
Rethinking aromaticity of reduced thienoquinoids: insights from an S-Pechmann dye dianion†
Takumi
Shiokawa
a and
Aiko
Fukazawa
 *b
*b
aDepartment of Energy and Hydrocarbon, Graduate School of Engineering, Kyoto University, Japan
bInstitute for Integrated Cell-Material Science (WPI-iCeMS), Institute for Advanced Study, Kyoto University, Japan. E-mail: afukazawa@icems.kyoto-u.ac.jp
First published on 28th May 2025
Abstract
A dianionic species of a thienoquinoid derivative bearing a cross-conjugated γ-thiolactone, the so-called S-Pechmann dye, has been synthesized and characterized. Comparative analysis with a pentafulvalene dianion with an isoelectronic structure revealed that the S-Pechmann dye dianion exhibits significantly reduced aromatic character due to preferential charge delocalization over the C4O units. This work provides an in-depth understanding of the fundamental electronic nature of thienoquinoid-based compounds.
Thienoquinoids, a class of compounds that consist of a quinoid-type thiophene skeleton, have consistently garnered attention as versatile building blocks for functional dyes and semiconductors.1 Since the initial report of quinoidal thiophenes in the 1970s, inspired by the molecular structure of TCNQ, early studies on thienoquinoids focused on oligothiophenes and fused thiophenes bearing dicyanomethylene moieties at both ends (Fig. 1a).2–4 In recent years, γ-thiolactone-based thienoquinoids, such as thieno[3,2-b]thiophene-2,5-dione (TTD),5 3,3′-bithiophene-2,2′-dione (S-Pechmann dye),6 and 2,2′-bithiophene-5,5′-dione (BTD),7 have drawn increasing interest (Fig. 1b). These scaffolds have been utilized as electron-accepting building blocks for organic small-molecule and polymer semiconductors.7–9 Key common features of these thienoquinoids are their high electron affinity and exceptional stability against multi-electron reduction, with the latter being particularly distinctive even compared to other electron-accepting π-conjugated systems. This high stability toward reduction processes has been qualitatively attributed to the acquisition of aromatic character in the reduced state.10 However, quantitative insights into the aromatic character of the reduced thienoquinoids remain unexplored. Given the markedly weaker aromatic character of thiophene compared to benzene, a difference that significantly influences the properties of thiophene-containing π-electron systems,11–13 quantitative assessment of the aromatic character employing multiple criteria14 in the reduced thienoquinoids is essential to elucidate the role of aromatic stabilization in their dianionic states.
 | ||
| Fig. 1 (a) Structures of TCNQ and its thiophene analogue, thiophene-TCNQ. (b) Structures of typical thienoquinoid scaffolds that embed cross-conjugated γ-thiolactone rings (filled blue). (c) Two-electron reversible redox processes of the S-Pechmann dye (1, top panel) shown alongside the formally isoelectronic pentafulvalene (2, bottom panel). | ||
To investigate whether the electron-accepting character of thienoquinoids is indeed derived from the aromatic character of the sulfur-containing five-membered rings, we evaluated their aromatic character in the reduced state based on both experimental and theoretical approaches. Specifically, we selected S-Pechmann dye 1, a γ-thiolactone-based thienoquinoid previously reported by our group,6,9 as a model system. We herein report the synthesis of the dianionic form of S-Pechmann dye 12−via chemical reduction and the quantitative assessment of its aromatic character. Throughout this investigation, we focused on the fact that the cross-conjugated γ-thiolactone framework of 1 can be regarded as isoelectronic with pentafulvalene (2), a cross-conjugated hydrocarbon with 5π electrons in each cycle that acquire aromatic character upon reduction, by approximating endocyclic sulfur atoms as 2π-electron units and carbonyl carbons as 0π-electron units equivalent to carbocations (Fig. 1c).15 We envisioned that the comparative analysis of 12− and pentafulvalene dianion 22− would elucidate the distinctive features of thienoquinoids and the contribution of aromatic character in their reduced states.
