistry, Metalorganics and Inorganic Meterials, Technische Universität Belrin, Strasse des 17. Juni 135, Sekr. C2, 10623 Berlin, Germany. E-mail: shenglai.yao@tu-berlin.de; matthias.driess@tu-berlin.de
bDepartment of Ecology, Lishui University, Lishui, 323000, P. R. China
cSchool of Science and Engineering, Chinese University of Hong Kong, Shenzhen, No. 2001 Longxiang Blvd., Longgang Dist., Shenzhen, Guangdong 518172, P. R. China
First published on 15th May 2025
Abstract
The first silylone-3d-metal complexes, LSiCu(NacNacM) (2) [L = 1,2-(RSi)2-1,2-C2B10H10, R = PhC(NtBu)2; NacNacM = HC(CMeNMes)2, Mes = 2,4,6-Me3-C6H2] and LSiNi(NacNacD) (3) [NacNacD = HC(CMeNDipp)2, Dipp = 2,6-iPr2-C6H3], are reported, resulting from the reaction of the strongly σ-donating and chelating bis(silylenyl)-ortho-carborane silylone LSi0 with [(NacNacMCu)2benzene] and [(NacNacDNi)2toluene], respectively. Density Functional Theory (DFT) analyses reveal that complex 2 features a Si0→CuI dative bond, while 3 exhibits a Si0→NiI bond. Oxidation of the Si0–NiI species 3 with [Cp2Fe]+ occurs at the Ni site to form the [3]+ cation with a Si0→NiII coordination.
Tetrylenes, the heavier homologues of carbenes, have emerged as promising alternative ligands to phosphines and carbenes for coordination to transition-metals and metal-mediated catalysis.1 Moving beyond tetrylene species, tetrylones,2 which feature two lone pairs at the central monatomic tetrel (group 14) element, remain a relatively nascent field.3,4 Among tetrylones, silylones, that is, zero-valent monoatomic silicon complexes, have attracted significant attention due to their unique electronic properties. The first molecular Si0 species, featuring a >Si
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SiSi< skeleton, was reported by Kira et al. in 2003 and initially described as a trisilaallene.5 However, density functional theory (DFT) calculations later suggested that this compound is better described as a silylone species supported by two silylene ligands.6 Over the past decade, silylone chemistry has rapidly expanded, with several isolable silylones stabilised by intricately designed, strong donor ligands-striking a delicate balance between electronic properties and steric protection.7
SiSi< skeleton, was reported by Kira et al. in 2003 and initially described as a trisilaallene.5 However, density functional theory (DFT) calculations later suggested that this compound is better described as a silylone species supported by two silylene ligands.6 Over the past decade, silylone chemistry has rapidly expanded, with several isolable silylones stabilised by intricately designed, strong donor ligands-striking a delicate balance between electronic properties and steric protection.7
Despite these advances, pioneering studies have highlighted the fascinating ability of silylones for small molecule activation. However, their potential for transition-metal coordination and catalytic applications remains largely unexplored.
The construction of complexes between low-valent p-block compounds and transition-metals has recently garnered increasing attention.8 With its symmetric LP(σ) and antisymmetric LP(π) electron orbitals at the Si centre, silylones represent a promising new class of ligands for the formation of electron-rich transition-metal complexes due to the strong σ-donating character of Si0.9 Recently, So and co-workers reported the coordination of a spiro-type disilylone to W(CO)5 via its σ-type lone pair,7i while Tan and co-workers described the coordination of a bis(mesoionic carbene)-supported silylone to two equivalents of CuCl.7h Moreover, Liang and Liu introduced a 1,4,2-diazasilole system incorporating both a mesoionic silylene and a silylone, which was capable of forming a silylone–diiron complex.10 After isolating the first cyclic NHC-supported silylone, we demonstrated its coordination with Lewis acidic metal salts such as GaCl3 and ZnCl2.2a To further explore silylone coordination chemistry and to gain insight into its bonding interactions with metal centres, we investigated the reactivity of the bis(silylenyl)-ortho-carborane silylone LSi: (1)7f [L = 1,2-(RSi)2-1,2-C2B10H10, R = PhC(NtBu)2] towards CuI, NiI and NiII complexes. Herein, we report the synthesis, characterization, and computational analysis of the first Si0–CuI, Si0–NiI and Si0–NiII complexes.
