
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePharmaceutical salts of venetoclax with dicarboxylic and sulfonic acids: solid-state characterization and dissolution performance†
Tereza
Havlůjová
 a,
Erika
Hriňová
a,
Eliška
Zmeškalová
ac,
Monika
Kučeráková
c,
Luděk
Ridvan
b and
Miroslav
Šoóš
*a
a,
Erika
Hriňová
a,
Eliška
Zmeškalová
ac,
Monika
Kučeráková
c,
Luděk
Ridvan
b and
Miroslav
Šoóš
*a
aDepartment of Chemical Engineering, University of Chemistry and Technology in Prague, Technická 3, 16628, Prague 6, Czech Republic. E-mail: miroslav.soos@vscht.cz
bZentiva k.s, U Kabelovny 130, 10237, Prague 10, Czech Republic
cInstitute of Physics of the Czech Academy of Sciences, Na Slovance 2, 182 00, Prague 8, Czech Republic
First published on 15th February 2025
Abstract
This study focuses on enhancing the aqueous dissolution of venetoclax through salt formation. Venetoclax, a BCS class IV B-cell lymphoma-2-selective inhibitor, exhibits very low solubility and bioavailability. Given its multiple protonable groups, salt formation was explored to improve its dissolution properties. Dicarboxylic and sulfonic acids were selected as counterions for salt screening. Ten salts were synthesized and characterized using powder X-ray diffraction, nuclear magnetic resonance spectroscopy, thermogravimetric analysis, and differential scanning calorimetry. Intrinsic dissolution rate measurements demonstrated that all salts dissolve faster than the parent drug. The crystal structures of venetoclax, venetoclax fumarate, venetoclax oxalate, and venetoclax napsylate, which all solvated with acetonitrile, and unsolvated venetoclax tosylate were elucidated and described.
1. Introduction
Venetoclax is a selective inhibitor of B-cell lymphoma-2 (BCL-2) protein used for the treatment of chronic lymphocytic leukemia, small lymphocytic lymphoma, or acute myeloid leukemia.1 This compound is classified as BCS class IV; therefore, it exhibits low solubility in water and low bioavailability in the human body. The absolute bioavailability of venetoclax is estimated to be only 5.4%.2 This poses a significant challenge for clinical application and pharmaceutical formulation. However, in the venetoclax structure, multiple protonable groups can be found (Fig. 1); therefore, the preparation of salts is a possible route to overcome venetoclax's low aqueous solubility. Due to the aforementioned possible multivalence, the stoichiometric ratio of both components needs to be taken into consideration.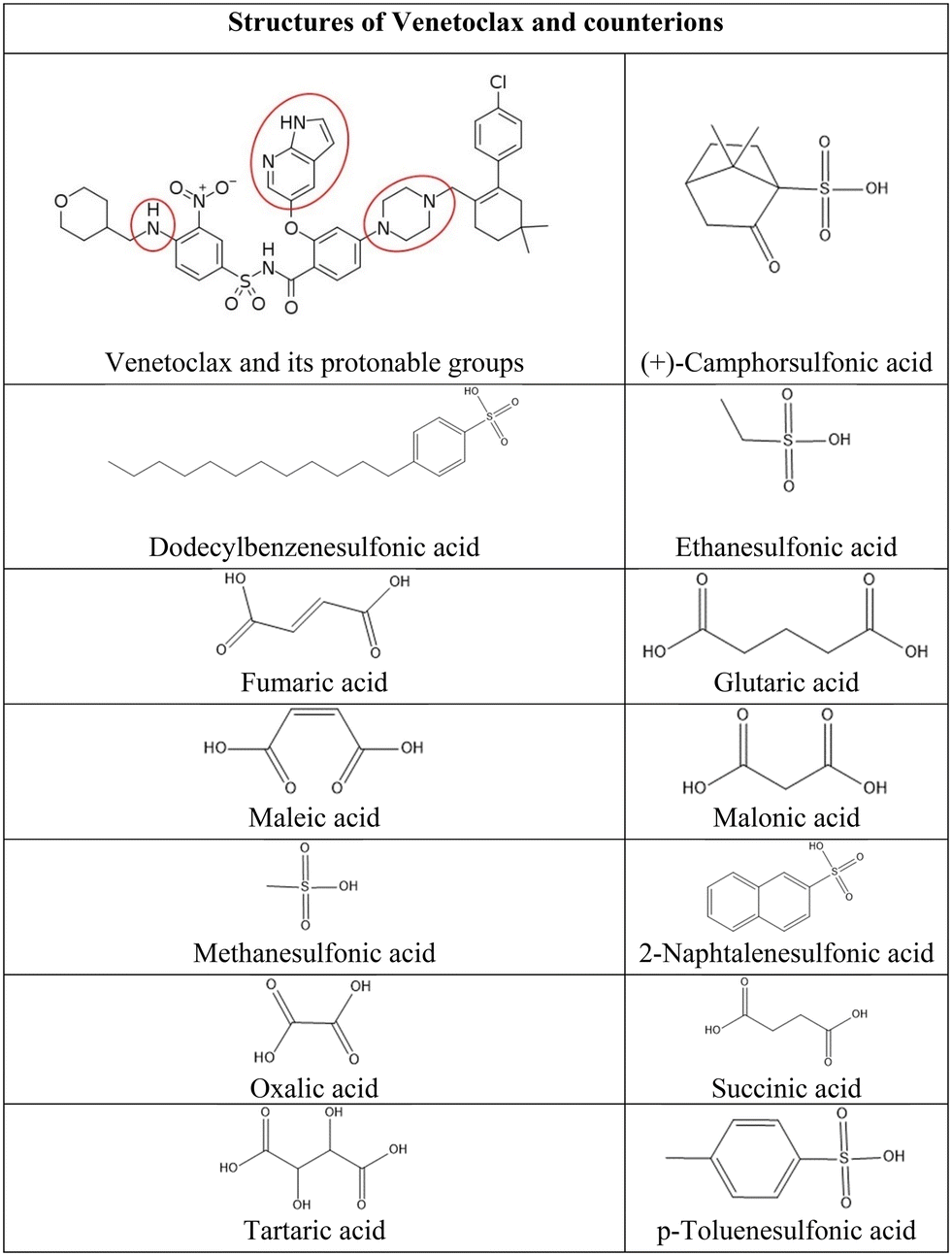 | ||
| Fig. 1 Structures of venetoclax and counterion acids. | ||
Converting active pharmaceutical ingredients (APIs) into salts is a widely used and effective practice for modifying the solubility of molecules containing ionisable functional groups. Salts generally exhibit better solubility than their free parent drug.3–7 The relative increase in intrinsic solubility is reported to be up to 1000 times that of the original compound.8 The drug can pose as either a base or an acid component depending on its functional groups; its partnering counterion is chosen accordingly. To efficiently choose suitable counterions the drug can form salts with, an empirical approach called the pKa rule of salt formation is often employed. The rule is based on the difference in pKa values (ΔpKa) between the acid and the base pair. Generally, a ΔpKa greater than 4 favors salt formation, while a ΔpKa lesser than −1 leans towards co-crystal formation. Intermediate ΔpKa values can lead to either outcome, being influenced by factors such as solvation and crystal packing.9,10 Another limitation in counterion selection is the pharmaceutical suitability as it needs to be a non-toxic agent ideally without any pharmacological effect. Therefore, only a limited number of acids and bases satisfy these criteria. One of the comprehensive lists of such compounds is the Orange Book published by the Food and Drug Administration, which compiles all approved drugs and pharmaceuticals in the USA.11–13 Moreover, this approach has already proved effective for various BSC II and IV drugs. In the case of miconazole and ketoconazole, both BCS II with extremely low solubility, formation of salt with naphthalene disulfonic acid not only led to enhanced dissolution and improved thermal stability but, due to the possibility of lowering the dosage at the same efficacy, also minimized host toxicity.14 Antipsychotic drug olanzapine (BCS II) is also a very good example; it forms salts with a wide range of different counterions. These not only exhibit improved dissolution properties but due to the large quantity of crystallographic data also serve as an interesting platform for examination of intramolecular interactions and molecular packing.15–18 Another interesting example is BCS IV fluoroquinolone ciprofloxacin which forms salts with various dicarboxylic acids without any detrimental effect on its antibacterial activity and, most notably, in glutarate and maleate salt demonstrate solubility improvement over commercially available forms.19,20
After careful consideration of all the possible anionic counterions, we have chosen 13 organic acids due to their safety, stability, propensity to form crystalline salts, and suitable ΔpKa value (Fig. 1). Specifically, dicarboxylic acids were chosen on the basis of their use in pharmaceutical products and FDA approvals.21 Moreover, their multivalence could be favorable in combination with the multiple protonation sites of venetoclax and lead to the formation of salts of various stoichiometries. Sulfonic acids were selected because they have been shown to greatly increase the solubility of drugs with extremely poor aqueous solubility,14,22–26 as is the case of venetoclax.