Dianionic S-Pechmann dye 12− was synthesized by chemical reduction of the corresponding charge-neutral species 1. To ensure the stability, solubility, and crystallinity of the dianionic species, 4-t-butylphenyl-substituted derivative 1a was selected and synthesized based on the previously reported methods (see the ESI†).6,9 Treatment of 1a with two equivalents of KC8 in THF-d8 at an ambient temperature resulted in a pronounced color change from dark blue to yellowish brown (Fig. 2a). The 1H NMR spectrum of the filtrate after removal of residual graphite revealed the complete consumption of 1a and the formation of other closed-shell species (Fig. 2b). Slow diffusion of a solution of [2.2.2]cryptand in hexane into a THF solution of the reaction mixture produced dark-red single crystals, which were unequivocally identified to be [(K[2.2.2]cryptand)+]21a2− by X-ray crystallographic analysis as shown in Fig. 3a (vide infra). While all the resonance signals in the 1H NMR spectrum of 1a2− were noticeably upfield shifted compared with those of the charge-neutral species 1a, the degree of the shift for the signals attributable to the protons on the central thiolactone framework (8.30 ppm and 6.86 ppm for 1a and 1a2−, respectively) was much more pronounced than those attributable to the other protons (Fig. 2b). The 13C NMR spectra also showed similar trends, with the resonance signals attributed to the carbon atoms in the thiolactone framework showing a greater degree of upfield shift than those of the terminal benzene rings (Fig. S10, ESI†). These characteristic upfield shifts of 1H and 13C resonance signals upon reduction should be attributable to the more pronounced increase of electron density in the central thiolactone framework compared to that in the terminal benzene rings.
 | ||
| Fig. 2 (a) Chemical reduction of 1a with two equivalents of KC8. (b) 1H NMR spectra of 1a (top) and 1a after the addition of two equivalents of KC8 in THF-d8 (bottom). | ||
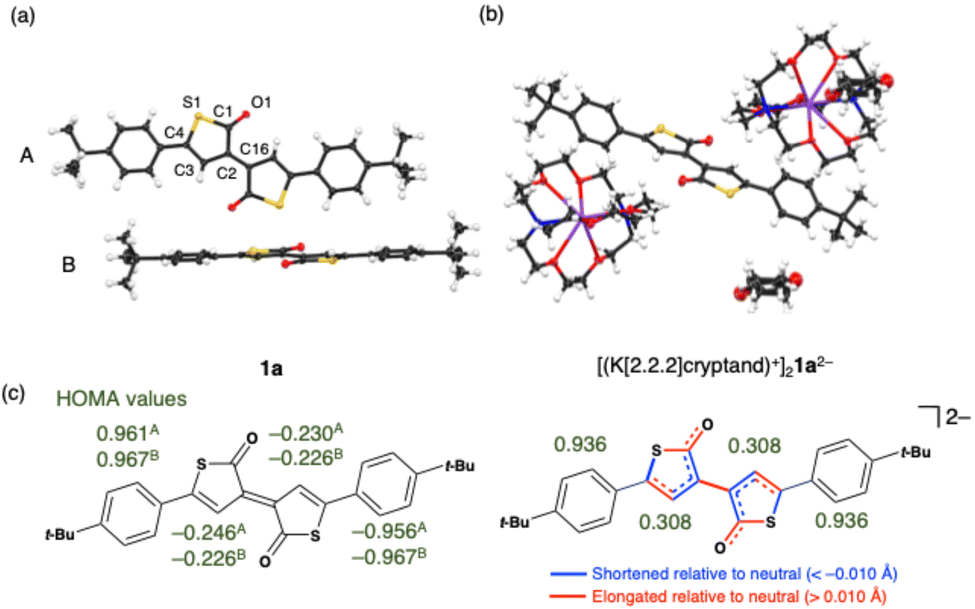 | ||
| Fig. 3 X-ray crystal structure of (a) 1a and (b) [(K[2.2.2]cryptand)+]21a2− in thermal ellipsoid plots at 50% probability. The charge-neutral 1a consists of two crystallographically independent units A and B in the crystal lattice. One THF molecule is included in each crystal lattice of [(K[2.2.2]cryptand)+]21a2−. (c) HOMA values of 1a and 1a2− and bond length changes based on the crystal structures. HOMA values for 1a were calculated for two independent crystals, A and B, respectively. | ||
Comparison of bond lengths of 1a and 1a2− in their X-ray crystal structures provided clear-cut insights into the structural change upon reduction (Fig. 3 and Table S1, ESI†). Firstly, all the bond lengths in the γ-thiolactone framework underwent substantial alternations, either elongating or shortening by more than 0.01 Å, while those of the other components changed by less than 0.01 Å. These structural changes are consistent with the results of 1H and 13C NMR spectra, suggesting that the additional electron density acquired by the reduction is predominantly localized on the central cross-conjugated thiolactone skeleton. Notably, the central exocyclic C–C double bond connecting the two γ-thiolactone moieties (C2–C16) exhibited the most significant elongation from 1.382(2) Å and 1.383(2) Å (for the crystallographically independent units A and B in 1a, respectively) to 1.486(3) Å in 1a2−, a length comparable to that of a typical Csp2–Csp2 single bond (e.g. 1.478(2) Å in 4, see Table S1, ESI†). This elongation of the exocyclic C–C bond, a phenomenon also observed in the two-electron reduction of pentafulvalene derivatives,15 reflects the uptake of the negative charge by five-membered rings upon reduction.