Treatment of silylone 1 with half an equivalent of [(NacNacMCu)2benzene]11 [NacNacM = HC(CMeNMes)2, Mes = 2,4,6-Me3C6H2] in Et2O at room temperature immediately yields a brownish-yellow solution (Scheme 1). Upon solvent removal, the desired complex LSiCu(NacNacM) (2) is obtained as a brownish-yellow powder in nearly quantitative yield. Notably, the 29Si{1H} NMR spectrum of 2 displays a single resonance at δ 44.2 ppm, attributed to the SiII atoms, which is slightly upfield-shifted compared to the free silylone 1 (δ 49.6 ppm).7f However, the signal corresponding to the Cu-bound central Si0 atom remains undetectable, even when employing an expanded spectrum width (1000 ppm) and prolonged acquisition time.
 | ||
| Scheme 1 Synthesis of the silylone–copper(I) complex 2. | ||
A single-crystal X-ray diffraction (scXRD) analysis reveals that 2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The molecular structure features a Si–Cu core, where both the Si (Σangles: 359.35°) and Cu (Σangles: 360.00°) atoms adopt a nearly trigonal-planar coordination geometry (Fig. 1 left). The six-membered C3N2Cu and five-membered C2Si3 rings are individually planar but not coplanar with the dihedral angle of 62.17°. The Si1–Cu1 distance of [2.2127(10)] Å is shorter than the SiII–CuI length observed in a three-coordinate silylene–CuI complex [2.289(4) Å],12 but longer than those in four-coordinate amidinato-silylene–CuI complexes (2.1716(12) to 2.244(8) Å).13
. The molecular structure features a Si–Cu core, where both the Si (Σangles: 359.35°) and Cu (Σangles: 360.00°) atoms adopt a nearly trigonal-planar coordination geometry (Fig. 1 left). The six-membered C3N2Cu and five-membered C2Si3 rings are individually planar but not coplanar with the dihedral angle of 62.17°. The Si1–Cu1 distance of [2.2127(10)] Å is shorter than the SiII–CuI length observed in a three-coordinate silylene–CuI complex [2.289(4) Å],12 but longer than those in four-coordinate amidinato-silylene–CuI complexes (2.1716(12) to 2.244(8) Å).13
 | ||
| Fig. 1 Molecular structures of 2 (left) and 3 (right). Thermal ellipsoids are drawn at 50% probability level. H atoms are omitted for clarity. | ||
Additionally, the Si1–Si2 [2.2060(11) Å] and Si1–Si3 [2.2093(12) Å] bond lengths in 2 are notably shorter than those in 1 [2.2272(6) and 2.2225(6) Å],7f indicating an enhanced donor ability of the silylene moiety toward the Cu-bound central Si0 atom. Complex 2 is highly sensitive to air and moisture but remains stable under an inert atmosphere in both solid and solution states. Upon exposing a pentane solution to CO, ligand exchange occurs, leading to the formation of [(NacNacM)Cu–CO] and liberation of 1 (see ESI†).
Encouraged by the successful isolation of 2, we sought to develop another silylone-transition-metal complex. As a precursor, we selected the NacNac-supported masked-NiI complex [(NacNacDNi)2toluene],14 where NacNacD = HC(CMeNDipp)2 (Dipp = 2,6-iPr2C6H3). Upon addition of toluene to a mixture of 1 and the Ni compound, a dark brown solution formed within minutes (Scheme 2). After workup, complex LSiNi(NacNacD) (3) was isolated as a dark brown powder in quantitative yield. Complex 3 is paramagnetic, and its magnetic susceptibility, determined by the Evans method,15 reveals μeff = 1.80, consistent with a d9-NiI centre. It exhibits good solubility in ethereal solvents and toluene. Single-crystals suitable for an XRD analysis were obtained by slow evaporation of a toluene solution at room temperature overnight. Complex 3 crystallizes in the monoclinic space group C2/c. Similar to 2, its molecular structure features a three-coordinate Si and Ni atom, respectively, both adopting trigonal-planar geometries, as indicated by the sum of bond angles around Si0 (360.00°) and Ni (360.01°) (Fig. 1 right). The five-membered C2Si3 and six-membered C3N2Ni rings are individually planar but not coplanar with a dihedral angle of 60.45°. The Si1–Ni1 bond length [2.2803(8) Å] is comparable to the calculated distance for a diphosphino silylone–Ni0 complex [2.261 Å],9b but is notably longer than those observed in selected SiII–Ni0 complexes [2.0751(5) to 2.2254(4) Å].16 The equal Si–Si bond distances [2.2309(7) Å] in 3 are slightly longer than those in 2 [2.2060(11) Å and 2.2093(12) Å].
