
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnexpected cyclization of β-(hydroxymethyl)phosphole into 1-phospha-1,6a-dihydrophosphapentalene: a fused 1,3-butadiene-based luminophore†
Tomohiro
Higashino
 *a,
Riku
Minobe
a,
Tomoya
Machino
a and
Hiroshi
Imahori
*abc
*a,
Riku
Minobe
a,
Tomoya
Machino
a and
Hiroshi
Imahori
*abc
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyoto, 615-8510, Japan. E-mail: t-higa@scl.kyoto-u.ac.jp; imahori@scl.kyoto-u.ac.jp
bInstitute for Integrated Cell-Material Sciences (iCeMS), Kyoto University, Kyoto, 606-8501, Japan
cInstitute for Liberal Arts and Sciences (ILAS), Kyoto University, Kyoto, 606-8316, Japan
First published on 22nd May 2025
Abstract
During the planned synthesis of carbon-bridged fused phospholes, an unexpected intramolecular cyclization of β-(hydroxymethyl)phosphole yielded 1-phospha-1,6a-dihydropentalene, besides the expected cyclized product. The solid-state emission of 1-phospha-1,6a-dihydropentalene indicates its potential as a 1,3-butadiene-based solid-state luminophore.
Incorporating bridging structures into π-systems can modulate their optical and electronic properties by introducing structural constraints and electronic perturbations via heteroatoms. Phosphorus-bridged π-conjugated molecules, especially fused phosphole derivatives, exhibit remarkable physicochemical properties, such as highly electron-accepting ability and intense emission with high fluorescence quantum yields.1–9 In addition, carbon-bridged structures offer optimal conjugation due to their highly planar and rigid conformation.10–15 In this context, a rational synthetic methodology for carbon-bridged fused phospholes would be useful toward effectively π-extended phosphole-based functional materials.
Although β-(hydroxymethyl)phospholes serve as key precursors to carbon-bridged fused phospholes, successful synthetic protocols for β-substituted 2,5-diarylphospholes remain limited.16,17 Given the proposed reaction mechanism for the n-BuLi mediated synthesis of 2,5-diarylphospholes from 1,3-butadiynes and phenylphosphine (PhPH2), we envisioned that treating 1,3-butadiynes with a stoichiometric amount of n-BuLi could generate β-lithiated intermediates.16–18 Subsequent reaction with ketones and oxidation of phosphorus atom would yield β-(hydroxymethyl)phosphole P-oxides, which could be transformed into carbon-bridged fused phosphole P-oxides via intramolecular Friedel–Crafts cyclization. Following this strategy, we pursued the straightforward synthesis of carbon-bridged fused phospholes and unexpectedly discovered the formation of 1-phospha-1,6a-dihydropentalene (Scheme 1). Though Latscha and co-workers reported the synthesis of a 1-phospha-1,6a-dihydropentalene derivative in 1991,19 further exploration and detailed analysis of their properties have not been conducted. Herein, we report the intramolecular Friedel–Crafts cyclization reaction of β-(hydroxymethyl)phospholes, yielding 1-phospha-1,6a-dihydropentalene as well as anticipated carbon-bridged fused phospholes.