On the other hand, a more detailed examination of the bond-length changes within the five-membered rings upon reduction revealed the characteristics of the cross-conjugated γ-thiolactone distinct from pentafulvalene derivatives. Specifically, the C1–C2 and C2–C3 bonds, which exhibited single bond character in 1a, shortened by 0.08 Å and 0.02 Å, respectively, and the C3–C4 bond with double bond character in 1a lengthened by 0.011 Å upon reduction, resulting in a small bond length alternation (BLA). This small BLA extended to the exocyclic C1–O1 bonds, elongating from 1.212(1) Å and 1.214(1) Å in 1a (for the units A and B, respectively) to 1.261(2) Å in 1a2−. Conversely, the endocyclic C1–S1 bonds elongated significantly from 1.792(1) Å and 1.796(1) Å (for the units A and B in 1a, respectively) to 1.839(2) Å in 1a2−, reaching values comparable to those of Csp3–S bonds (e.g. 1.827 Å in tetrahydrothiophene16). These results indicate that the negative charge in 1a2− is delocalized over the C4O moieties, excluding sulfur atoms, rather than entire sulfur-containing five-membered rings. Reflecting these bond lengths, the harmonic oscillator model of aromaticity (HOMA)17 value for the γ-thiolactone rings of 0.308 in 1a2− indicates their weak aromatic character based on the structural criteria.
To gain quantitative insights into the degree of aromaticity of dianionic species based on magnetic criteria, the diatropicity strength of dianionic cross-conjugated thiolactones of 12− was validated in comparison with pentafulvalene dianion 22−, which is considered an isoelectronic counterpart. To reduce the computational costs, the diphenyl-substituted S-Pechmann dye without t-butyl groups, 1b, and its pentafulvalene counterpart 3,3′-diphenyl-substituted pentafulvalene 2b were selected as model compounds, and their nucleus-independent chemical shift (NICS) values,18 the anisotropy of the current density (ACID),19 and the gauge including magnetically induced current (GIMIC)20 for the corresponding dianions 1b2− and 2b2− were computed using the optimized geometries at the ωB97X-D/6-31+G(d) level of theory (Fig. 4a). The NICS(1)zz value of the thiolactone ring in 1b2− was calculated to be −6.51 ppm. The ACID plot also showed a clockwise induced ring current along the thiolactone rings, suggesting that the thiolactone rings in 1b2− exhibit a certain extent of diatropic ring current attributable to their aromatic character. However, the absolute values of the NICS(1)zz and the current density integration (|χ| = 3.91 nA T−1) of the thiolactone framework were significantly smaller compared to those of the five-membered rings in 2b2− (NICS(1)zz = −13.94 ppm, |χ| = 7.65 nA T−1), an isoelectronic counterpart of 1b2−. These comparisons along with the results obtained by X-ray crystallographic analysis clearly indicate that the aromatic character is considerably weak in 12−.
 | ||
| Fig. 4 (a) Molecular structures of 1b and 2b. (b) NICS(1)zz values of 1b2− and 2b2− calculated at the GIAO-ωB97X-D/6-31+G(d) level of theory. (c) ACID plots (isovalue surface: 0.030) of 1b2− and 2b2− calculated at the CSGT-ωB97X-D/6-31+G(d) level of theory. The red arrows show the clockwise ring current flow. | ||
Comparison of dianionic S-Pechmann dye 12− and pentafulvalene dianion 22− by natural resonance theory (NRT) analysis21 provided insights into why π-electron delocalization along the C4O backbone of 12− surpasses the contribution of aromatic stabilization in the sulfur-containing five-membered rings (Fig. 5).‡ To reduce computational cost and simplify interpretation, model compounds without terminal aryl substituents, namely, 1c2− and 2c2−, were subjected to geometry optimization at the ωB97X-D/6-31+G(d) level, followed by the NRT analysis at the same level. The NRT analyses revealed that 2c2− exists primarily as a resonance hybrid of six major contributors (Fig. 5b), each featuring a cyclopentadienyl anion dimer with negative charge delocalized over C2, C3, C4, and C5. The NRT weights of these six resonance contributors range from 3.31% to 3.40%, collectively accounting for 53.7% of the total. These findings suggest that the pentafulvalene dianion can be described as a dimer of cyclopentadienyl anions with evenly delocalized negative charges across the five-membered rings, as illustrated in the structure 2c2−-R. This effective charge delocalization results in the acquisition of Hückel aromaticity upon two-electron reduction, as widely accepted (Fig. 5b). In contrast, a similar analysis of 1c2− identified numerous resonance contributors, as shown in Fig. 5c. Among these, only four resonance contributors (type I in Fig. 5c) manifest the Hückel aromaticity of the thiolactone rings, with a total NRT weight of merely 19.5%. This substantially lower NRT weight in 1c2− compared to that in 2c2− quantitatively indicates a lesser degree of π-electron delocalization over the five-membered ring, supporting the conclusion that 12− exhibits weaker aromatic character than 22−, as discussed based on the NICS values and the ACID plots. Notably, three of the four type I resonance contributors do not have double-bond characteristics in the S1–C1 bond. Moreover, all four type I contributors show localization of negative charge on the carbonyl oxygen atom. These results indicate that the weak donation of the lone pair electrons on the sulfur atoms to the carbonyl carbons, along with the strong electron-withdrawing nature of the carbonyl groups, promote the charge delocalization in 1c2− along the C4O unit rather than the five-membered rings.
