
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHelical ammonium halide framework constituting polar conglomerate crystals of 2-ethylanilinium chloride†
Shohei
Tashiro

Department of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: tashiro@chem.s.u-tokyo.ac.jp
First published on 27th December 2024
Abstract
Crystal engineering, which periodically aligns supramolecular synthons through intermolecular interactions, is a key technology for creating a variety of functional crystalline materials such as porous, polar, chiral, and elastic/plastic crystals. Organic ammonium halides, composed of readily available amines and hydrogen halides, are typical crystalline framework motifs called long-range synthon aufbau modules. In contrast to ammonium halide frameworks such as ladder and sheet structures, chiral helical frameworks have received limited attention, despite their significant potential to exhibit useful functions owing to their unique structures. Herein, the facile synthesis of chiral and polar fibrous crystals with space group P32 by neutralizing 2-ethylaniline with hydrogen chloride is reported. Single-crystal X-ray diffraction reveals that the resultant 2-ethylanilinium chloride formed a helical framework through consecutive hydrogen bonding between NH3+ and Cl− moieties along the fiber axis, which presents helical arrays of 2-ethylanilinium moieties with the same handedness. The gas adsorption isotherms suggest that the crystals are essentially nonporous, whereas molecular hydrogen is slightly adsorbed. The fibrous crystals are elastic to some extent, which is consistent with the noncovalent packing of helices perpendicular to the fiber axis. Furthermore, the preparation of mixed crystals of 2-ethylaniline and other 2-substituted anilines is demonstrated. The study promotes the rational design of tunable crystalline materials based on helical frameworks of polar conglomerate fibrous crystals.
Introduction
Crystal engineering is a fundamental technology for the design of supramolecular materials,1,2 which periodically align supramolecular synthons through intermolecular interactions. A variety of supramolecular synthons, including short- and long-range modules, have been utilized to construct distinctive crystals exhibiting multiple functions.3,4 The most typical example is porous crystals, which are used in storage, separation, ion conductance, and catalysis.5,6 Crystals belonging to non-centrosymmetric space groups are known to potentially exhibit piezoelectricity and second harmonic generation, and among them, polar crystals exhibit pyroelectricity.7,8 The modulation of mechanical properties such as elasticity and plasticity is also attracting significant attention in crystal engineering.9,10 Therefore, the development and discovery of novel supramolecular synthons continues to be a critical focus in materials science.Among a variety of intermolecular interactions that constitute the synthons, hydrogen bonding is extensively utilized in crystal engineering.11–16 For instance, the carboxylic acid dimer is a typical example of supramolecular synthons; it is utilized to construct various porous structures such as hydrogen-bonded organic frameworks.17–20 Charge-assisted hydrogen bonding is another effective synthon for designing porous organic salts exhibiting high stability.21–23 In addition to porous structures, designing helical frameworks is an appealing objective for providing chiral functionality to crystalline materials.24–26 In such structures, organic moieties are helically connected through hydrogen bonding, and homochiral helical arrangement is realized with chiral components.26–28 In contrast, even achiral molecules may occasionally arrange helically through spontaneous resolution to form chiral crystal structures with enantiomorphic space groups.29–32
Consecutive synthons, called long-range synthon aufbau modules, are important structural motifs for designing hierarchical crystal structures.33 Organic ammonium halides, comprising charge-assisted hydrogen bonds, are excellent candidates for such consecutive synthons.34–36 Because they are composed of protonated amines and their counter halides, they can be easily prepared with readily available or commercial amines. One prominent example is diisopropylammonium halides, which form networked polar crystals with excellent ferroelectric properties.37–39 However, identifying new structural motifs of ammonium halides using simple organic amines remains a challenging task. The structures of organic ammonium halide frameworks have been comprehensively summarized in previous reviews by Bond, wherein ladder, sheet, and ring-stacking structures have been primarily obtained (Fig. 1).40–42 Other unique structures, such as cubane-type clusters, have also been reported;43 however, examples of helical framework crystals with enantiomorphic space groups are very limited and present scope for new findings.42,44