 | ||
| Scheme 2 Syntheses of the complexes 3 and [3]+[BArF]−. | ||
Cyclic voltammetry (CV) was performed in an N2-saturated THF solution of 3 containing 0.1 M TBAPF6 (Fig. S6, ESI†). A reversible redox peak at approximately −0.5 V was observed, which can be attributed to the NiI/NiII redox couple.17 Inspired by the electrochemical redox properties, we explored the chemical redox behavior of 3. Attempts to reduce 3 using various reducing agents resulted in either no reaction or cleavage of the Si–Ni bond, leading to the formation of an SiI–SiI coupling product.7f However, oxidation of 3 with one equivalent of [Cp2Fe][BArF] {BArF = B[C6H3(CF3)2]4} in toluene produced an oily green mixture, from which only a small amount of crystalline [3][BArF] could be isolated (Scheme 2). A scXRD analysis of [3][BArF] was conducted, but the structural data lacked sufficient accuracy due to disorder in the anion, as compared to the DFT-optimized geometry (Table S10, ESI†). The optimized structure of [3]+ closely resembles that of its precursor 3, particularly in the overall geometry, yet exhibits a significantly shorter Si–Ni bond length [2.205 Å vs. 2.2803(8) Å in 3]. In contrast, the SiII–Si0 bond in [3]+ shows a slight elongation relative to 3 [2.288 Å vs. 2.2309(7) Å]. This geometric change is consistent with oxidation of the nickel centre from NiI to NiII, which enhances its interaction with the Si0 atom, drawing it closer. Unfortunately, the poor yield and challenging purification process prevented further characterization.
To gain deeper insight into the electronic structures of 2 and 3, DFT calculations were performed using the Gaussian 16 software package.18 Geometry optimizations were conducted at the TPSS-D3BJ/Def2-SVP∼ma-TZVP level of theory, chosen based on its optimal performance in reproducing the XRD structural data. A complete set of calculated geometries and energies are given in ESI.†
The nature of Cu–Si bond in 2: both principal interacting orbital (PIO) analysis19 with PIO-based bonding index (PBI) value (0.78) and natural adaptive orbitals (NAdO) analysis20 with eigenvalue (1.174) suggest a single Cu–Si bond in 2. The primary interaction between the Cu and Si atoms is σ-type donation from the lone pair of Si into the vacant s orbital of Cu (Fig. 2a–c). This interaction contributes 1.79e from Si and 0.21e from Cu, indicating a dative bonding nature. The first bonding principal interacting molecular orbital (PIMO) closely resembles the first NAdO, which has the highest eigenvalue 0.769 (Fig. S16, ESI†). Additionally, there is a π-backdonation from dxy (dyz) orbitals of Cu into π* orbitals of Si atoms. The contributions amount to 1.91e (1.96e) from Cu and 0.09e (0.04e) from Si (Fig. 2b and c). The slight decrease in dxy (dyz) electron population from 2.0e to approximately 1.9e reflects partial delocalization. In line with the PIO analysis, the second and third bonding NAdOs illustrate similar interactions between Cu dxy/dyz and Si π* orbitals, with eigenvalues of 0.216 and 0.153, respectively (Fig. S16, ESI†).
