
Synthesis of a twisted azananographene featuring a diquinoxaline-fused pyrene†
Zhiyu
Zhang
 a,
Zhenxun
Xu
a,
Aihui
Zhang
a,
Fenghua
Bai
*a,
Yoshifumi
Hashikawa
*b and
Chaolumen
*a
a,
Zhenxun
Xu
a,
Aihui
Zhang
a,
Fenghua
Bai
*a,
Yoshifumi
Hashikawa
*b and
Chaolumen
*a
aCollege of Chemistry and Chemical Engineering, Inner Mongolia University, Hohhot, 010021, China. E-mail: chaolumen@imu.edu.cn
bInstitute for Chemical Research, Kyoto University, Uji, Kyoto 611-0011, Japan. E-mail: hashi@scl.kyoto-u.ac.jp
First published on 24th June 2025
Abstract
π-Extension from a diquinoxaline-fused pyrene led to a twisted azananographene (azaNG) exhibiting red emission. Although the azaNG was highly contorted, the electronic nature of the diquinoxaline-fused pyrene was confirmed to be retained, and thus it exhibited three-electron accepting ability as well as a low-lying LUMO level.
Doping heteroatom(s) such as B, N, O, P, and S into the π skeletons of nanocarbons is a widely studied strategy to alter their electronic nature including orbital energy levels, redox properties, and chemical reactivity without significant changes in their topological structures.1,2 As a doping element, nitrogen has been most intensively studied because it plays either an electron-donating or an electron-withdrawing role, depending on its incorporation mode. Thus, replacing benzene rings in nanocarbons with N-containing aromatic units such as pyrrole, pyridine, pyrimidine, or pyrazine is an available strategy to access azananocarbons. With this strategy, N-containing nanocarbons with various structures including planar,3 helical,4 bowl,5,6 wavy,7 and spherical8–10 shapes have been constructed to explore their distinctive nature like coordination to metals, molecular dynamics, chiroptical properties, and supramolecular assembly behaviour (Fig. 1).
 | ||
| Fig. 1 Structures of selected azaNGs and twisted azaNG studied herein. | ||
Employing a twisted geometry renders the structures of nanographenes (NGs) unique,11–13 while the role of the twist operation in their electronic nature has not been fully understood.14–21 In 2016, Sato and co-workers reported a diazonium derivative of hexabenzoperylene,22 whose stereostructure was confirmed as a twisted conformer through a comprehensive study involving 1H NMR, UV-vis absorption, and theoretical calculations (Fig. 1). Recently, we reported a C76-NG with orthogonally arranged two-fold unidirectionally twisted skeletons23–25 and, shortly thereafter, an alternatively twisted C52-NG.26 For both NGs, the pyrene cores are significantly contorted, which is the origin of their narrowed energy gaps and high emissivity. Herein, we focus on a twisted N-containing NG. As an aza-aromatic core, we chose diquinoxalinopyrene to explore how electron deficiency is altered or retained after being embedded into NGs. In this paper, we present the synthesis of an alternatively twisted C64N4-azaNG featuring the electronic nature of diquinoxaline-fused pyrene.
The synthesis of azaNG 4 was commenced with the oxidation of diaryl pyrene 1 by NaIO4 in the presence of RuCl3, which yielded tetracarbonyl analogue 2 in 9% isolated yield (Fig. 2).27,28 Subsequently, 2 was condensed with 1,2-benzenediamine to construct the diquinoxaline-fused pyrene 3. Finally, the Scholl cyclization was performed using DDQ (2,3-dichloro-5,6-dicyano-p-benzoquinone) in CH2Cl2/TfOH (Tf: CF3SO2), furnishing 4 in 20% yield.