| API | Counterion | Acetonitrile | |
|---|---|---|---|
| Camsylate | 1 | 2 | — |
| Dodecylbesylate | 1 | 2 | — |
| Esylate | 1 | 2 | — |
| Fumarate | 1 | 1 | — |
| Maleate | 1 | 1 | — |
| Malonate | 1 | 1 | — |
| Mesylate | 1 | 2 | 0.02 |
| Napsylate | 1 | 2 | 0.033 |
| Oxalate | 1 | 1 | — |
| Tosylate | 1 | 2 | — |
| Acetonitrile solvate | 1 | — | 1 |
Information about venetoclax salts is scarce in peer-reviewed literature; only one salt has been reported, namely, a lipophilic venetoclax docusate intended to improve lipid solubility and subsequent lymphatic transport.27 So far, only one crystal structure of venetoclax is known and available in CSD; it is a hydrate.28
2. Materials and methods
2.1. Materials
Venetoclax was provided by Zentiva k.s. Most of the material available to us was amorphous. A small amount was crystalline. Acetonitrile and organic acids were obtained from various suppliers and used as delivered without any modifications.2.2. Salt screening
Thirteen organic acids from two different groups were selected. Sulfonic acids were represented by (+)-camphorsulfonic (camsylate), dodecylbenzenesulfonic (dodecylbesylate), ethanesulfonic (esylate), methanesulfonic (mesylate), 2-naphtalenesulfonic (napsylate), and p-toluenesulfonic (tosylate) acid. Dicarboxylic acids were represented by fumaric (fumarate), glutaric (glutarate), maleic (maleate), malonic (malonate), anhydrous oxalic (oxalate), succinic (succinate), and tartaric (tartrate) acid. 30 mg of venetoclax and 3 molar equivalents of counterion were dissolved in acetonitrile, then the mixture was heated to 70 °C and stirred for 2 hours. The solutions were cooled and stirred overnight. The non-precipitated samples were put into an ice bath and stirred for an additional 2 hours. All resulting suspensions were filtered and dried at 50 °C under reduced pressure. All dried powder samples were then analysed by Raman spectroscopy. The samples displaying shifts when compared to those of API and the corresponding acid were then analysed by nuclear magnetic resonance, differential scanning calorimetry, and powder diffraction.2.3. X-ray powder diffraction (XRPD)
Diffraction patterns were collected using an X'Pert PRO MPD powder diffractometer (PANalytical): X-ray beam Cu Kα (λ = 1.542 Å), measured range: 4–40° 2θ, excitation voltage: 45 kV, anodic current: 40 mA, step size: 0.016° 2θ, time per step 0.3 s. Measurement was carried out on a flat sample with an area/thickness ratio equal to 10/0.5 mm. 2.5° Soller slits with a fixed slit width of 0.6 mm and automatic antiscatter slits were used to correct the primary beam. The irradiated area of the sample was 10 mm. Secondary optics consisted of 2.5° Soller slits and 5.0 mm antiscattering slits to correct the secondary beam. The detector was LYNXEYE_XE_T (1D mode). HighScore Plus software was used to process the diffraction patterns.2.4. Raman spectroscopy
The samples in glass vials were measured in an FTRaman RFS100/S spectrometer with a germanium detector (Bruker Optics, Germany). The wavelength of the Nd:YAG laser was 1064 nm. The measurement range was 4000 to 200 cm−1 with the spectral resolution of 4.0 cm−1. 64 scans were accumulated for a single measurement. The software OMNIC and OPUS were used to process the Raman spectra.2.5. Solution nuclear magnetic resonance (1H NMR)
Solution 1H NMR was used to confirm the stoichiometric ratio of both components as well as to determine the amount of residual solvent. Samples were dissolved in d6-DMSO, and 1H NMR spectra were measured by a Bruker Avance III 500 MHz NMR spectrometer equipped with a Prodigy probe and with a repetition delay of 10 s.2.6. Thermogravimetric analysis (TGA)
The samples were weighed in an aluminium pan (10 mg). All measurements were performed on the TGA 6 instrument (PerkinElmer, USA). The range of investigated temperatures ranged from 20 °C to 300 °C with a heating rate of 10 °C min−1.2.7. Differential scanning calorimetry (DSC)
The samples were weighed in an aluminium pan (10 mg). The pan was covered, and the measurement was carried out under a nitrogen gas flow of 50 ml min−1. All measurements were performed on the TA Instruments Discovery DSC. The range of investigated temperatures ranged from 0 to 300 °C with a heating rate of 10 °C min−1.2.8. Intrinsic dissolution rate (IDR)
Intrinsic dissolution rate (IDR) was determined using a Sirius inForm (Pion Inc., USA) device. IDR discs of 6 mm diameter were prepared by the direct compression of approximately 50 mg of API or venetoclax salt. The material was compressed at a constant load of 100 kg, relaxed for 1 min, and compressed again at the same pressure for another minute. IDR measurements were performed in 50 mL of phosphate buffer solution (pH 6.8) with the addition of 0.1% sodium dodecyl sulphate (SDS) at a stirring speed of 100 rpm. UV spectra were recorded every 30 s using a probe with an optical path length equal to 20 mm. Absorbance between wavelengths of 300–500 nm was used to evaluate the amount of API released at each time point. IDR was obtained from the linear fit of the first 10 min of the measurement. The first three points were excluded because they usually represent the dissolution of the free powder captured on the disc during the preparation. Measurements were done in triplicates.2.9. Single-crystal X-ray diffraction (XRD)