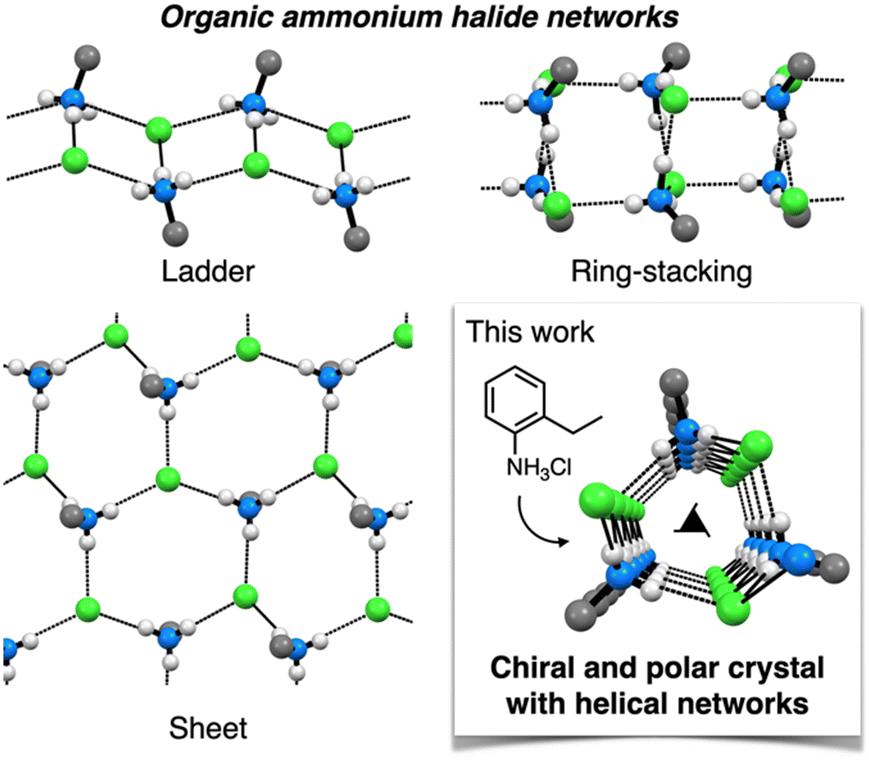 | ||
| Fig. 1 Schematic of the conventional organic ammonium halide frameworks such as ladder, sheet, and ring-stacking structure, and an unconventional helical framework of 1 revealed in this study. Hydrogen bonds are represented as doted lines. Color code: grey C, white H, blue N, green Cl. | ||
This paper reports that a helical ammonium halide framework can be easily synthesized from simple commercial 2-ethylaniline (Fig. 1). When neutralized with hydrogen chloride, 2-ethylaniline forms fibrous polar crystals with space group P32 as a conglomerate. X-ray crystallographic analysis revealed that NH3+ moieties and Cl− anions of 2-ethylanilinium chloride (1) constructed a consecutive helical framework along the fiber axis with the same handedness. The novelty of the helical framework was verified on the basis of literature and database surveys. The crystal structure of 1 was also investigated through gas adsorption analysis and on the basis of the elastic nature of the crystal. The formation of mixed crystals with other 2-substituted anilines was also investigated. The significance of this study is not only the novelty of the crystal structure, but also the finding of a method for easily preparing polar and chiral crystals through self-assembly of a simple ammonium salt into a helical network structure. Therefore, the results of this study extend the possibilities of using helical ammonium halide frameworks as functional crystalline materials.
Experimental section
Materials and methods
Commercially available solvents and organic and inorganic reagents were used without further purification.Single-crystal X-ray diffraction (ScXRD) was performed using a Rigaku XtaLAB P200 diffractometer with CuKα radiation. The obtained data were analyzed using the Olex2 crystallographic software package,45 while refinement was performed using the SHELXL-2015 program suite.46 Hydrogen atoms were placed at the calculated positions with AFIX instructions and refined using a riding model. X-ray structures were displayed using the Mercury47 or PyMOL programs. Hirshfeld surface analysis was conducted using CrystalExplorer (version 17.5).48
Nuclear magnetic resonance (NMR) spectroscopy was performed using a Bruker AVANCE 500 spectrometer (500 MHz for 1H). Chemical shifts are reported in parts per million (ppm) on the δ scale and referenced to dimethyl sulfoxide (DMSO)-d5 (δ = 2.50 ppm for 1H NMR) in DMSO-d6.
Fourier-transform infrared (FTIR) spectra were recorded on a JASCO FT/IR-4200 spectrometer using the ZnSe-attenuated total reflection (ATR) method.
Diffuse reflectance UV-visible (DRUV-vis) absorption spectroscopy was performed using a JASCO V-770 spectrometer with an integrating sphere attachment.
Elemental analysis was conducted at the Microanalytical Laboratory, Department of Chemistry, School of Science, The University of Tokyo, using a Vario MICRO Cube elemental analyzer with the addition of MgO. Calibration curves were created, and analytical values were calculated in Excel.
Thermogravimetry-differential thermal analysis (TG-DTA) was performed using a Rigaku Thermoplus EVO2 TG-DTA8122 thermal analyzer under a N2 flow at a heating rate of 2 °C min−1 in a temperature range (30–170 °C).
Powder X-ray diffraction (PXRD) was conducted using a Rigaku Miniflex 600 diffractometer with CuKα radiation at room temperature (25–28 °C).
The isotherms of N2, CO2, and H2 on the crystals of 1 were recorded using a MICROTRAC BELSORP MINI X (under ambient pressure) or BELSORP HP (under high pressure) analyzer, which uses the volumetric adsorption method to calculate the quantity of the adsorbed gas by measuring vapor pressures. Prior to each analysis, the samples were activated using BELPREP VAC II apparatus in a vacuum at 50 °C for 12 h and then weighed (0.1911, 0.1624, or 0.1548 g for N2, CO2, or H2 isotherms under atmospheric pressure, respectively, and 0.2203 or 0.3012 g for H2 isotherms at high pressures of 77 or 298 K, respectively). Dried N2 (99.99995%), CO2 (99.995%), and H2 (99.99999%) were used for the analyses.