 | ||
| Fig. 5 (a) Chemical structures of 1c2− and 2c2− with atom numbering. (b) and (c) Dominant resonance contributors and their NRT weights [%] calculated at the ωB97X-D/6-31G+(d) level. The contributors with weights <2% were omitted. The numbers in the parentheses indicate degeneracy due to molecular symmetry. Pink-filled five-membered rings denote Hückel aromaticity. Resonance contributors of 1c2− are classified as type I (aromatic ring present) and type II (nonaromatic). Resonance hybrids (2c2−-R and 1c2−-R) based on the dominant resonance contributors are shown to reflect realistic bond lengths and charge distributions. | ||
In summary, a dianionic species of S-Pechmann dye 1a2−, a carbonyl-substituted thienoquinoid π-electron system, was synthesized by chemical reduction using KC8. NMR spectroscopy, X-ray crystallography, and theoretical analysis revealed both similarities and differences between γ-thiolactone-based thienoquinoid 1a and pentafulvalene 2. Electron density introduced upon two-electron reduction was predominantly localized in the central γ-thiolactone moieties, exhibiting aromatic character. The conceptual analogy between thienoquinoids and pentafulvalene illustrates that the isoelectronic relationships provide intuitive guidance for molecular design, akin to analogies previously drawn between tropone and benzene,22 or tetrathiafulvalene (TTF) and heptafulvalene.23 However, quantitative analysis showed significantly lower aromatic character in thienoquinoid dianion 1a2− than in pentafulvalene dianion 22−. This diminished aromatic character arises from preferential charge delocalization over the C4O units rather than the sulfur-containing five-membered rings, driven by the strong electron-withdrawing nature of the carbonyl groups and weak orbital interactions between sulfur and carbon-based π-frameworks. These findings deepen the understanding of the fundamental electronic structure of thienoquinoid compounds.
This work was supported by JSPS KAKENHI grant nos. JP20H05864, 21H01916, and 24H00458 from MEXT, Japan (for A. F.). The authors thank the iCeMS Analysis Center for providing access to the NMR spectrometer. The synchrotron single-crystal X-ray diffraction measurements were performed at the BL02B1 beamline of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; project nos. 2023A1785, 2023A1925, and 2024B2124). The authors are also grateful to Dr S. Hasegawa and Dr Z. Wang (Kyoto University) for their help with the crystallographic analyses and the quantum chemical calculations, respectively.
Data availability
Data supporting this article are included in the ESI.† Crystallographic data have been deposited at the CCDC with deposition numbers 2446608 (1a), 2446610 (([K[2.2.2]cryptand)+]21a2−), and 2446609 (4).†Conflicts of interest
There are no conflicts to declare.Notes and references
- K. Takimiya and K. Kawabata, J. Synth. Org. Chem. Jpn., 2018, 76, 1176–1184 CrossRef CAS.
- S. Gronowitz and B. Uppström, Acta Chem. Scand., 1974, 981–985 CrossRef CAS.
- M. L. Kaplan, R. C. Haddon, F. B. Bramwell, F. Wudl, J. H. Marshall, D. O. Cowan and S. Gronowitz, J. Phys. Chem., 1980, 84, 427–431 CrossRef CAS.
- S. Yoshida, M. Fujii, Y. Aso, T. Otsubo and F. Ogura, J. Org. Chem., 1994, 59, 3077–3081 CrossRef CAS.
- S. Chen, A. Bolag, J. I. Nishida and Y. Yamashita, Chem. Lett., 2011, 40, 998–1000 CrossRef CAS.