All single crystals were obtained by solvent evaporation under ambient conditions. The vial cap was pierced with a sterile needle with a 1.2 mm diameter to slow down solvent evaporation. Analyses of venetoclax acetonitrile solvate and venetoclax fumarate acetonitrile solvate were performed at 95 K using a SuperNova diffractometer with a microfocus sealed tube, mirror-collimated Cu-Kα radiation (λ = 1.54184) and an Atlas S2 CCD detector. Venetoclax oxalate acetonitrile solvate was measured at 120 K on an Xcalibur Gemini ultra diffractometer using Mo-Kα radiation (λ = 0.71073) from a fine-focus sealed X-ray tube with a graphite monochromator and an Atlas S2 CCD detector. Data reduction and absorption correction were performed with CrysAlisPro software.29 The structures were solved by charge-flipping methods using Superflip software and refined by full-matrix least squares on squared value using Crystals30,31 and Jana2020 software.32 MCE software was used for the visualisation of residual electron density maps.33 All H atoms were placed from the residual electron density map, and the C–H atoms were constrained to ideal geometries. In the structure of venetoclax napsylate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 acetonitrile solvate, the solvent was partially occupied. It was modelled isotropically with an occupancy of 0.5. Structures were compared using the CrystalCMP30,31 software.
:2 acetonitrile solvate, the solvent was partially occupied. It was modelled isotropically with an occupancy of 0.5. Structures were compared using the CrystalCMP30,31 software.
2.10. Molecular electrostatic potential (MEP) calculation
Molecular electrostatic potential (MEP) calculations were performed using the Avogadro software.34 The settings used for generating the MEP surface included selecting the van der Waals surface type and colouring it by electrostatic potential. A medium resolution of 0.18 Å was chosen to balance computational efficiency and surface detail, with the iso value set to 0. This configuration accurately represents the spatial extent of the molecule and provides insights into the charge distribution.3. Results and discussion
The salt screening was done by crystallization of venetoclax together with the respective acid in acetonitrile (described in detail in section 2.2). As recrystallization of just the API could lead to a different polymorph or solvate, it was tested as well. The resulting crystalline sample had a distinct diffraction pattern. Later we will show it to be acetonitrile solvate. For the salt screening evaluation, both the original crystal form and the acetonitrile solvate were used as references for the identification of new salt forms.Precipitation occurred at room temperature for all samples except dodecylbenzenesulfonic acid, which precipitated only after an additional 2 h long stirring in an ice bath. Additional precipitation was observed in camsylate and napsylate samples upon refrigeration. Raman spectroscopy was employed to identify potential salt formation (Fig. S1†); the group frequency region was checked for specific functional groups present in both venetoclax and the corresponding acid and compared to the Raman spectra of each component searching for potential shifts signifying bonding in the sample; the same was done for the fingerprint region. Even though the liquid state of several counterions (dodecylbenzenesulfonic, ethanesulfonic and methanesulfonic acid) and the poor visibility of the sulfonic functional group in Raman spectra complicated this process, Raman spectroscopy was preferred over infrared spectroscopy due to its non-destructive nature. To further confirm the salt formation of samples exhibiting shifts in Raman spectra, a combination of 1H NMR and XRD was employed. The presence of the counterion in the samples was confirmed by 1H solution NMR and the molar ratio of both components was calculated (Table 1). All dicarboxylic salts crystallize in a 1:1 stoichiometry (API:acid), while all sulfonic salts crystallize in the 1:2 ratio (API:acid). In venetoclax mesylate and napsylate traces of residual acetonitrile were detected and the amount was estimated from 1H solution NMR (Table 1). The estimated molar equivalents were then used to calculate the amount of residual solvent for each salt using the daily dose of venetoclax, which is 400 mg.1 This amounts approximately to 0.38 mg of acetonitrile in venetoclax mesylate and 0.62 mg in venetoclax napsylate. After consulting the ICH guidelines for residual solvents, we found that the permitted daily exposure of acetonitrile amounts to 4.1 mg per day; both our salts are well within the permitted limit.35 Therefore, both samples were considered of sufficient quality for further work. The transcript of each spectrum can be found in the ESI.†
Powder diffraction patterns were compared with the starting material as well as venetoclax acetonitrile solvate and the corresponding acid for possible overlap indicating mixture of pure components or different phases in the sample (Fig. 2 and S2 in the ESI†). XRPD confirmed the formation of 10 new crystalline salts with 13 possible counterions. This indicates a high propensity of venetoclax for salt formation.
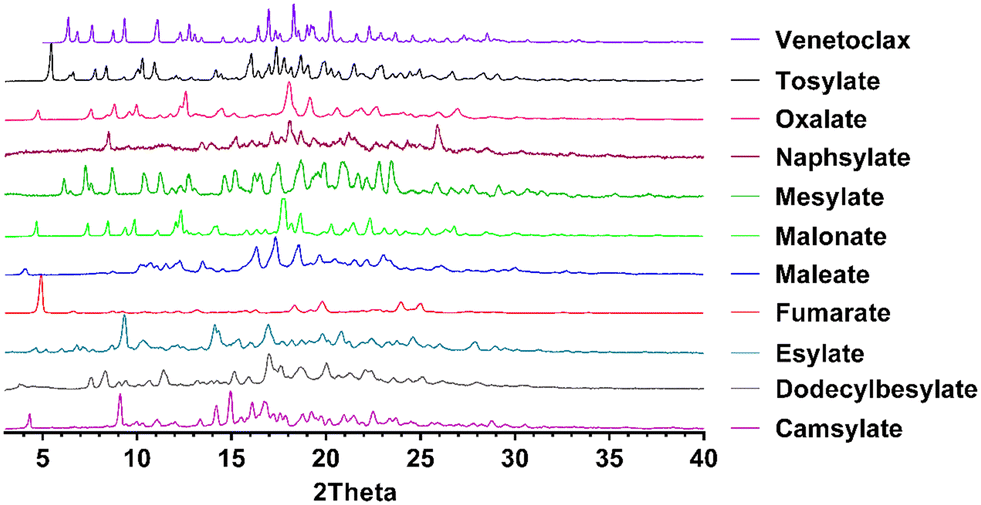 | ||
| Fig. 2 Powder diffraction patterns of novel salts of venetoclax. | ||
To evaluate the thermal behaviour and study the thermodynamic properties of the novel salts, DSC and TGA were performed. From the DSC, we can obtain information about the melting points and any possible solid-form transformations.