 | ||
| Fig. 2 Synthesis of azaNG 4. | ||
In the 1H NMR spectrum, a single conformation with six characteristic well-separated signals was observed in the aromatic region (Fig. S7, ESI†), indicating a highly symmetrical structure of the product. The X-ray diffraction analysis revealed that 4 adopts an alternatively twisted geometry along the terropyrene moiety, wherein the end-to-end angle is 0°, while the repeating twist angle (θtw) was measured to be 24.7° (Fig. 3a). In contrast, a double bent geometry was observed at the centre of diquinoxalinopyrene where the bend angle of each phenazine moiety was measured to be 158°. The torsion angle along the helical inner rims was measured to be 〈φ〉 = 23.3°, which is comparable to that observed for pristine [5]helicene (22.1°).29 Within the crystal, 4 is arranged in a slipped π-staking mode at diquinoxaline moieties between nearest molecules with a distance of 3.385 Å (Fig. 3a). The studies on aromaticity suggested benzenoid character for rings A, C, F, and G (NICS(0) −9.8 to −7.3 ppm), while D is a weak antiaromatic ring (NICS(0) +3.3 ppm), which differs from that found in pristine pyrene (D, −4.7 ppm) (Fig. 3b). According to theoretical calculations (B3LYP-D3/6-31G(d), 298 K), the experimentally observed alternatively twisted geometry is most stable compared with other conformations including unidirectionally twisted (ΔG = +6.6 kcal mol−1), cis-wavy (+4.8), trans-wavy (+20.0), and mixed (+7.2) forms (Fig. S13, ESI†).
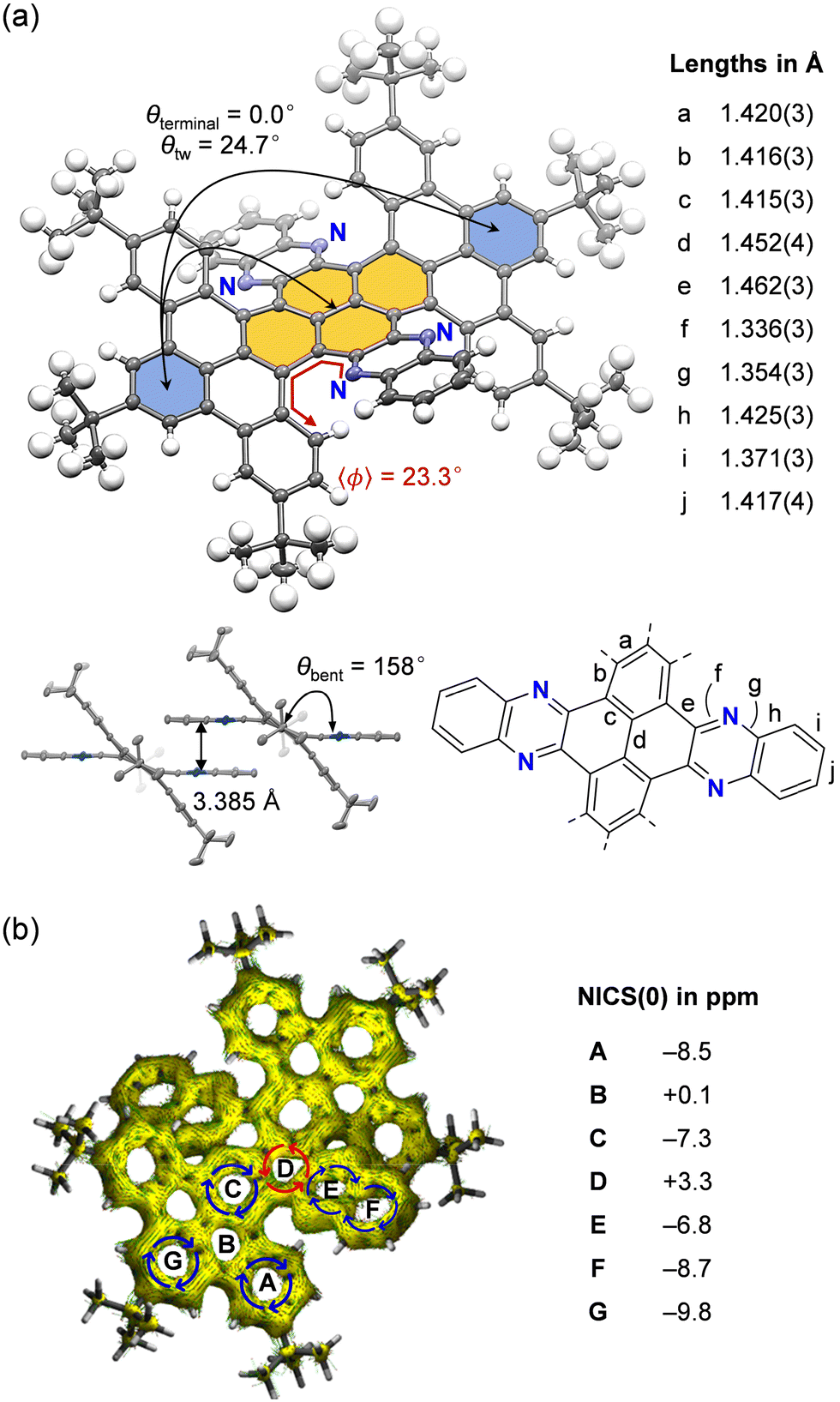 | ||
| Fig. 3 (a) Crystal structure of 4 with the packing mode and selected bond lengths. (b) ACID (anisotropy of current-induced density) plots with NICS(0) (nucleus-independent chemical shift) values (HF/6-31+G(d,p)//B3LYP-D3/6-31G(d)). | ||
Diquinoxalinopyrene derivatives have been reported to show very weak and/or irreversible reduction waves.27 The cyclic voltammogram of 4, however, exhibited stable electrochemical processes with three reversible reduction waves at E1/2 of −1.54, −1.83, and −2.12 V as well as three oxidation waves at Epa of +0.73, +0.93, and +1.15 V (Fig. 4a) by means of effective π-conjugation. The longest absorption band was observed at 526 nm for 4, which was bathochromically shifted from λabs = 419 nm (3) (Fig. 4b). Likewise, emission bands were also shifted from 486 nm (3) to 591 nm (4), where the fluorescence quantum efficiency (ΦF) was measured to be 0.07 and 