Preparation of 1
An ethanol solution of hydrogen chloride (1.0 mol L−1, 2.6 mL, 2.6 mmol) was carefully added to 2-ethylaniline (310 mg, 2.56 mmol). Diethyl ether was added to the mixture (10–15 mL) until precipitation occurred. After 2 h, the precipitate was collected, washed by filtration, and dried in vacuo for 1 h to create 1 as colorless needles (271 mg, 1.72 mmol, 67%). The crystals for ScXRD analysis were prepared using a similar procedure. Note that similar crystals were also obtained using the vapor diffusion technique with diethyl ether, but the size and quality of the crystals did not improve. IR (ATR, cm−1): 2805 (s), 2717 (w), 2595 (m), 1978 (m), 1625 (w), 1597 (m), 1568 (w), 1525 (w), 1493 (s), 1448 (w), 1136 (m), 795 (w), and 765 (vs). DRUV-vis (5 wt% of 1 with BaSO4, rt): 262 nm. Elemental analysis results (%) for C8H12NCl: C 60.95, H 7.67, N 8.89; however, the following values (%) were obtained: C 60.52, H 7.40, N 8.91. The slight deviation of C was probably due to the adsorption of a very small amount of water on the crystal surface: calcd (%) for C8H12NCl·(H2O)0.01: C 60.88, H 7.68, N 8.88.Crystal data for 1: C8H12ClN, Fw = 157.64, crystal dimensions 0.176 × 0.046 × 0.029 mm3, trigonal, space group P32, a = 21.7201(11), c = 4.8629(3) Å, V = 1986.8(2) Å3, Z = 9, ρcalcd = 1.186 g cm−3, μ = 3.234 cm−1, T = 93 K, λ(CuKα) = 1.54184 Å, 2θmax = 133.098°, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 699/4608 reflections collected/unique (Rint = 0.0793), R1 = 0.0526 (I > 2σ(I)), wR2 = 0.1310 (for all data), GOF = 0.979, largest diff. peak and hole 0.298/−0.252 e Å−3. Flack parameter = −0.02(2). The structure was refined as a two-component twin with twin law [0 −1 0 −1 0 0 0 0 −1] and domain fractions of 0.620(3) and 0.380(3), respectively. CCDC deposit number 2386826.
699/4608 reflections collected/unique (Rint = 0.0793), R1 = 0.0526 (I > 2σ(I)), wR2 = 0.1310 (for all data), GOF = 0.979, largest diff. peak and hole 0.298/−0.252 e Å−3. Flack parameter = −0.02(2). The structure was refined as a two-component twin with twin law [0 −1 0 −1 0 0 0 0 −1] and domain fractions of 0.620(3) and 0.380(3), respectively. CCDC deposit number 2386826.
Preparation of 2-ethylanilinium bromide
An ethanol solution of hydrogen bromide (1.2–2.5 mol L−1, 0.5 mL, 0.60–1.3 mmol) was carefully added to 2-ethylaniline (121 mg, 1.0 mmol). Diethyl ether was added to the mixture (15–20 mL) until precipitation occurred. After several minutes, the precipitate was collected, washed by filtration, and dried in vacuo to create 2-ethylanilinium bromide as colorless blocks (95 mg, 0.47 mmol, 47%).Reaction of 2-ethylaniline and hydroiodic acid
Hydroiodic acid (6.9–7.3 mol L−1, 0.14 mL, 0.97–1.0 mmol) was carefully added to 2-ethylaniline (120 mg, 1.0 mmol). Diethyl ether was then added to the mixture (ca. 40 mL). However, no precipitation occurred.Results and discussion