The melting point of pure venetoclax is around 139 °C.36 The acetonitrile solvate desolvates in the interval of 100–125 °C with a mass loss of 2.95% and then melts at 153.1 °C (see Fig. 3). As can be seen in Fig. 4, all the prepared salts, except dodecylbesylate (Tm = 109 °C), have a significantly higher melting point, ranging from 165 °C to 226 °C. In general, the dicarboxylic acid salts have higher Tm values than the sulfonic acid salts. There is an exception, though, with tosylate and its Tm of 216 °C. In some samples, a decrease in mass is observed within the interval of 25–100 °C (Fig. 4). As the presence of residual solvent in any significant quantity was already negated by solution 1H NMR, we suspect the presence of sorbed (unbound) water due to the steeper decrease in mass in the 25–50 °C region and, to some extent, the 50–100 °C region (Table S11 in ESI†). The sorbed water can influence stability and even facilitate solid-state changes such as hydrate formation.37,38 Even through our best efforts to mitigate the hygroscopicity of some of the salts (thorough drying and storage in airtight containers), it could not be avoided altogether.
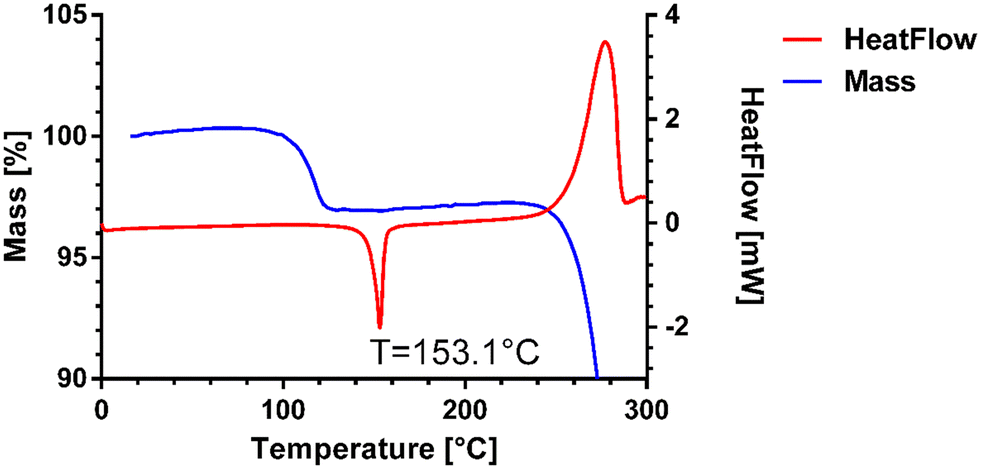 | ||
| Fig. 3 DSC and TGA of venetoclax acetonitrile solvate. | ||
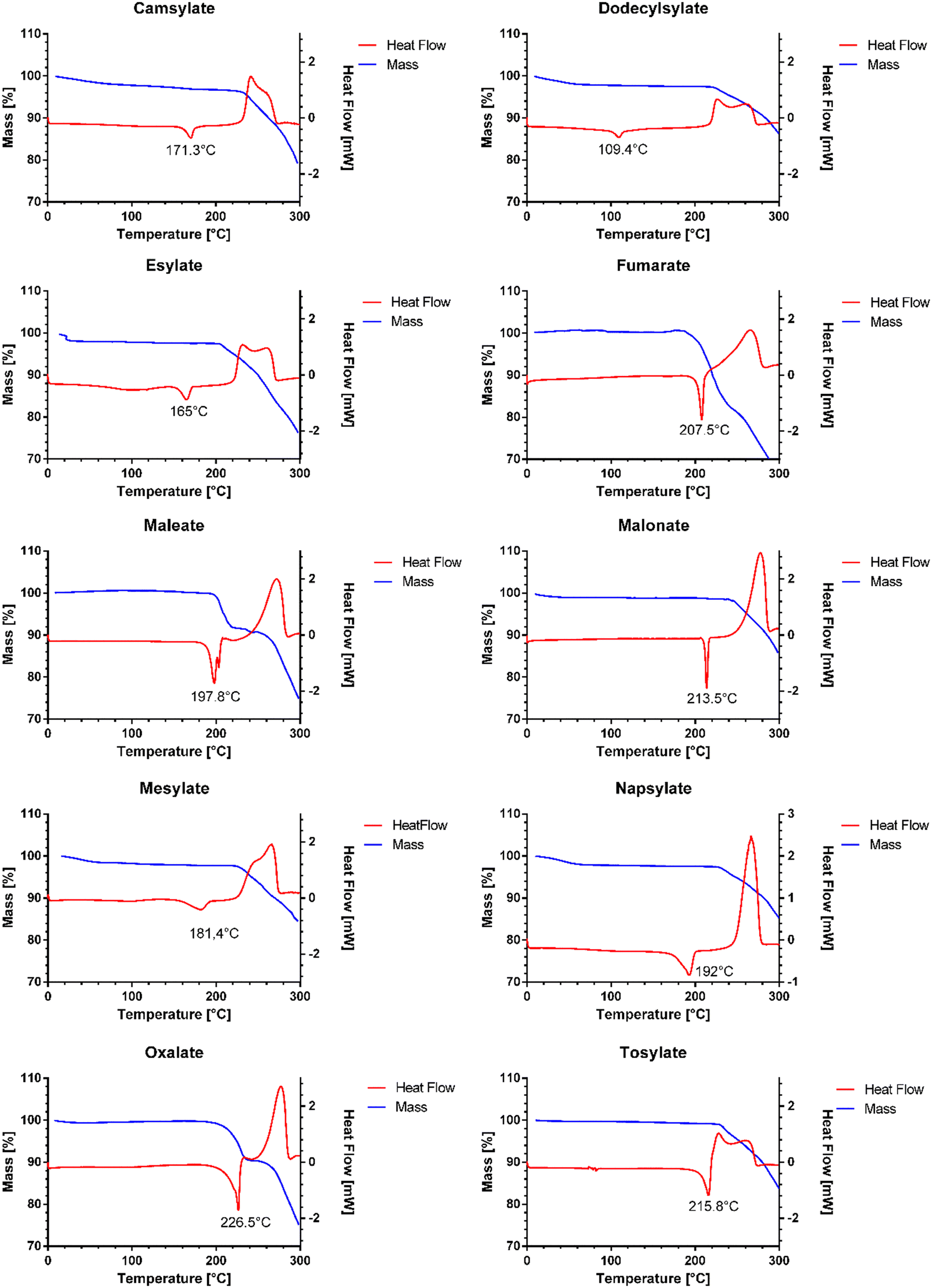 | ||
| Fig. 4 DSC and TGA of new salts. | ||
In some respects, the dissolution properties are the most important characteristic of novel pharmaceutical salts. Acquiring an improvement in the aqueous solubility is the reason for studies like this one. Therefore, we have performed intrinsic dissolution rate (IDR) measurements. For IDR, the powder studied is compressed into a disc with a known surface. Using this type of analysis, we obtain information about the dissolution of the investigated solid form without any potential effect of differing particle sizes. However, as we use UV-vis detection of broad wavelength interval (200 nm) to calculate the concentration and three of our counterions contain an aromatic ring, there is another arising concern, possible overlap with absorption maxima of venetoclax and subsequent false increase in concentration. As such, spectra of venetoclax and all aromatic sulfonic acids were measured, and the concentration of acid solutions was doubled compared to that of venetoclax for better comparability. Upon investigation of these spectra, a slight increase in absorbance was observed in the interval of 300–325 nm of naphtalenesulfonic acid spectra (see Fig. S3 in ESI†), confirming an overlap. However, because of the lower absorbance for longer wavelengths and still fairly low solubility of salts, the interval could not be shortened without the introduction of significant variability as some of the measured values were near the detection limit of the UV-vis probe. Nevertheless, based on good data fit, the effect should be minimal, and the obtained data serve the purpose of characterizing new crystalline solid forms of venetoclax (see Fig. S4 in ESI†).