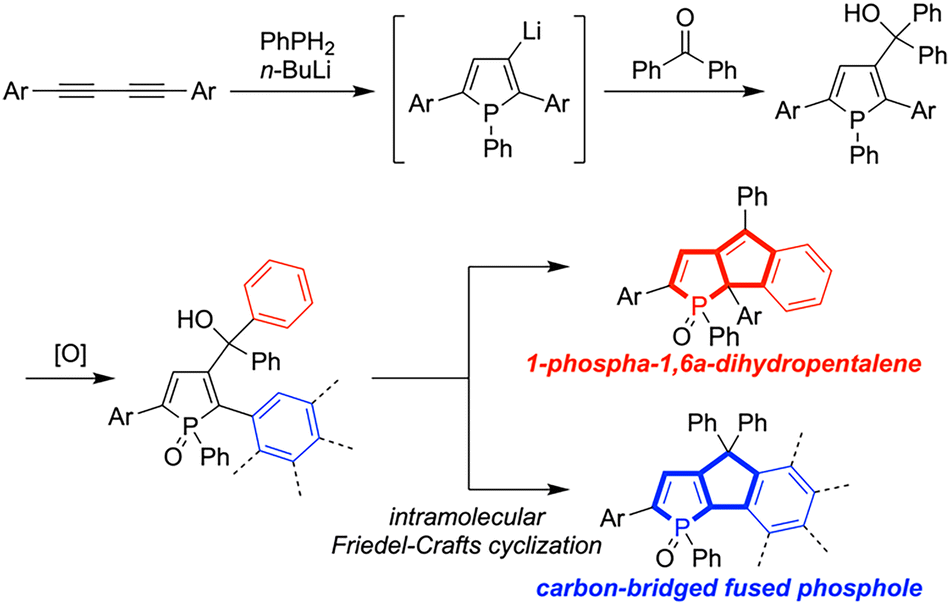 | ||
| Scheme 1 n-BuLi mediated synthesis of β-substituted phospholes and intramolecular Friedel–Crafts cyclization of β-(hydroxymethyl)phospholes. | ||
First, we embarked on synthesizing β-substituted 2,5-diarylphospholes (Scheme 2). Treating PhPH2 with a stoichiometric amount of n-BuLi (1 equiv.) and subsequently adding 1,4-diphenylbutadiyne generated the β-lithiated phosphole intermediate at −78 °C. Reacting this intermediate with benzophenone at −45 °C then afforded the desired β-(hydroxymethyl)phosphole 1a. Due to the instability of the σ3,λ3-phosphole 1a under ambient conditions, it was difficult to isolate using conventional silica-gel column chromatography. Therefore, the crude 1a was converted into the stable phosphole oxide 2a by oxidation with an aqueous H2O2 solution. Additionally, we synthesized the β-substituted phospholes 1b and 2b in a similar manner using 1,4-di(1-naphthyl)butadiyne.
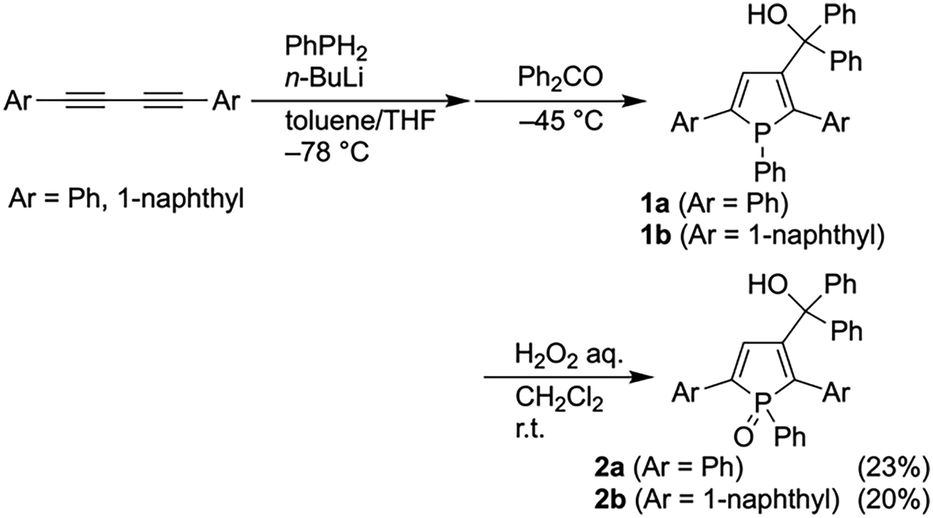 | ||
| Scheme 2 Synthesis of β-(hydroxymethyl)phospholes 1 and 2. | ||
Next, we conducted the intramolecular Friedel–Crafts cyclization reaction of phosphole P-oxides 2 in the presence of BF3·Et2O as reported by Yamaguchi and co-workers (Scheme 3).20–23 When 2a was treated with 1 equivalent of BF3·Et2O, it was completely recovered after conventional aqueous workup, suggesting that the stoichiometric amount of BF3 was consumed to form a phosphine oxide-BF3 adduct.24,25 Conversely, treating 2a with an excess amount of BF3·Et2O (5 equiv.) proceeded smoothly, yielding a new product 3 in high yield (87%). Surprisingly, the crystal structure of 3 unambiguously revealed a 1-phospha-1,6a-dihydropentalene skeleton, not the anticipated carbon-bridged fused phosphole (Fig. 1a and Table S1, ESI†). Notably, the two phenyl groups at the 1- and 6a-positions adopted a syn-configuration, and the anti-isomer was not obtained. The selective formation of the syn-isomer can be explained by the steric hindrance of the phenyl