- A. Fukazawa, M. Adachi, K. Nakakura, S. Saito and S. Yamaguchi, Chem. Commun., 2013, 49, 7117–7119 RSC.
- K. Kawabata, M. Saito, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 7725–7732 CrossRef CAS PubMed.
- T. Mikie and I. Osaka, J. Mater. Chem. C, 2020, 8, 14262–14288 RSC.
- T. Mikie, M. Hayakawa, K. Okamoto, K. Iguchi, S. Yashiro, T. Koganezawa, M. Sumiya, H. Ishii, S. Yamaguchi, A. Fukazawa and I. Osaka, Chem. Mater., 2021, 33, 8183–8193 CrossRef CAS.
- M. Nakatsuka, K. Nakasuji, I. Murata, I. Watanabe, G. Saito, T. Enoki and H. Inokuchi, Chem. Lett., 1983, 905–908 CrossRef CAS.
- R. R. Jones, J. M. Brown and F. Sondheimer, Tetrahedron Lett., 1975, 16, 4183–4186 CrossRef.
- (a) C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827–16838 CrossRef CAS PubMed; (b) J. L. Marshall, K. Uchida, C. K. Frederickson, C. Schütt, A. M. Zeidell, K. P. Goetz, T. W. Finn, K. Jarolimek, L. N. Zakharov, C. Risko, R. Herges, O. D. Jurchescu and M. M. Haley, Chem. Sci., 2016, 7, 5547–5558 RSC; (c) J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Nat. Chem., 2018, 10, 1134–1140 CrossRef CAS PubMed; (d) G. I. Warren, K. Młodzikowska-Pieńko, S. Jalife, I. S. Demachkie, J. I. Wu, M. M. Haley and R. Gershoni-Poranne, Chem. Sci., 2024, 16, 575–583 RSC.
- (a) A. Fukazawa, D. Kishi, Y. Tanaka, S. Seki and S. Yamaguchi, Angew. Chem., Int. Ed., 2013, 52, 12091–12095 CrossRef CAS PubMed; (b) H. Oshima, A. Fukazawa and S. Yamaguchi, Angew. Chem., Int. Ed., 2017, 56, 3270–3274 CrossRef CAS PubMed; (c) J. Usuba, M. Hayakawa, S. Yamaguchi and A. Fukazawa, Chem. – Eur. J., 2021, 27, 1638–1647 CrossRef CAS PubMed; (d) J. Usuba and A. Fukazawa, Chem. – Eur. J., 2021, 27, 16127–16134 CrossRef CAS PubMed.
- G. Merino, M. Solà, I. Fernández, C. Foroutan-Nejad, P. Lazzeretti, G. Frenking, H. L. Anderson, D. Sundholm, F. P. Cossío, M. A. Petrukhina, J. Wu, J. I. Wu and A. Restrepo, Chem. Sci., 2023, 14, 5569–5576 RSC.
- (a) N. S. Mills and M. Benish, J. Org. Chem., 2006, 71, 2207–2213 CrossRef CAS PubMed; (b) F. G. Brunetti, X. Gong, M. Tong, A. J. Heeger and F. Wudl, Angew. Chem., Int. Ed., 2010, 49, 532–536 CrossRef CAS PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. Guy Orpen and R. Taylor, J. Chem. Soc. Perkin Trans. 2, 1987, S1–S19 RSC.
- (a) T. M. Krygowski, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS; (b) T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385–1419 CrossRef CAS PubMed.
- (a) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed; (b) P. V. R. Schleyer, H. Jiao, N. J. R. van, E. Hommes, V. G. Malkin and O. L. Malkina, J. Am. Chem. Soc., 1997, 119, 12669–12670 CrossRef CAS.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- (a) J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef PubMed; (b) H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, Phys. Chem. Chem. Phys., 2011, 13, 20500–20518 RSC.
- E. D. Glendening and F. Weinhold, J. Comput. Chem., 1998, 19, 593–609 CrossRef CAS.
- (a) M. J. S. Dewar, Nature, 1950, 166, 790–791 CrossRef CAS PubMed; (b) M. J. S. Dewar, Nature, 1945, 155, 50–51 CrossRef CAS.
- F. Wudl, G. M. Smith and E. J. Hufnagel, J. Chem. Soc. D, 1970, 1453–1454 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, copies of NMR spectra for all new compounds, and DFT calculation data including the output files. CCDC 2446608–2446610. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02327k |
| ‡ NPA charge analysis was also performed and is presented in Tables S2 and S3 in the ESI.† The results support general charge accumulation in the central γ-thiolactone moieties, but do not resolve resonance pathways as clearly as NRT. |
| This journal is © The Royal Society of Chemistry 2025 |