0.01, respectively. Notably, both 3 and 4 showed significant Stokes shifts of 19 and 18 eV, respectively. According to theoretical calculations, this is ascribed to a structural relaxation at the S1 state by ΔE = −2.15 kcal mol−1 for 3 and −1.27 kcal mol−1 for 4 though the computed Stokes shifts were slightly overestimated to be 25 and 28 eV, respectively (Fig. 4c and Fig. S12, ESI†). Curiously, the origin of the structural deviation at S1 relative to that at S0 was confirmed to be different between 3 and 4. Thus, the central C–C bond of the pyrene moiety in 3 is shortened at S1, rendering the pyrene moiety bent by Δθ = 2.8°. In contrast, the central bond in 4 remains intact at S1, while the torsion angle at the helical units becomes smaller by Δ〈φ〉 = −1.1°.
 | ||
| Fig. 4 (a) Cyclic voltammograms of 3 and 4 (1 mM in CH2Cl2, 0.1 M n-Bu4N·PF6, 100 mV s−1). (b) Absorption (10 μM) and fluorescence (1.0 μM) spectra of 3 and 4. (c) Structural overlay of 3 and 4 at S0 (cyan) and S1 (red) (B3LYP-D3/6-31G(d)). | ||
Further computations were performed to study optical characteristics (TD-CAM-B3LYP-D3/6-31G(d)//B3LYP-D3/6-31G(d)). From the results, the absorption bands of 4 at 492 and 526 nm were suggested to be assigned to the S0 → S2 and S0 → S1 transitions, respectively. The S1 → S2 transition arises from the HOMO → LUMO+1 excitation with a high oscillator strength (f = 0.6751) (Fig. 5). In contrast, the S0 → S1 transition is a forbidden HOMO → LUMO excitation, which becomes allowed in part (f = 0.0084) when the symmetry is disrupted by the rotation of the peripheral t-Bu groups (Fig. S11, ESI†). To gain deeper insights, we investigated pristine and twisted diquinoxalinopyrenes, the latter of which was generated from the coordinates of optimized 4 (Fig. 5). The low-lying LUMO level of 4 (−2.32 eV) originated from pristine diquinoxalinopyrene (−2.32 eV), while the twisting effect is minimal. The twist operation on diquinoxalinopyrene causes a slight elevation of the HOMO level from −5.91 to −5.81 eV, thus contributing to a red shift of the S0 → S1 transition (λ = 386 to 398 nm). The HOMO level of 4 at −4.95 eV is, however, far higher than those of twisted and pristine diquinoxalinopyrenes. This is due to the effective π-conjugation as found in the HOMO coefficients distributed outside the diquinoxalinopyrene core in 4 (Fig. 5), indicating that π-extension is a major contributor to the alteration of the HOMO level.