group on the phosphorus atom in the transition state (vide infra). The C–C bond lengths of C2–C3 (1.5260(18) Å) and C2–C8 (1.5218(18) Å) clearly indicate that 3 possesses an sp3-carbon atom at the 6a-position (C2 atom in Fig. 1a). The C4–C5 (1.3584(18) Å) and C3–C6 (1.3553(18) Å) bond lengths are almost comparable to a typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond length (1.35 Å) in 1,3-butadiene.26 Thus, the crystal structure clearly corroborates that 3 possesses a 1,3-butadiene skeleton.
C double bond length (1.35 Å) in 1,3-butadiene.26 Thus, the crystal structure clearly corroborates that 3 possesses a 1,3-butadiene skeleton.
 | ||
| Scheme 3 Intramolecular cyclization reaction of phosphole P-oxides 2 in the presence of BF3·Et2O. | ||
 | ||
| Fig. 1 X-Ray crystal structures of (a) 3, (b) 4, and (c) 5. Thermal ellipsoids represent 50% probability. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
In contrast to the reaction of 2a, the reaction of 2b with BF3·Et2O resulted in two new compounds, 4 (16%) and 5 (11%), which were successfully separated by HPLC-GPC (Scheme 3). The structures of 4 and 5 were confirmed by single crystal X-ray diffraction analysis, revealing them to be the expected carbon-bridged 1,8- and 1,2-fused phospholes, respectively (Fig. 1b, c and Table S1, ESI†). The slightly higher yield of 4 compared to 5 is attributed to the higher reactivity at the α-position than the β-position of naphthalene.
To elucidate the reaction mechanism of the intramolecular Friedel–Crafts cyclization reactions and understand the selectivity of the products, we conducted the density functional theory (DFT) calculations at the ωB97XD/6-311G++(d,p)//B3LYP/6-31+G(d,p) level with the polarizable continuum model (PCM) using CH2Cl2 as a solvent (Tables S2 and S3, ESI†). Given that one equivalent of BF3 is consumed to form the phosphine oxide-BF3 adduct (vide supra), we set the phosphine oxide-BF3 adducts S1 and S2 as starting materials for the intramolecular cyclization.