The crystals of 1 were prepared by reprecipitation in a poor solvent. Equimolar amounts of 2-ethylaniline and an ethanolic solution of hydrogen chloride were mixed to prepare a solution of the hydrochloride salt, which was then diluted with an excess amount of diethyl ether at room temperature (25–28 °C).49 The precipitate immediately formed was collected, washed by filtration, and then dried in vacuo to give 1 as colorless fibrous crystals (Fig. 2a), which were insoluble in nonpolar solvents such as n-hexane, ethyl acetate, and toluene.50 The products were characterized by FTIR and DRUV-vis spectroscopy and elemental analysis. In addition, TG-DTA results indicated that the crystals begin to decompose at approximately 90 °C. The phase purity of the crystalline product was examined by PXRD, and its high purity was confirmed by comparison with the simulated PXRD pattern of the crystal structure described below (Fig. 2b).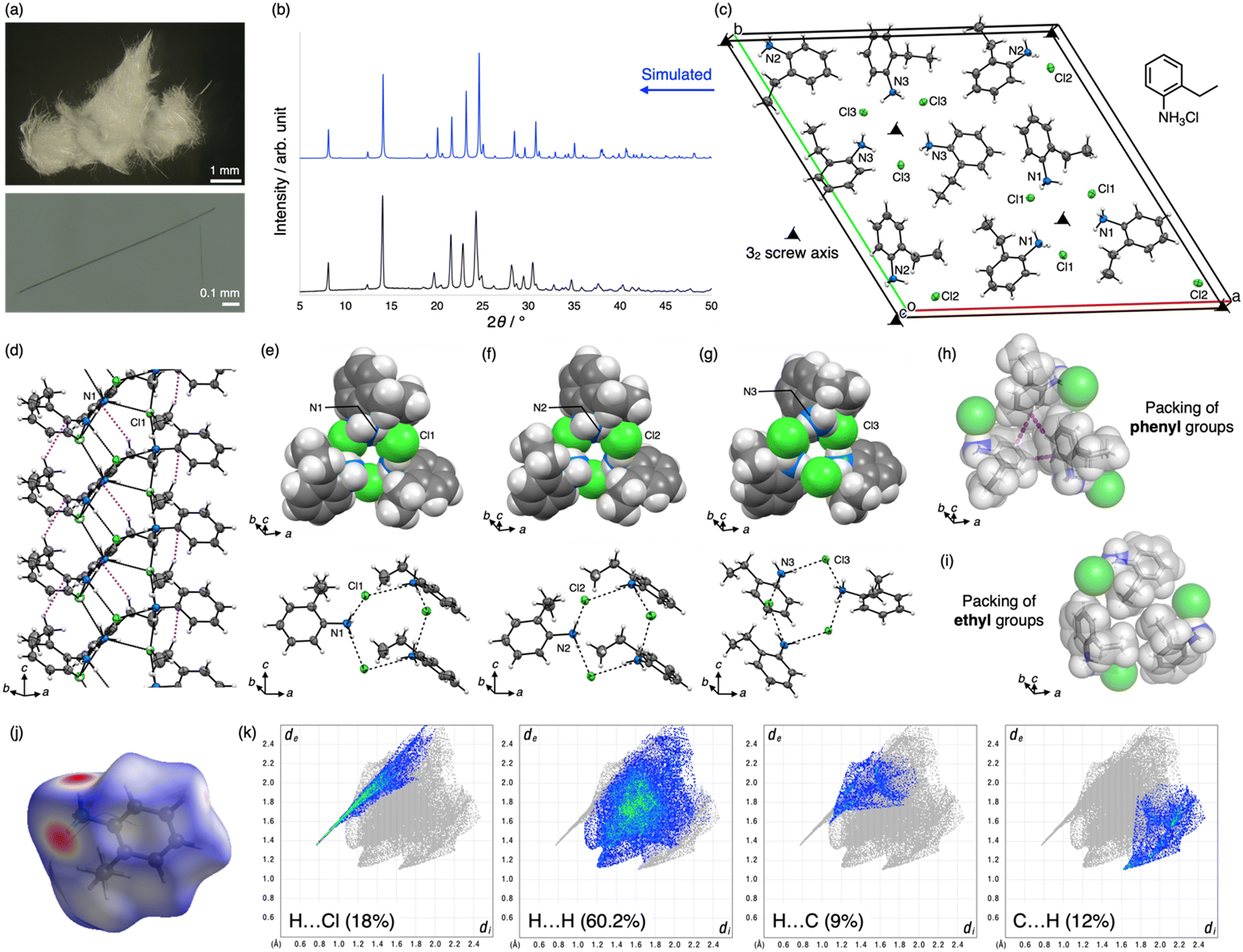 | ||
| Fig. 2 Helical ammonium halide framework of 1. (a) Photographs of the fibrous crystals of 1. (b) PXRD pattern (CuKα, rt) of the fibrous crystals (bottom) and a simulated pattern of the helical framework structure determined by ScXRD (top). (c) Unit-cell structure of 1. (d) Side view of a helix composed of 2-ethylanilinium moieties with N1 and Cl1 atoms. (e)–(g) Top view of the helices and hexagonal hydrogen bonding units composed of N1 and Cl1, N2 and Cl2, and N3 and Cl3 atoms. (h) and (i) Packing structures of phenyl or ethyl parts in the crystal structure. Hydrogen bonds and CH–π interactions are represented as black and purple dots, respectively. Color code: grey C, white H, blue N, green Cl. (j) Hirshfeld surface, mapped with dnorm, of the 2-ethylanilinium cation with the N1 atom, and (k) its 2D fingerprint plots, where the di and de values are the closest internal and external distances from given points on the Hirshfeld surface. | ||
The fibrous crystals of 1, which often tend to form merohedral twins, were structurally analyzed by ScXRD to reveal that the fibrous crystal of 1 belongs to space group P32 and comprises nine molecules of 1 in the unit cell. Notably, the three molecules of 1 in the asymmetric unit, labeled N1, Cl1, N2, Cl2, N3, and Cl3, form individual infinite helices around 32 screw axes (Fig. 2c). The helices comprise a core part of NH3+ and Cl− moieties, which form consecutive hydrogen bond networks, and a shell part of 2-ethylphenyl groups, which stack through CH–π interactions between methylene and phenyl moieties (Fig. 2d). In the core, NH3+ and Cl− moieties make three hydrogen bonds with each other (N⋯Cl distances are 3.07(1)–3.176(8) Å); thus, both moieties are regarded as 3-connected nodes in the network and form a (3,3)-connected net with a Schläfli symbol of {6,3} (Fig. 2d). Therefore, the unit of the network is a hexagon with alternate arrangements of NH3+ and Cl− moieties although the positions of the moieties are reversed in helices with N3 and Cl3 atoms, and the hexagonal shape is deformed with the right side raised when viewed from outside the helices (Fig. 2e–g). As a result, the deformed hexagons were helically aligned around the 32 screw axes to form chiral helical frameworks, which were surrounded by a shell of helically arranged 2-ethylphenyl moieties. Next, focusing on the structure of the shells, all ethyl groups facing upward were oriented in a clockwise direction when viewed from the [001] direction, indicating that their helical handedness was the same in all helices.