 | ||
| Fig. 5 Molecular orbitals and their energy levels with selected optical transitions. The transition energies were calibrated by 0.88 (TD-CAM-B3LYP-D3/6-31G(d)//B3LYP-D3/6-31G(d)). | ||
Finally, we tested the availability of 4 as a fluorescence probe for ion sensing since phenazine is often utilized as a sensory core.29 The titration of 4 by AgClO4 was performed in tetrahydrofuran. From the results, a significant decrease in the emission intensity at 581 nm was observed, while a new band appeared at 624 nm, suggesting the formation of 4·Ag+ (Fig. 6a). The apparent binding constant was estimated to be K = 866 M−1. By the addition of N(n-Bu)4I to the solution containing 4·Ag+, the fluorescence band was almost completely recovered back to the original one, demonstrating the excellent repeatability of 4 as a fluorescence probe toward Ag+ (Fig. 6b).
 | ||
| Fig. 6 (a) Florescence titration of 4 (10 μM) by AgClO4. (b) Sensor repeatability (500 equiv. ion source for each step). | ||
In summary, facile synthesis of red-emissive azaNG 4 was performed through the Scholl cyclization of diaryl diquinoxalinopyrene 3. The X-ray crystallographic analysis revealed an alternatively twisted geometry for 4, which is the most stable conformation as supported computationally. Owing to the electron-deficient nature of the diquinoxalinopyrene moiety, 4 exhibited stable three-electron accepting ability. Although 4 is highly contorted, the nature of the key structure, i.e., diquinoxalinopyrene, was shown to remain in the nanographene. The effect of the twist operation is minimal to the LUMO level, while it caused a greater effect on the HOMO level as opposed to an all-carbon system bearing a contorted electron-rich pyrene core, which resulted in a narrowed HOMO–LUMO gap by the alteration of both MO levels.23 This azaNG exhibited sensing ability toward Ag+ with good repeatability.
Financial support was provided by the National Natural Science Foundation of China (No. 22161035), the Education Department of Inner Mongolia Autonomous Region (NJYT22089), the Funding Scheme for High-Level Overseas Chinese Students' Return, the International Collaborative Research Program of Institute for Chemical Research (ICR), Kyoto University (2025-37), the ISHIZUE 2024 of Kyoto University, the JACI Prize for Encouraging Young Researcher, the Yazaki Memorial Foundation for Science and Technology, the Asahi Glass Foundation, the Takahashi Industrial and Economic Research Foundation, and The Kyoto University Foundation. We are also grateful to Mr He Meng, Mr Renmandufu Sha, and Prof. Jianguo Wang (Inner Mongolia University) for their support with the measurement of NMR spectra and quantum yields.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Notes and references
- M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed
.