First, we examined the reaction mechanism for 2,5-diphenylphosphole S1 (Fig. 2). The hydroxy group of S1 is activated by additional BF3, generating the cation intermediate INT1viaTS1. Intramolecular C–C bond formation then occurs viaTS2a–c, and the deprotonation of the resultant intermediates INT2a–c affords the products. The energy barrier for TS2a (ΔG‡ = +17.9 kcal mol−1) to produce a 1-phospha-1,6a-dihydropentalene with syn-configuration P1a is smaller than those for TS2b (ΔG‡ = +20.2 kcal mol−1) and TS2c (ΔG‡ = +20.6 kcal mol−1), which yield a 1-phospha-1,6a-dihydropentalene with anti-configuration P1b and a carbon-bridged fused phosphole P1c, respectively. In TS2a, the C3–C6 bond length (1.406 Å) is significantly shorter than the non-conjugated C(sp2)–C(sp2) single bond length (1.48 Å),27 implying the contribution of a CC double bond (Fig. S1, ESI†). The small torsion angle of C2–C3–C6–C7 (19°) is consistent with the double bond character of the C3–C6 bond. TS2a thus possesses an allylic cation-like structure. Furthermore, the short H8⋯F1 distance in TS2a (2.13 Å) implies activation of the C8 atom by the CH⋯F hydrogen bond interaction (2.20–2.26 Å).28 Although the structural features of TS2b (C2–C3: 1.459 Å; C3–C6: 1.404 Å; ∠C2–C3–C6–C7: 18°) also suggest an allylic cation-like structure, the larger torsion angle of C2–P1–O1–B1 for TS2b (73°) compared to TS2a (58°) suggests significant steric repulsion between the BF3 moiety and the phenyl ring at the 2-position, destabilizing TS2b.
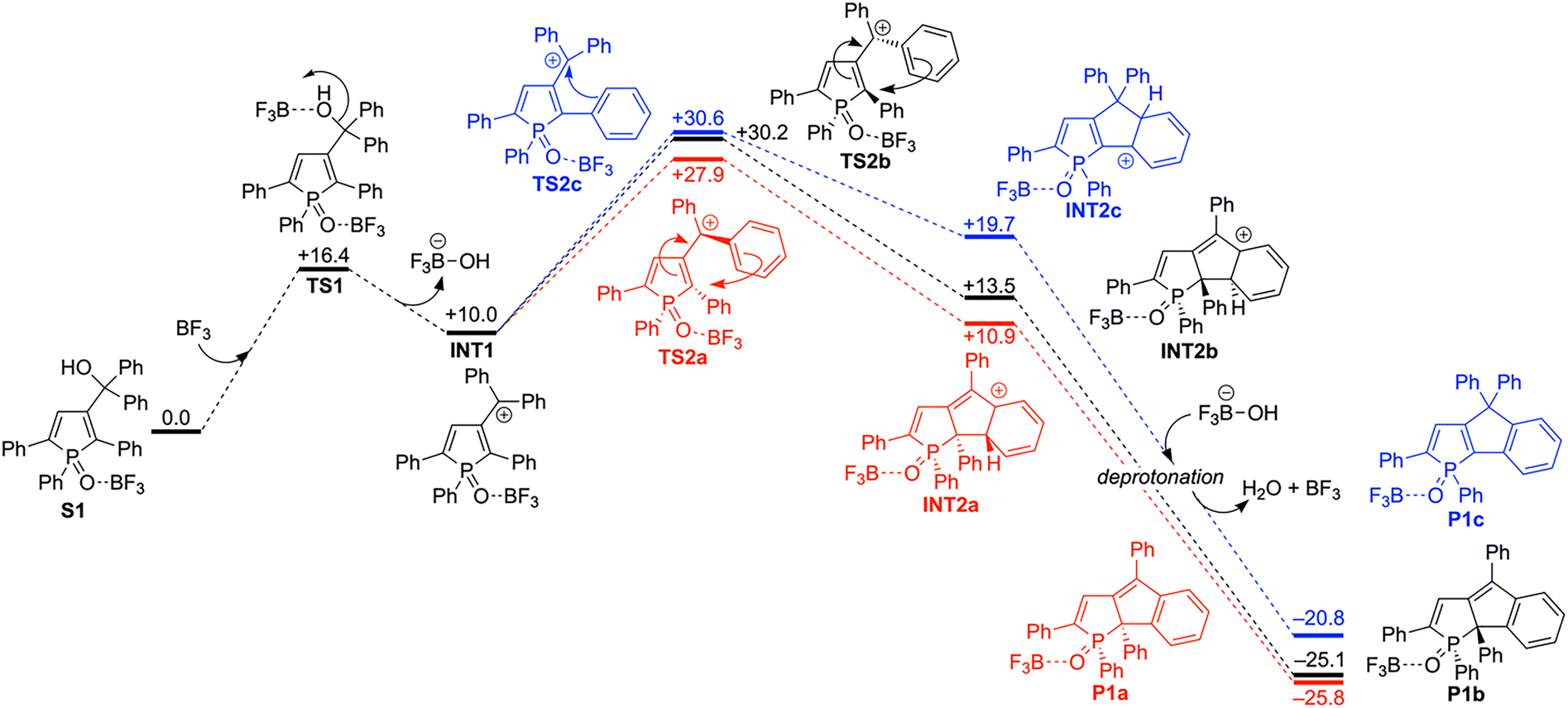 | ||
| Fig. 2 Mechanistic studies of intramolecular cyclization for 2,5-diphenylphosphole S1 using DFT methods at the ωB97XD/6-311G++(d,p)//B3LYP/6-31+G(d,p) level with polarizable continuum model (PCM) using CH2Cl2 as the solvent. The relative Gibbs free energy values are given in kcal mol−1 units. | ||
In contrast to TS2a and TS2b, the long C3–C6 bond length (1.497 Å) and the large torsion angle of C2–C3–C6–C8 (81°) agree with the single bond character of the C3–C6 bond. As a result, the localized positive charge on the C6 atom destabilizes TS2c. Overall, TS2a is stabilized by the delocalized positive charge over the allylic cation-like structure, minimal unfavorable steric repulsion, and activation through intramolecular CH⋯F hydrogen bond interaction. Since the C–C bond formation should be the rate-determining step, the smallest ΔG‡ value of TS2a clearly supports the selective formation of 1-phospha-1,6a-dihydropentalene with syn-configuration.