The shells of each helix interact with each other through noncovalent interactions such as CH–π interactions and van der Waals (vdW) contacts to constitute the crystal structure. For instance, three helices are interlocked with each other through interhelical CH–π interactions among phenyl moieties (Fig. 2h). In addition, the ethyl groups of the three helices are packed via vdW contacts (Fig. 2i). The importance of noncovalent interactions is also supported by the Hirshfeld surface analysis (Fig. 2j and k).48 Pores are not formed among the helices owing to the dense packing of helices. By contrast, very narrow tubular voids are found within the helices when a smaller probe radius (0.7 Å) is applied in the void analysis on the Mercury program (Fig. 3a).47
 | ||
| Fig. 3 Gas adsorption properties of 1. (a) Voids in the unit cell visualized using a Mercury program with a probe radius of 0.7 Å. (b) N2 adsorption isotherms for crystals of 1 at 77 K. (c) H2 and CO2 adsorption isotherms for the crystals at 77 K and 298 K, respectively. Full and open symbols represent adsorption and desorption, respectively. | ||
Belonging to enantiomorphic space group P32 with a significantly low Frack parameter (−0.02(2)), the single crystals of 1 do not contain any enantiomers of the helices. In contrast, the crystal structure was refined as a merohedral twin, as mentioned above, using twin law [0 −1 0 −1 0 0 0 0 −1] (domain fractions of 0.620(3) and 0.380(3)), indicating that the other domain was composed of helical enantiomers. This result is reasonable because no chiral sources or materials were provided during crystallization, and it is also suggested that the sample was an equimolar mixture of enantiomeric crystals with space group P31 and P32. Since the merohedral twinning of the polar crystal may hinder the measurement of certain physical properties and practical applications, the development of a different crystallization method to solve the twinning will be a future issue.
The Cambridge Structural Database (CSD) was examined using ConQuest Version 2024.1.0 to confirm the novelty of the crystal structure.51 For instance, five previous structures of 2-ethylanilinium salts were found in the database; however, they were not halides but oligophosphates or carboxylate salts with different hydrogen bonding networks (refcodes BEFYAN, BIJSIX, UCEJES, OZIFOU, and LIGLEU).52–56 By contrast, while searching for the same hydrogen bonding networks of NH3+ and X− (X = F, Cl, Br, or I) in the database, only one example of the same network structure composed of (R)-1-(naphthalen-2-yl)but-3-en-1-amine hydrochloride with space group P31 was found (refcode SETREQ). However, the network structure was not discussed because the crystal structure was analyzed to determine the absolute configuration of chiral amines synthesized by catalytic asymmetric reactions.57 Thus, this report marks the first identification of this helical hydrogen bonding framework in the structural series of ammonium halide frameworks.
Furthermore, gas adsorption, which is one of the basic properties of crystalline materials, was investigated. N2 adsorption at 77 K exhibited a type-II isotherm, suggesting that 1 is a nonporous material (Fig. 3b). This result is consistent with the void analysis of 1 using the Mercury program, which showed 0% porosity with a probe radius of 1.2 Å. In contrast to the almost negligible adsorption of CO2, the uptake of H2 (0.02 mmol g−1) at 77 K appears to be slightly higher (Fig. 3c) and increased to 0.2 mmol g−1 under high pressure (500 kPa). This uptake is significantly smaller than that of metal–organic frameworks and other porous materials owing to its very low porosity,58,59 although the narrow pores in 1 possibly contribute to the adsorption of H2 (Fig. 3a).