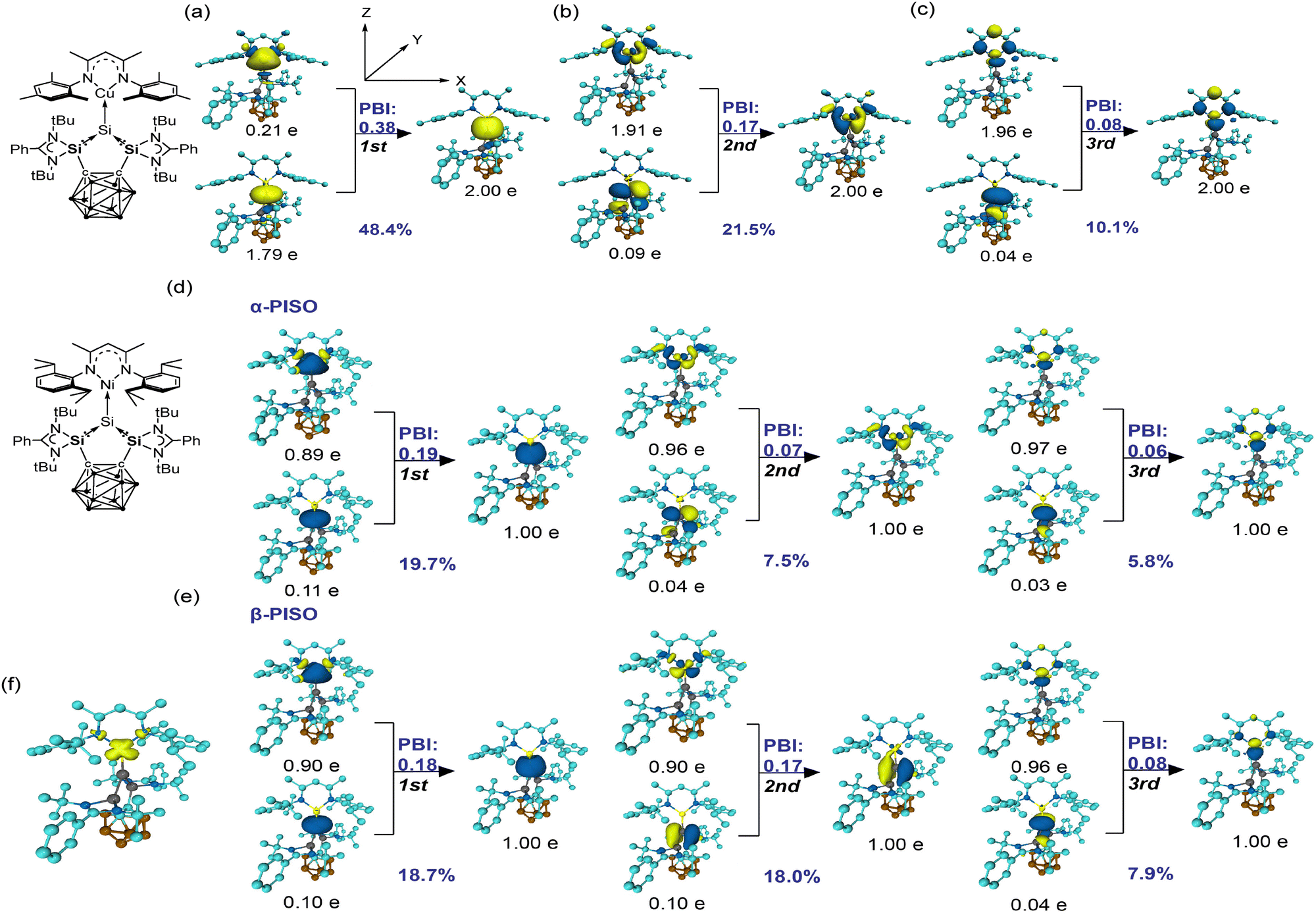 | ||
| Fig. 2 (a)–(c) PIO analysis on the bonding modes of Cu–Si in compound 2. (d) and (e) PISO analysis on the bonding modes of Ni–Si in compound 3: three dominating contributing PISO pairs of α system (d) and β system (e). In each case, the compound was split into two fragments along the Cu–Si or Ni–Si axis, with the orientation indicated by the respective Lewis structure on the left. The isosurface 0.030 a.u. is plotted for PIO and PISO, 0.005 a.u. for spin density. (f) The spin density plot for the molecule 3: the spin population in Ni atom (0.743) is labelled in yellow-green. Hydrogen atoms are omitted for clarity. | ||
The nature of Ni–Si bond in 3. Compound 3 has a doublet ground state (Table S10, ESI†), with an unpaired α electron primarily localised on the Ni centre (Fig. 2f). This electron resides in an orbital resembling dxy and has minimal effect on the Ni–Si bonding. Consequently, it is unsurprising that three similar dominant principal interacting spin orbital (PISO) pairs21 are observed for both α and β systems (Fig. 2d and e). Specifically, both systems exhibit one σ-type and two π-type interactions, with total PBI of 0.43 (α) and 0.54 (β). The close-to 1 sum of these PBIs (0.97), closely aligning with the combined NAdO eigenvalues for α and β (1.214, Fig. S17, ESI†), strongly supports the presence of a single Ni–Si bond. Among these interactions, the σ-donating interaction is the strongest, as indicated by both PISO and NAdO analyses, confirming a dative Si–Ni bonding character. Of the two π-type interactions, one shows nearly identical PBI (0.06 vs. 0.08) and NAdO eigenvalues (0.104 vs. 0.102, Fig. S17, ESI†) in both spin systems. However, the other π-type interaction differs between α and β due to the presence of the unpaired α electron in the Ni d orbital, which reduces its ability to accept electron donation. In contrast, the empty β dxy orbital of Ni can effectively accept π electron density from the Si atom, leading to a slightly higher NAdO eigenvalue (0.146 vs. 0.114, Fig. S17, ESI†).