The calculated reaction mechanism for 2,5-dinaphthyl-phosphole S2 is depicted in Fig. S2 (ESI†). We examined three pathways from the cation intermediate INT3 to form the possible products, the 1-phospha-1,6a-dihydropentalene P2a and two carbon-bridged fused phospholes P2b and P2c. In contrast to S1, the ΔG‡ value of TS4a for forming a 1-phospha-1,6a-dihydropentalene P2a (ΔG‡ = +22.0 kcal mol−1) is considerably higher than those of TS4b (ΔG‡ = +16.5 kcal mol−1) and TS4c (ΔG‡ = +17.0 kcal mol−1) for carbon-bridged fused phospholes P2b and P2c. The steric hindrance of the naphthyl group at the 2-positoin against the diphenylmethyl group at the 3-position destabilizes TS4a (Fig. S3 and S4, ESI†). Moreover, the higher reactivity of the naphthyl group compared to the phenyl group toward electrophilic substitution reactions promotes the formation of carbon-bridged fused phospholes. Therefore, the selectivity of the products can be rationalized by the steric hindrance and reactivity of the aryl substituents at the 2-position on the phosphole skeleton. Additionally, the slightly smaller ΔG‡ value of TS4b compared to TS4c is attributed to the higher reactivity at the α-position than the β-position of naphthalene, which aligns with the higher yield of 1,8-fused phosphole 4 compared to 1,2-fused phosphole 5 (vide supra).
We examined the optical properties of the products 3–5 (Fig. 3 and Table S4, ESI†). The blue-shifted absorption and fluorescence of 3 compared to the reference phosphole 6 can be attributed to the less effective interaction of σ*-orbital of the P–C bond and π*-orbital of the butadiene moiety in 3 because of the sp3 carbon atom neighboring the phosphorus atom. In addition, the s-trans configuration of the 1,3-butadiene skeleton in 3 may also contribute to the blue-shifted absorption.29 On the other hand, the carbon-bridged fused phospholes 4 and 5 exhibit red-shifted absorption and fluorescence in comparison with the non-fused phosphole 7. The sp3 carbon atom contributes to the effective π-extension resulting from the co-planarization of the naphthyl group.