When one end of a crystal of 1 was pulled with a metal pin in n-hexane, the crystal was observed to be curved (Fig. 4). The crystal returned to its original shape after the force was removed. This procedure is repeated several times. This observation suggested that the crystal of 1 was elastic. A similar reversible deformation of the crystal shape was observed in the dry state although the crystals became slightly more fragile. The observed elasticity can be explained by the crystal structure based on discussions in previous papers.9,60,61 The robust helices made by hydrogen bonding along the c-axis, which is parallel to the fiber axis as determined by face indexing, are packed in parallel through weak vdW and CH–π interactions of ethyl and phenyl groups, respectively, as discussed above. The weak interactions along the fiber axis presumably act as structural buffers against bending of the fibrous crystal. In contrast, the weak interaction columns are interlocked with each other and isotropically distributed perpendicular to the fiber axis, suggesting that the crystal does not have distinct slide planes, which are characteristic of plastic crystals; thus, the crystals of 1 are elastic rather than plastic.
 | ||
| Fig. 4 Bending of a crystal of 1. Photographs of the crystal (a) before and (b) after bending with a metal pin. | ||
One effective way to tune only the properties of an organic crystal without changing the packing structure is to form mixed crystals, i.e., organic alloys.62,63 Therefore, the formation of mixed crystals of 1 with 2-substituted anilines was investigated. When a non-substituted aniline was mixed with 2-ethylaniline under similar crystallization conditions, only physical mixtures of fibrous crystals of 1 and plate crystals of anilinium chloride having a sheet-like framework were obtained,64 as supported by the microscopic observations and PXRD results. In contrast, the formation of mixed crystals, rather than cocrystals, with o-toluidine (2-methylaniline) was suggested when the mixtures of 2-ethylanilinium and o-toluidinium chlorides were crystallized, despite the fact that o-toluidinium chloride itself forms a sheet-like framework.65 The incorporation of o-toluidine into the helical networks of 1 was also supported by the slight shift of the PXRD peaks to the higher angles, which is consistent with the shrinkage of the crystals due to the mixing of the less bulky methyl groups. Furthermore, although it does not crystallize as a chloride, 2-isopropylaniline was incorporated into the helical framework of 1 to form mixed crystals. In contrast to o-toluidine, bulkier 2-isopropylaniline resulted in a shift of the PXRD peak to a lower angle, suggesting the expansion of the crystals (Fig. 5). Notably, the elasticity of the mixed crystals was maintained. These results indicate that the mixed-crystal approach can be utilized to tune the properties of 1, while maintaining its helical structure.
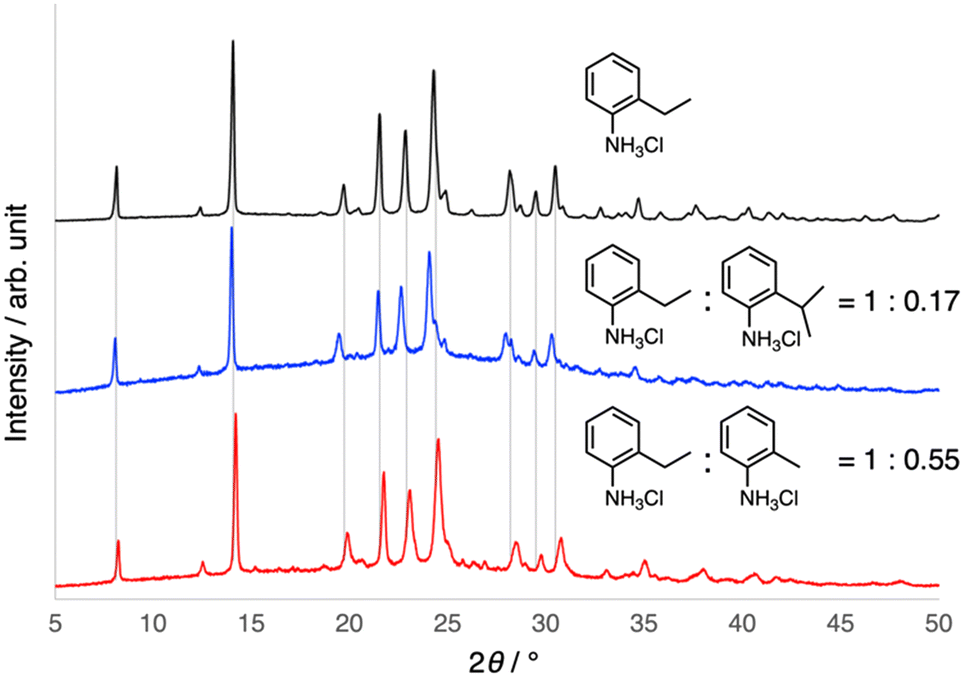 | ||
| Fig. 5 Formation of mixed crystals with 2-substituted anilinium chlorides. PXRD patterns (CuKα, rt) of 1 (top) and mixed crystals with o-toluidinium chloride (bottom) and 2-isopropylanilinium chloride (middle) with molar ratios shown in the figure, which is estimated through 1H NMR analysis of the crystals dissolved in DMSO-d6. | ||
Conclusions
A readily and commercially available organic amine, 2-ethylaniline, reacted with equimolar HCl to form fibrous crystals with enantiomorphic space group P32 (P31), which comprises a continuous helical hydrogen bonding framework along the fiber axis. Databases and literature surveys indicated that this crystal structure has not been reported to date despite its very simple components. The resultant helical network structure is a new structural series of organic ammonium halides. In addition, the fibrous crystals are elastic and can be modified by forming mixed crystals with other 2-substituted anilines. As described in the introduction, crystals with polarity and chirality exhibit several functions, which can also be tuned or controlled by elastic deformation and/or mixed crystal formation. Moreover, the helical ammonium halide framework found in this study will be a new structural motif on chiral supramolecular materials such as porous crystals, liquid crystals, and supramolecular polymers. Therefore, this study, which showed that such a structure can be prepared through a very simple operation of neutralizing a commercial amine, will contribute to the development of crystal engineering and materials science in the future.Data availability
Data supporting this study has been included in the ESI.† Crystallographic data for 1 has been deposited at the Cambridge Crystallographic Data Centre under CCDC no. 2386826 and can be obtained from https://www.ccdc.cam.ac.uk.Author contributions
S. T.: conceptualization, data curation, formal analysis, funding acquisition, investigation, project administration, resources, validation, visualization, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I acknowledge the financial support from JST PRESTO (grant number JPMJPR22A8). I would also like to thank Ms. Ai Inoue for preparing the mixed crystals and Dr. Motoharu Nakano and MicrotracBEL Corp. for gas adsorption measurements and helpful discussions. We would like to thank Editage (http://www.editage.jp) for English language editing.Notes and references
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef.