All of the novel salts have a faster dissolution rate compared to that of the pure API. Seven of the novel salts show only a minor improvement over pure API, increasing the IDR values approximately up to 1.5 times. Three salts, namely fumarate, maleate and napsylate, have a higher dissolution rate by a factor of 2 to 3. The most promising appears to be napsylate with an IDR of 34.13 ± 8.20 μg min−1 cm−2, which is approximately 3 times higher than that of the venetoclax-free base (Fig. 5). One of the reasons for such a high dissolution rate could be the amorphous content in the desolvated salt powders. However, except for the venetoclax acetonitrile solvate and tosylate, none of the venetoclax samples displayed a high level of crystallinity based on their XRPD results.
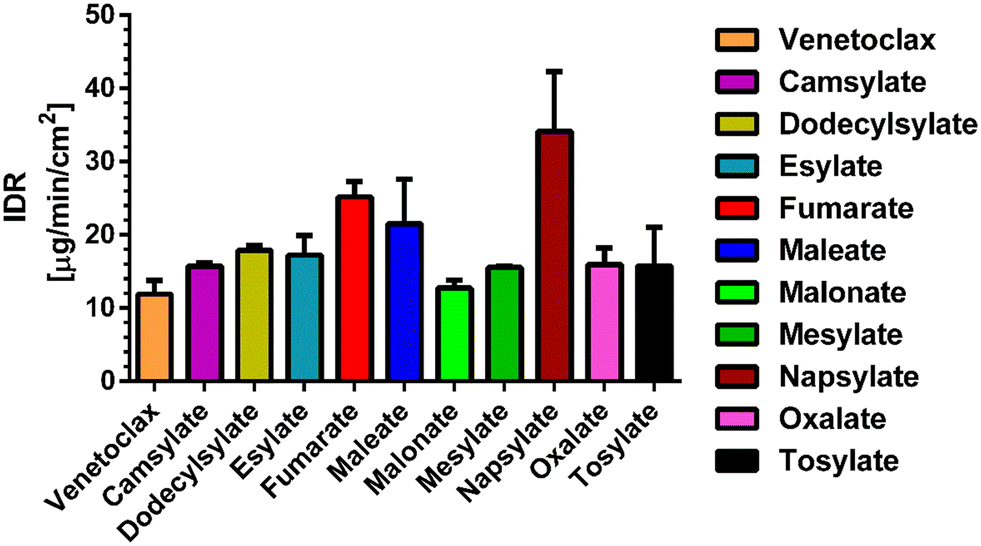 | ||
| Fig. 5 IDR values for venetoclax and all its salts. | ||
We have attempted to find trends to explain the dissolution behaviour of the novel venetoclax salts. In comparison to dicarboxylic and sulfonic salts, there is no clear trend to suggest the superiority of one group over the other. An intuitive negative correlation would be expected with the melting point of the salts. However, Fig. 6 shows no strong trend. It seems that the most successful candidates, dissolution-wise from the IDR point of view, are salts with melting points between 195 and 210 °C. When this temperature increases, a steep decrease in the IDR values can be seen. A similar trend is observed in salts with Tm ranging between 170 °C and 190 °C. Interestingly, salts with melting points below 170 °C exhibit a slight dissolution improvement over their slightly higher melting counterparts.
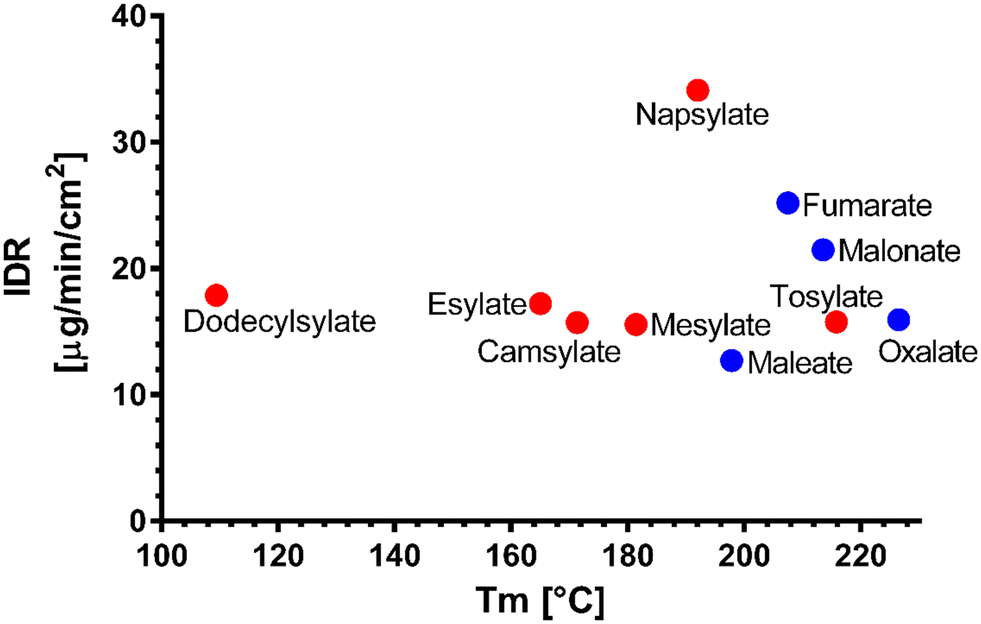 | ||
| Fig. 6 Melting points (Tm) of the novel salts vs. their dissolution IDR; sulfonic salts (red), dicarboxylic salts (blue). | ||
Upon correlation of IDR values of novel salts and Tm values of their parent acids published in the literature, again, no strong trend is observed (Fig. S5†).