In summary, we successfully synthesised the first bis(silylene) supported silylone-transition metal complexes, 2 and 3, featuring Si0→CuI and Si0→NiI coordination, respectively. These were obtained by reacting the bis(silylenyl)-ortho-carborane silylone 1 with [(NacNacMCu)2benzene] and [(NacNacDNi)2toluene]. Their structures were characterised with XRD and further analysed using DFT calculations. PISO and NAdO analyses revealed that the primary interaction between the silylone Si0 atom and the monovalent metal centres is σ-type donation from the lone pair of Si to the vacant orbital of the metal, confirming the dative bonding nature. Additionally, oxidation of 3 results in Ni oxidation, leading to the formation of a Si0→NiII complex.
Data availability
CCDC-2111076 (2), 2111077 (3), 2111078 ([3][BArF]) 2123117 ([(NacNacM)Cu–CO]) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk./data_request/cif. All other experimental and computational data associated with this work are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) S. Raoufmoghaddam, Y.-P. Zhou, Y. Wang and M. Driess, J. Organomet. Chem., 2017, 829, 2–10 CrossRef CAS; (b) Y.-P. Zhou and M. Driess, Angew. Chem., Int. Ed., 2019, 58, 3715–3728 CrossRef CAS PubMed.
- (a) S. Yao, Y. Xiong and M. Driess, Acc. Chem. Res., 2017, 50, 2026–2037 CrossRef CAS PubMed; (b) P. K. Majhi and T. Sasamori, Chem. – Eur. J., 2018, 24, 9441–9455 CrossRef CAS PubMed; (c) S. Yao, Y. Xiong, A. Saddington and M. Driess, Chem. Commun., 2021, 57, 10139–10153 RSC.
- F. Ramirez, N. B. Desai, B. Hansen and N. McKelvie, J. Am. Chem. Soc., 1961, 83, 3539–3540 CrossRef CAS.
- R. Tonner, F. Oxler, B. Neumuller, W. Petz and G. Frenking, Angew. Chem., Int. Ed., 2006, 45, 8038–8042 CrossRef CAS PubMed.
- S. Ishida, T. Iwamoto, C. Kabuto and M. Kira, Nature, 2003, 421, 725–727 CrossRef CAS PubMed.
- N. Takagi, T. Shimizu and G. Frenking, Chem. – Eur. J., 2009, 15, 3448–3456 CrossRef CAS PubMed.
- (a) K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, B. Niepotter, H. Wolf, R. Herbst-Irmer and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 2963–2967 CrossRef CAS PubMed; (b) Y. Xiong, S. Yao, S. Inoue, J. D. Epping and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 7147–7150 CrossRef CAS PubMed; (c) T. Sugahara, T. Sasamori and N. Tokitoh, Angew. Chem., Int. Ed., 2017, 56, 9920–9923 CrossRef CAS PubMed; (d) J. Keuter, A. Hepp, C. Muck-Lichtenfeld and F. Lips, Angew. Chem., Int. Ed., 2019, 58, 4395–4399 CrossRef CAS PubMed; (e) Y. Wang, M. Karni, S. Yao, A. Kaushansky, Y. Apeloig and M. Driess, J. Am. Chem. Soc., 2019, 141, 12916–12927 CrossRef CAS PubMed; (f) S. Yao, A. Kostenko, Y. Xiong, A. Ruzicka and M. Driess, J. Am. Chem. Soc., 2020, 142, 12608–12612 CrossRef CAS PubMed; (g) T. Koike, T. Nukazawa and T. Iwamoto, J. Am. Chem. Soc., 2021, 143, 14332–14341 CrossRef CAS PubMed; (h) M. Wu, Y. He, L. Zhang, R. Wei, D. Wang, J. Liu, L. Leo Liu and G. Tan, Eur. J. Inorg. Chem., 2022, e202200413 CrossRef CAS; (i) M. Y. Wee, S. Quek, C. S. Wu, M. D. Su and C. W. So, J. Am. Chem. Soc., 2024, 146, 14410–14415 CrossRef CAS PubMed.