- M. Köberl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Dalton Trans., 2011, 40, 6834–6859 RSC.
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef PubMed.
- K. M. Ok, E. O. Chi and P. S. Halasyamani, Chem. Soc. Rev., 2006, 35, 710–717 RSC.
- D. Zhang, H. Wu, C. R. Bowen and Y. Yang, Small, 2021, 17, 2103960 CrossRef.
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957–2967 CrossRef PubMed.
- T. Seki, N. Hoshino, Y. Suzuki and S. Hayashi, CrystEngComm, 2021, 23, 5686–5696 RSC.
- P. Brunet, M. Simard and J. D. Wuest, J. Am. Chem. Soc., 1997, 119, 2737–2738 CrossRef.
- Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef PubMed.
- C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- S. Subramanian and M. J. Zaworotko, Coord. Chem. Rev., 1994, 137, 357–401 CrossRef.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef PubMed.
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef PubMed.
- D. J. Duchamp and R. E. Marsh, Acta Crystallogr., Sect. B, 1969, 25, 5–19 CrossRef.
- O. Ermer, J. Am. Chem. Soc., 1988, 110, 3747–3754 CrossRef.
- I. Hisaki, S. Nakagawa, N. Tohnai and M. Miyata, Angew. Chem., Int. Ed., 2015, 54, 3008–3012 CrossRef PubMed.
- P. Li, P. Li, M. R. Ryder, Z. Liu, C. L. Stern, O. K. Farha and J. F. Stoddart, Angew. Chem., Int. Ed., 2019, 58, 1664–1669 CrossRef.
- V. A. Russell, C. C. Evans, W. Li and M. D. Ward, Science, 1997, 276, 575–579 CrossRef.
- A. Yamamoto, T. Hamada, I. Hisaki, M. Miyata and N. Tohnai, Angew. Chem., Int. Ed., 2013, 52, 1709–1712 CrossRef.
- G. Xing, D. Peng and T. Ben, Chem. Soc. Rev., 2024, 53, 1495–1513 RSC.
- P. Vishweshwar, R. Thaimattam, M. Jaskólski and G. R. Desiraju, Chem. Commun., 2002, 1830–1831 RSC.
- I. Hisaki, T. Sasaki, N. Tohnai and M. Miyata, Chem. – Eur. J., 2012, 18, 10066–10073 CrossRef.
- R. S. Giri and B. Mandal, CrystEngComm, 2022, 24, 10–32 RSC.
- P. Grosshans, A. Jouaiti, V. Bulach, J.-M. Planeix, M. W. Hosseini and J.-F. Nicoud, CrystEngComm, 2003, 5, 414–416 RSC.
- T. Friščič, L. Fábián, J. C. Burley, W. Jones and W. D. S. Motherwell, Chem. Commun., 2006, 5009–5011 RSC.
- W. Jaunky, M. W. Hosseini, J. M. Planeix, A. De Cian, N. Kyritsakas and J. Fischer, Chem. Commun., 1999, 2313–2314 RSC.
- I. Azumaya, D. Uchida, T. Kato, A. Yokoyama, A. Tanatani, H. Takayanagi and T. Yokozawa, Angew. Chem., Int. Ed., 2004, 43, 1360–1363 CrossRef PubMed.
- S. Mehrparvar, C. Wölper and G. Haberhauer, Angew. Chem., Int. Ed., 2023, 62, e202304202 CrossRef PubMed.
- J. Jacques, A. Collet and S. H. Wilen, Enantiomers, Racemates, and Resolutions, Wiley, New York, 1994 Search PubMed.
- P. Ganguly and G. R. Desiraju, CrystEngComm, 2010, 12, 817–833 RSC.