This lack of correlation can be explained by examining the work of de Moraes et al.,39 who studied 51 salts of methylephedrine. Although they found some correlations, they were mostly valid only in isostructural salt groups. The crystal structures are ultimately what governs the solid-state properties. To explain the behaviour of the novel salts, it would be best to obtain their crystal structures. Therefore, we have attempted to grow single crystals of these salts. The only single crystals of sufficient quality obtained were of venetoclax acetonitrile solvate, venetoclax fumarate acetonitrile solvate and venetoclax oxalate acetonitrile solvate, venetoclax tosylate and venetoclax napsylate acetonitrile solvate. The structure solution from powder diffraction data is not feasible because of the size and flexibility of venetoclax and the presence of one or two molecular counterions combined with the relatively poor crystallinity of the materials. By visually examining the XRPD patterns, we found that they are all dissimilar, suggesting a significantly different crystal arrangement, which is in line with our findings.
Due to fairly large ΔpKa values (Table 2) we expect the successful hits to be salts, and therefore we refer to them as such. However, to properly distinguish between salt and cocrystal, knowledge of crystal structure is essential. The correct classification is important not only to crystallographers but also to registration authorities.40,41
| pKa value | ΔpKa (strongest basic–strongest acidic) | |
|---|---|---|
| Venetoclax | 8.1 (strongest basic); 3.52 (second strongest basic) | — |
| (+)-Camphorsulfonic acid | −0.81 | 8.91 |
| Dodecylbenzenesulfonic acid | −1.84 | 9.94 |
| Ethanesulfonic acid | −1.3 | 9.4 |
| Fumaric acid | 3.55 (strongest acidic); 4.41 (second strongest acidic) | 4.55 |
| Glutaric acid | 4.46 (strongest acidic); 5.26 (second strongest acidic) | 3.64 |
| Maleic acid | 3.05 (strongest acidic); 5.91 (second strongest acidic) | 5.05 |
| Malonic acid | 2.43 (strongest acidic); 5.92 (second strongest acidic) | 5.67 |
| Methanesulfonic acid | −1.61 | 9.71 |
| 2-Naphtalenesulfonic acid | −2 | 10.1 |
| Anhydrous oxalic acid | 1.36 (strongest acidic); 4.11 (second strongest acidic) | 6.74 |
| Succinic acid | 3.55 (strongest acidic); 5.69 (second strongest acidic) | 4.55 |
| Tartaric acid | 2.72 (strongest acidic); 4.79 (second strongest acidic) | 5.38 |
| p-Toluenesulfonic acid | −2.14 | 10.24 |
Venetoclax acetonitrile solvate crystallises in the triclinic system in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . There is one molecule of API and one molecule of solvent in the asymmetric unit. Fig. S6† shows the asymmetric unit with the numbering of non C/H atoms.
. There is one molecule of API and one molecule of solvent in the asymmetric unit. Fig. S6† shows the asymmetric unit with the numbering of non C/H atoms.
The comparison between the calculated and the experimental XRPD suggests the same crystal structure for both samples (Fig. S7†). This is further confirmed by 1H NMR (transcript in the ESI†) as the calculated molar ratio of venetoclax:acetonitrile is 1:1 as well.
There are three strong H-bonds in the structure, and they are shown in Fig. 7. In both figures, there are two molecules of venetoclax. The amidic N(19)–H forms an intramolecular H-bond with O(42). The other two H-bonds are intermolecular. The amine N(27)–H connects to amidic O(18) and the azaindole N(47)–H forms a H-bond to one of the sulfonyl oxygens, O(22).
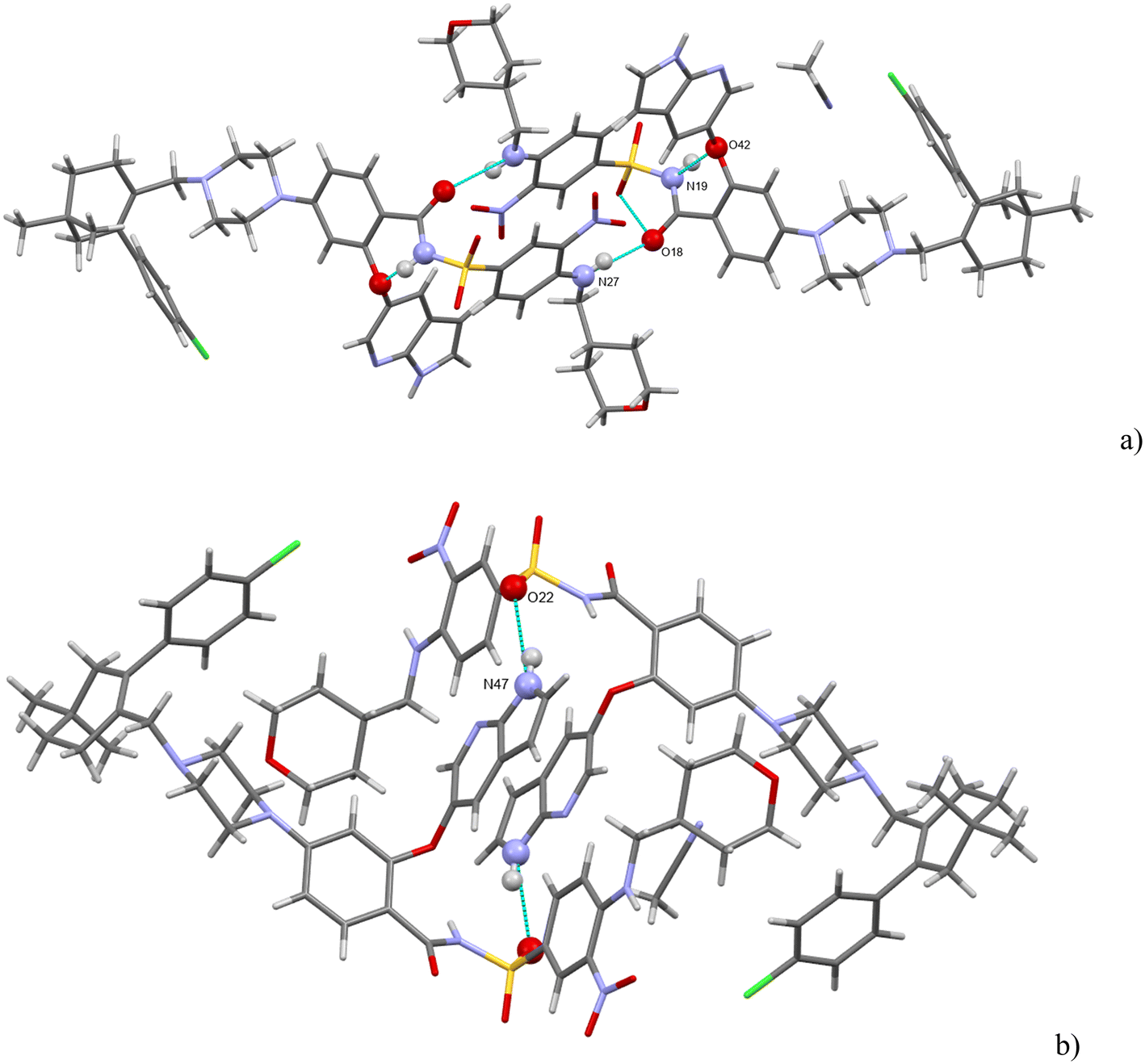 | ||
| Fig. 7 (a and b) The main H-bonding motifs in venetoclax acetonitrile solvate. | ||
Acetonitrile occupies discrete cavities in the structure. It is held together only by multiple weaker interactions like C–H⋯N(62) and C–H⋯N(50). A prominent feature in the molecular packing is the aromatic interaction between the two aromatic regions of the venetoclax molecule. The first (A) is the 7-azaindole (bicyclic functional group containing N47 and N50), and the second (B) is nitrophenyl (the benzene ring to which N37 connects). These form two parallel ribbons (visible in Fig. 8, for example, in the middle of the shown unit cell). These ribbons run parallel to the b axis and are both –A–B–A–B–. The ribbons are slipped with respect to each other, creating a zigzag pattern of the alternating aromatic groups.