- A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M. Żyła-Karwowska and M. Stępień, Chem. Rev., 2022, 122, 565–788 CrossRef CAS PubMed
- R. Rieger, M. Kastler, V. Enkelmann and K. Müllen, Chem. – Eur. J., 2008, 14, 6322–6325 CrossRef CAS PubMed
- F. Ammon, S. T. Sauer, R. Lippert, D. Lungerich, D. Reger, F. Hampel and N. Jux, Org. Chem. Front., 2017, 4, 861–870 RSC
- S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256–7260 CrossRef CAS PubMed
- H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef PubMed
- K. Oki, M. Takase, S. Mori, A. Shiotari, Y. Sugimoto, K. Ohara, T. Okujima and H. Uno, J. Am. Chem. Soc., 2018, 140, 10430–10434 CrossRef CAS PubMed
- J. C. Hummelen, B. Knight, J. Pavlovich, R. González and F. Wudl, Science, 1995, 269, 1554–1556 CrossRef CAS PubMed
- Y. Hashikawa, M. Murata, A. Wakamiya and Y. Murata, J. Am. Chem. Soc., 2016, 138, 4096–4104 CrossRef CAS PubMed
- Y. Hashikawa and Y. Murata, J. Am. Chem. Soc., 2017, 139, 18468–18471 CrossRef CAS PubMed
- Y.-F. Wu, L. Zhang, Q. Zhang, S.-Y. Xie and L.-S. Zhang, Org. Chem. Front., 2022, 9, 4726–4743 RSC
- H. V. Anderson, N. D. Gois and W. A. Chalifoux, Org. Chem. Front., 2023, 10, 4167–4197 RSC
- Y. Zhang, J. Guan, L. Luo, X. Han, J. Wang, Y. Zheng and J. Xu, Interdiscip. Mater., 2024, 3, 453–479 CAS
- S. Ma, J. Gu, C. Lin, Z. Luo, Y. Zhu and J. Wang, J. Am. Chem. Soc., 2020, 142, 16887–16893 CrossRef CAS PubMed
- R. K. Dubey, M. Melle-Franco and A. Mateo-Alonso, J. Am. Chem. Soc., 2022, 144, 2765–2774 CrossRef CAS PubMed
- S. T. Bao, H. Jiang, C. Schaack, S. Louie, M. L. Steigerwald, C. Nuckolls and Z. Jin, J. Am. Chem. Soc., 2022, 144, 18772–18777 CrossRef CAS PubMed
- B. Borrisov, G. M. Beneventi, Y. Fu, Z. Qiu, H. Komber, Q. Deng, P. M. Greißel, A. Cadranel, D. M. Guldi, J. Ma and X. Feng, J. Am. Chem. Soc., 2024, 146, 27335–27344 CrossRef CAS PubMed
- S. E. Penty, G. R. F. Orton, D. J. Black, R. Pal, M. A. Zwijnenburg and T. A. Barendt, J. Am. Chem. Soc., 2024, 146, 5470–5479 CrossRef CAS PubMed
- A. Swain, K. Radacki, H. Braunschweig and P. Ravat, Chem. Sci., 2024, 15, 11737–11747 RSC
- X. Liu, Z. Jin, F. Qiu, Y. Guo, Y. Chen, Z. Sun and L. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202407547 CrossRef CAS PubMed
- C. Wallerius, O. Erdene-Ochir, E. V. Doeselar, R. Alle, A. T. Nguyen, M. F. Schumacher, A. Lützen, K. Meerholz and S. H. Pun, Precis. Chem., 2024, 2, 488–494 CrossRef CAS PubMed
- K. Sato, Y. Seki, S. Suga, Y. Ikeda and M. Yamaguchi, J. Photochem. Photobiol., A, 2016, 331, 8–16 CrossRef CAS
- Y. Dong, Z. Zhang, Y. Hashikawa, H. Meng, F. Bai, K. Itami and Chaolumen, Angew. Chem., Int. Ed., 2024, 63, e202406927 CrossRef CAS PubMed
- P. Song, Y. Hashikawa and Chaolumen, Nanomaterials, 2024, 14, 1737 CrossRef CAS PubMed
- Z. Zhang, Y. Hashikawa and Chaolumen, Chem. Commun., 2024, 60, 13875–13878 RSC
- Z. Xu, S. Meng, Z. Zhang, S. Han, F. Bai, Y. Dong, Y. Hashikawa and Chaolumen, Precis. Chem., 2025, 3, 289–294 CrossRef CAS PubMed
- L. Ueberricke, D. Mizioch, F. Ghalami, F. Mildner, F. Rominger, T. Oeser, M. Elstner and M. Mastalerz, Eur. J. Org. Chem., 2021, 4816–4823 CrossRef CAS
- T. H. El-Assaad and D. V. McGrath, J. Org. Chem., 2024, 89, 1989–1992 CrossRef CAS PubMed
- Q. Xiao-Ni, L.-R. Dang, W.-J. Qu, Y.-M. Zhang, H. Yao, Q. Lin and T.-B. Wei, J. Mater. Chem. C, 2020, 8, 11308–11339 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data and optimized geometries. CCDC 2438323. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02120k |
| This journal is © The Royal Society of Chemistry 2025 |