- (a) P. W. Roesky, Dalton Trans., 2009, 1887–1893 RSC; (b) A. Doddi, M. Peters and M. Tamm, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS PubMed.
- (a) T. A. Nguyen and G. Frenking, Chem. – Eur. J., 2012, 18, 12733–12748 CrossRef CAS PubMed; (b) T. A. N. Nguyen, D. S. Tran, T. P. L. Huynh, T. H. Le, T. Q. Duong, T. T. Nguyen, T. C. Vo, V. T. Pham and T. H. Dang, Z. Anorg. Allg. Chem., 2017, 643, 826–838 CrossRef CAS; (c) H. T. P. Loan, H. V. Duc, D. T. Quang, P. V. Tat, D. T. Hiep and N. T. A. Nhung, Comput. Theor. Chem., 2018, 1131, 13–24 CrossRef CAS.
- X. Lan, H. Wang, Q. Liang and L. L. Liu, Angew. Chem., Int. Ed., 2025, 64, e202415246 CrossRef CAS PubMed.
- W. Xie, J. Heo, D. Kim and S. Chang, J. Am. Chem. Soc., 2020, 142, 7487–7496 CrossRef CAS PubMed.
- A. G. Avent, B. Gehrhus, P. B. Hitchcock, M. F. Lappert and H. Maciejewski, J. Organomet. Chem., 2003, 686, 321–331 CrossRef CAS.
- (a) G. Tan, B. Blom, D. Gallego and M. Driess, Organometallics, 2014, 33, 363–369 CrossRef CAS; (b) N. Parvin, J. Hossain, A. George, P. Parameswaran and S. Khan, Chem. Commun., 2020, 56, 273–276 RSC.
- G. Bai, P. Wei and D. W. Stephan, Organometallics, 2005, 24, 5901–5908 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- (a) G. Tavcar, S. S. Sen, R. Azhakar, A. Thorn and H. W. Roesky, Inorg. Chem., 2010, 49, 10199–10202 CrossRef CAS PubMed; (b) W. Wang, S. Inoue, S. Yao and M. Driess, J. Am. Chem. Soc., 2010, 132, 15890–15892 CrossRef CAS PubMed; (c) Y. Wang, A. Kostenko, S. Yao and M. Driess, J. Am. Chem. Soc., 2017, 139, 13499–13506 CrossRef CAS PubMed; (d) T. A. Schmedake, M. Haff, B. J. Paradise, D. Powell and R. West, Organometallics, 2000, 19, 3263–3265 CrossRef CAS; (e) T. J. Hadlington, T. Szilvasi and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 7470–7474 CrossRef CAS PubMed.
- S. Yao, Y. Xiong, M. Vogt, H. Grützmacher, C. Herwig, C. Limberg and M. Driess, Angew. Chem., Int. Ed., 2009, 48, 8107–8110 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- (a) J. X. Zhang, F. K. Sheong and Z. Lin, Chem. – Eur. J., 2018, 24, 9639–9650 CrossRef CAS PubMed; (b) J. X. Zhang, F. K. Sheong and Z. Lin, WIREs Comput. Mol. Sci., 2020, e1469 CrossRef CAS.
- J. L. Casals-Sainz, A. Fernandez-Alarcon, E. Francisco, A. Costales and A. Martin Pendas, J. Phys. Chem. A, 2020, 124, 339–352 CrossRef CAS PubMed.
- F. K. Sheong, J. X. Zhang and Z. Lin, Phys. Chem. Chem. Phys., 2020, 22, 10076–10086 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental, calculations, and characterizations as well as Tables S1, S12 and Fig. S1–S17. CCDC 2111076–2111078 and 2123117. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02098k |
| This journal is © The Royal Society of Chemistry 2025 |