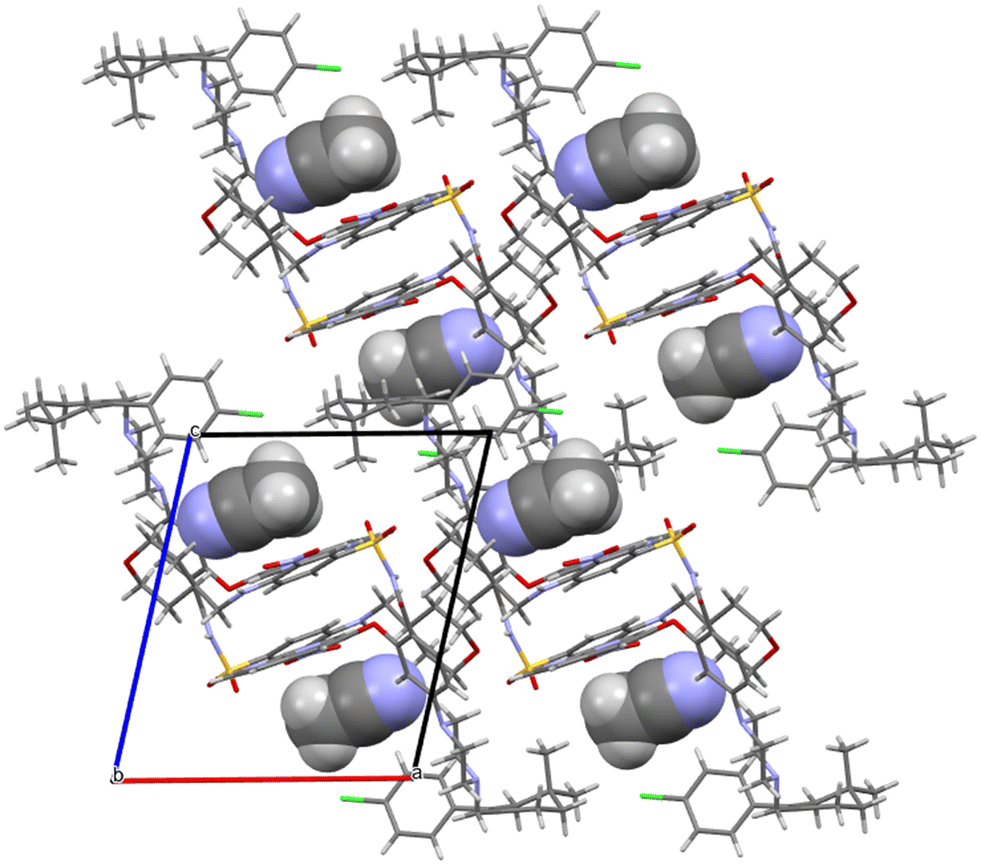 | ||
| Fig. 8 Molecular packing of venetoclax acetonitrile solvate. The solvent is shown in the spacefill mode. | ||
Venetoclax oxalate acetonitrile solvate crystallises in the triclinic system in the space group P. There is one molecule of API, one molecule of the oxalate ion, and one molecule of the solvent in the asymmetric unit. Fig. S8† shows the asymmetric unit with the numbering of non C/H atoms.
H-bonding of venetoclax oxalate acetonitrile solvate is shown in Fig. 9. Six strong interactions can be identified. There is a proton transfer indicating that the material is truly a salt between the oxalate carboxylate O(62) and the API N(9). The other carboxyl of oxalic acid is not deprotonated and forms a self-complementary homosynthon with another molecule of oxalate. There are two intramolecular H-bonds within the API: the amino N(39)–H connects to the adjacent nitro O(51) and the amidic N(31)–H connects to the ether O(16). Cl(1) forms a halogen bond with O(34). The azaindole moiety forms a self-complementary homosynthon N(21)–H⋯N(19).