 | ||
| Fig. 3 UV/Vis absorption (solid lines) and normalized fluorescence (dashed lines) spectra of (a) 3 (red) and 6 (black), and (b) 4 (red), 5 (blue), and 7 (black) in CH2Cl2. For fluorescence measurements, the samples were excited at λex = 370 nm for 3, λex = 430 nm for 4, λex = 450 nm for 5, and λex = 395 nm for 6 and 7. | ||
Notably, 1-phospha-1,6a-dihydropentalene 3 exhibits distinct fluorescence even in the solid state whereas phosphole derivatives 4–7 show no emission in the solid state (Fig. S5, ESI†). Importantly, the ΦF value of 3 in the solid state (0.25) is considerably higher than that in solution (0.04). Given that aryl-substituted 1,3-butadiene structure have emerged as effective scaffolds for solid-state emission owing to their aggregation-induced emission (AIE) features,30–34 a 1-phospha-1,6a-dihydropentalene structure can also be a promising platform for fused 1,3-butadiene-based solid-state fluorophores.
In summary, we pursued a rational synthetic protocol for carbon-bridged fused phospholes and unexpectedly discovered an intramolecular cyclization reaction of β-(hydroxymethyl)phosphole 2a, resulting in the formation of 1-phospha-1,6a-dihydropentalene 3. Theoretical investigations into the reaction mechanism described plausible pathways, suggesting that the product selectivity is governed by the substituents at the 2-position on the phosphole skeleton. Importantly, we revealed the solid-state emissive nature of 3 for the first time, indicating its potential as a 1,3-butadiene-based solid-state luminophore. Thus, we believe that further investigation on the intramolecular cyclization reaction of β-(hydroxymethyl)phospholes will pave the way for developing 1-phospha-1,6a-dihydropentalenes and carbon-bridged fused phospholes as promising organic functional materials.
This work was supported by the JSPS (KAKENHI Grant Numbers JP20H05841, JP22K05066, and JP25K01874 (T. H.), JP20H05832 and JP23H00309 (H. I.)).
Data availability
Data supporting this article is included in the ESI.† Crystallographic data have been deposited at the CCDC with deposition numbers 2424364 (3), 2424363 (4), and 2424362 (5).Conflicts of interest
There are no conflicts to declare.Notes and references
- Y. Matano and H. Imahori, Org. Biomol. Chem., 2009, 7, 1258–1271 RSC.
- T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 Search PubMed.
- D. Joly, P.-A. Bouit and M. Hissler, J. Mater. Chem. C, 2016, 4, 3686–3698 Search PubMed.
- M. P. Duffy, W. Delaunay, P.-A. Bouit and M. Hissler, Chem. Soc. Rev., 2016, 45, 5296–5310 Search PubMed.
- P. Hibner-Kulicka, J. A. Joule, J. Skalik and P. Bałczewski, RSC Adv., 2017, 7, 9194–9236 Search PubMed.
- N. Asok, J. R. Gaffen and T. Baumgartner, Acc. Chem. Res., 2023, 56, 536–547 CrossRef CAS PubMed.
- T. Higashino, K. Ishida, T. Sakurai, S. Seki, T. Konishi, K. Kamada, K. Kamada and H. Imahori, Chem. – Eur. J., 2019, 25, 6425–6438 CrossRef CAS PubMed.
- K. Ishida, T. Higashino, Y. Wada, H. Kaji, A. Saeki and H. Imahori, ChemPlusChem, 2021, 86, 130–136 Search PubMed.
- K. Zhang, X. Wang, Z. Zhou, J. Guo, H. Liu, Y. Zhai, Y. Yao, K. Yang and Z. Zeng, Angew. Chem., Int. Ed., 2025, 64, e202418520 Search PubMed.
- U. Scherf, J. Mater. Chem., 1999, 9, 1853–1864 Search PubMed.
- H. Tsuji and E. Nakamura, Acc. Chem. Res., 2019, 52, 2939–2949 Search PubMed.