- S. Feng, Q. Huang, S. Yang, Z. Lin and Q. Ling, Chem. Sci., 2021, 12, 14451–14458 RSC.
- W. Lv, Y. Cai, Y. Wang, C. Sun, X. Chen, S. Liu, C. Han, Y. Tang, L. Xu, D. Lin and R. Chen, Chem. Mater., 2024, 36, 7929–7939 Search PubMed.
- M. Owczarek, A. Miniewicz, P. Szklarz and R. Jakubas, Chem. Phys. Lett., 2016, 665, 31–35 CrossRef.
- D.-W. Fu, W. Zhang, H.-L. Cai, J.-Z. Ge, Y. Zhang and R.-G. Xiong, Adv. Mater., 2011, 23, 5658–5662 CrossRef PubMed.
- D.-W. Fu, H.-L. Cai, Y. Liu, Q. Ye, W. Zhang, Y. Zhang, X.-Y. Chen, G. Giovannetti, M. Capone, J. Li and R.-G. Xiong, Science, 2013, 339, 425–428 CrossRef PubMed.
- A. Piecha, A. Gągor, R. Jakubas and P. Szklarz, CrystEngComm, 2013, 15, 940–944 RSC.
- A. D. Bond, Chem. – Eur. J., 2004, 10, 1885–1898 CrossRef.
- A. D. Bond, Cryst. Growth Des., 2005, 5, 755–771 CrossRef.
- A. D. Bond, Coord. Chem. Rev., 2005, 249, 2035–2055 CrossRef.
- A. D. Bond and E. L. Doyle, Chem. Commun., 2003, 2324–2325 RSC.
- J. Weinstock, H.-J. Oh, C. W. DeBrosse, D. S. Eggleston, M. Wise, K. E. Flaim, G. W. Gessner, J. L. Sawyer and C. Kaiser, J. Med. Chem., 1987, 30, 1303–1308 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- In a similar reaction using an ethanol solution of HBr, polycrystalline blocks were obtained instead of fibrous crystals. PXRD analysis of crystals obtained from different crystallization batches showed two different peak patterns, due to the formation of polymorphs or pseudopolymorphs. These complicated behaviors are outside the scope of this paper and will be reported elsewhere. In contrast, no precipitation occurred when HI was used as an acid. These results indicate that HCl is a suitable acid for forming this crystal structure.
- The ethanol solution of hydrogen chloride can be replaced with hydrochloric acid to obtain similar needle crystals with the identical crystal structure, although its preparation procedure needed to be modified. See the ESI† for details.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B:Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- S. Akriche and M. Rzaigui, Mater. Res. Bull., 2001, 36, 375–382 CrossRef.
- W. Smirani, C. B. Nasr and M. Rzaigui, Mater. Res. Bull., 2004, 39, 1103–1111 CrossRef.
- H. Hemissi, S. Abid and M. Rzaigui, Phosphorus, Sulfur Silicon Relat. Elem., 2006, 181, 543–551 CrossRef.
- H. Marouani, S. S. Al-Deyab and M. Rzaigui, ISRN Mater. Sci., 2011, 457924 Search PubMed.
- O. Ugono, N. P. Rath and A. M. Beatty, CrystEngComm, 2011, 13, 753–758 RSC.
- S. Gandhi and B. List, Angew. Chem., Int. Ed., 2013, 52, 2573–2576 CrossRef.
- B. Schmitz, U. Müller, N. Trukhan, M. Schubert, G. Férey and M. Hirscher, ChemPhysChem, 2008, 9, 2181–2184 CrossRef PubMed.
- Y. Ming, J. Purewal, D. Liu, A. Sudik, C. Xu, J. Yang, M. Veenstra, K. Rhodes, R. Soltis, J. Warner, M. Gaab, U. Müller and D. J. Siegel, Microporous Mesoporous Mater., 2014, 185, 235–244 CrossRef.
- S. Ghosh and C. M. Reddy, Angew. Chem., Int. Ed., 2012, 51, 10319–10323 CrossRef PubMed.
- S. Ghosh, M. K. Mishra, S. B. Kadambi, U. Ramamurty and G. R. Desiraju, Angew. Chem., Int. Ed., 2015, 54, 2674–2678 CrossRef.
- M. Dabros, P. R. Emery and V. R. Thalladi, Angew. Chem., Int. Ed., 2007, 46, 4132–4135 CrossRef PubMed.
- M. Lusi, CrystEngComm, 2018, 20, 7042–7052 RSC.
- K. M. Anderson, A. E. Goeta, K. S. B. Hancock and J. W. Steed, Chem. Commun., 2006, 2138–2140 RSC.
- P. McArdle, K. Gilligan, D. Cunningham, R. Dark and M. Mahon, CrystEngComm, 2004, 6, 303–309 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data for 2-ethylanilinium chloride and mixed crystals. CCDC 2386826. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org10.1039/d4ce01084a |
| This journal is © The Royal Society of Chemistry 2025 |