- Y. Kurumisawa, T. Higashino, S. Nimura, Y. Tsuji, H. Iiyama and H. Imahori, J. Am. Chem. Soc., 2019, 141, 9910–9919 Search PubMed.
- Y. Zhang, T. Higashino, I. Nishimura and H. Imahori, ACS Appl. Mater. Interfaces, 2024, 16, 67761–67770 CrossRef CAS PubMed.
- J. Wang and X. Zhan, Acc. Chem. Res., 2021, 54, 132–143 Search PubMed.
- K. Wang, S. Jinnai, T. Urakami, H. Sato, M. Higashi, S. Tsujimura, Y. Kobori, R. Adachi, A. Yamakata and Y. Ie, Angew. Chem., Int. Ed., 2024, 63, e202412691 CrossRef CAS PubMed.
- D. Klintuch, K. Krekić, C. Bruhn, Z. Benkő and R. Pietschnig, Eur. J. Inorg. Chem., 2016, 718–725 Search PubMed.
- F. Roesler, M. Kovács, C. Bruhn, Z. Kelemen and R. Pietschnig, Organometallics, 2023, 42, 793–802 CrossRef CAS.
- G. Märkl and R. Potthast, Angew. Chem., Int. Ed. Engl., 1967, 6, 86 CrossRef.
- J. Silberzahn, H. Pritzkow and H. P. Latscha, Z. Naturforsch. B, 1991, 46, 197–201 Search PubMed.
- A. Fukazawa, Y. Ichihashi, Y. Kosaka and S. Yamaguchi, Chem. – Asian J., 2009, 4, 1729–1740 CrossRef CAS PubMed.
- C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 15213–15217 Search PubMed.
- R. A. Adler, C. Wang, A. Fukazawa and S. Yamaguchi, Inorg. Chem., 2017, 56, 8718–8725 CrossRef CAS PubMed.
- C. Wang, M. Taki, Y. Sato, A. Fukazawa, T. Higashiyama and S. Yamaguchi, J. Am. Chem. Soc., 2017, 139, 10374–10381 Search PubMed.
- N. Burford, R. E. V. H. Spence, A. Linden and T. S. Cameron, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 92–95 Search PubMed.
- V. M.-Y. Leung, H.-C. F. Wong, C.-M. Pook, Y.-L. S. Tse and Y.-Y. Yeung, Chem. Sci., 2023, 14, 12684–12692 Search PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- K. Kveseth, R. Seip and D. A. Kohl, Acta Chem. Scand., 1980, 34a, 31–42 Search PubMed.
- E. Kryachko and S. Scheiner, J. Phys. Chem. A, 2004, 108, 2527–2535 CrossRef CAS.
- M. E. Squillacote, R. S. Sheridan, O. L. Chapman and F. A. L. Anet, J. Am. Chem. Soc., 1979, 101, 3657–3659 CrossRef CAS.
- M. K. Bera, C. Chakraborty and S. Malik, J. Mater. Chem. C, 2017, 5, 6872–6879 Search PubMed.
- Y. Zhang, H. Mao, W. Xu, J. Shi, Z. Cai, B. Tong and Y. Dong, Chem. Eur. J., 2018, 24, 15965–15977 CrossRef CAS PubMed.
- M. K. Bera, P. Pal and S. Malik, J. Mater. Chem. C, 2020, 8, 788–802 Search PubMed.
- P. Pal, A. Datta, S. Mukherjee, A. Perumal and S. Malik, J. Mater. Chem. C, 2023, 11, 16594–16604 RSC.
- P. Pal, A. Datta, R. Mondal and S. Malik, ACS Appl. Polym. Mater., 2024, 6, 6001–6009 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, synthetic details, X-ray crystallographic details, computational investigation into reaction mechanism, optical properties, HR-MS, and NMR spectra. CCDC 2424364 (3), 2424363 (4), and 2424362 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5cc02428e |
| This journal is © The Royal Society of Chemistry 2025 |