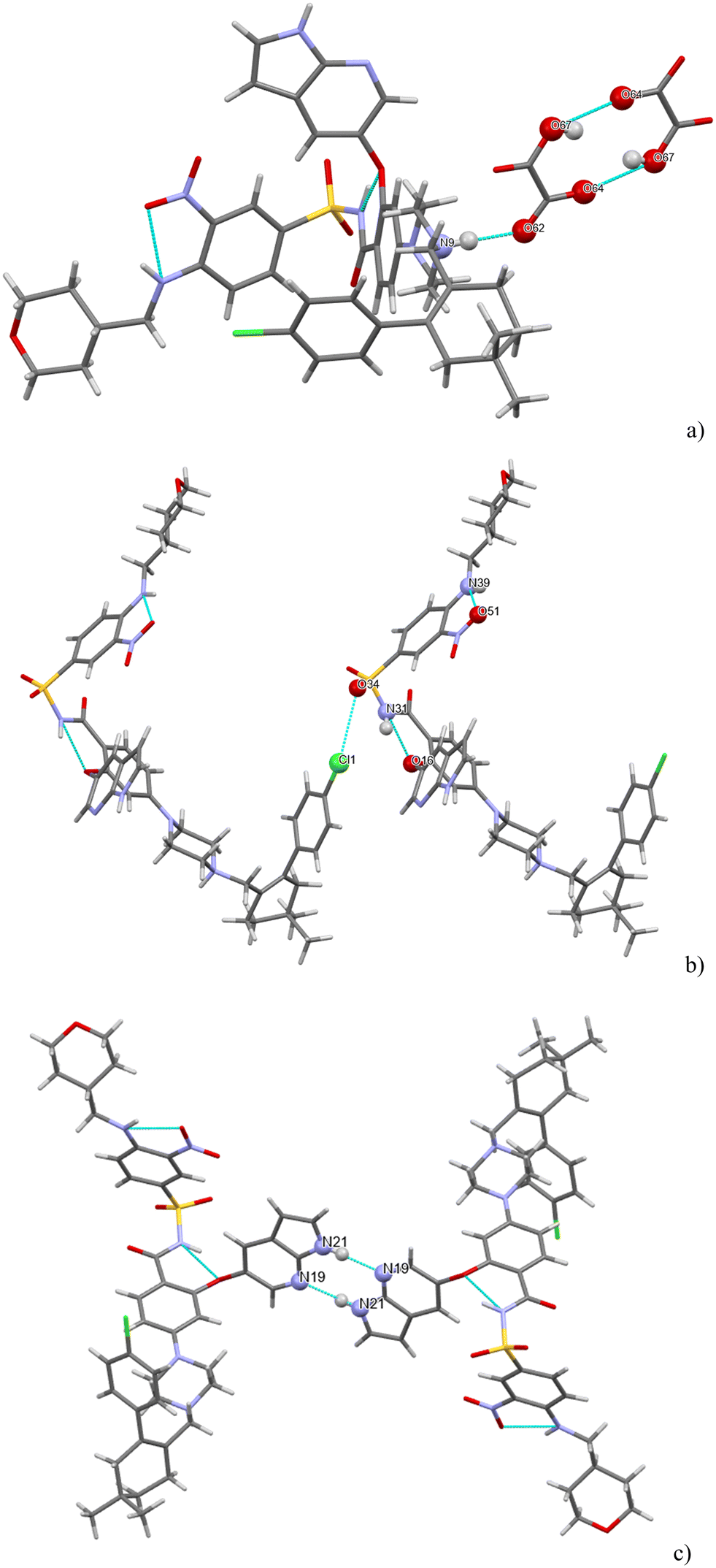 | ||
| Fig. 9 (a–c) The main H-bonding motifs in the structure of venetoclax oxalate acetonitrile solvate. | ||
Oxalates form a flat zigzag ribbon onto which acetonitrile molecules attach perpendicularly (Fig. 10). The –A–B–A–B– aromatic interaction described in detail above for venetoclax acetonitrile solvate is also present.
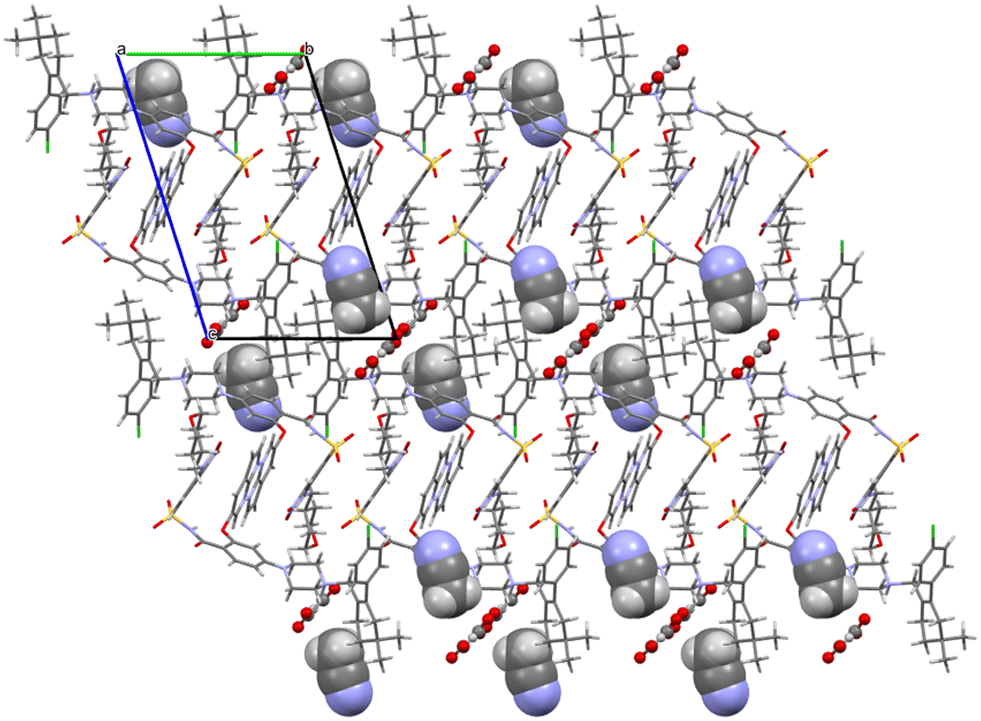 | ||
| Fig. 10 Molecular packing of venetoclax oxalate acetonitrile solvate. The solvent is shown in the spacefill mode, and the counterion is shown in ball-and-stick mode. | ||
The structure of venetoclax oxalate acetonitrile solvate contains one equivalent of solvent. However, in the bulk sample, no solvent was detected by 1H NMR. This suggests a full desolvation under our drying conditions. Comparison between the calculated and the bulk powder XRPD suggests similarities in the structure (Fig. S9†); however, not all experimental peak positions are in accordance with the calculated ones. This is expected as both 1H NMR and TGA confirm full desolvation of the bulk powder.
Venetoclax fumarate acetonitrile solvate crystallises in the monoclinic system in the space group P21/c. There is one molecule of API, one molecule of the fumarate ion, and three molecules of the solvent in the asymmetric unit. Fig. S10 in ESI† shows the asymmetric unit with the numbering of non C/H atoms.
H-bonding of venetoclax fumarate acetonitrile solvate is shown in Fig. 11. Six strong interactions can be identified in the structure. There is a proton transfer indicating that the material is truly a salt between the fumarate carboxylate O(68) and the API N(9). The other carboxyl of fumaric acid is not deprotonated and also forms a H-bond with O(68). There are two intramolecular H-bonds within the API: the amino N(37)–H connects to the adjacent nitro O(48) and the amidic N(29)–H connects to the ether O(16). N(29) is also an acceptor of a halogen bond from Cl(1). The azaindole moiety forms a self-complementary homosynthon N(22)–H⋯N(24). The H-bonding is almost completely different in both structures. The only exceptions are a common H-bond and an intramolecular amidic N(H)⋯ether (O), and that in both structures, the solvent is held only by weak interactions.
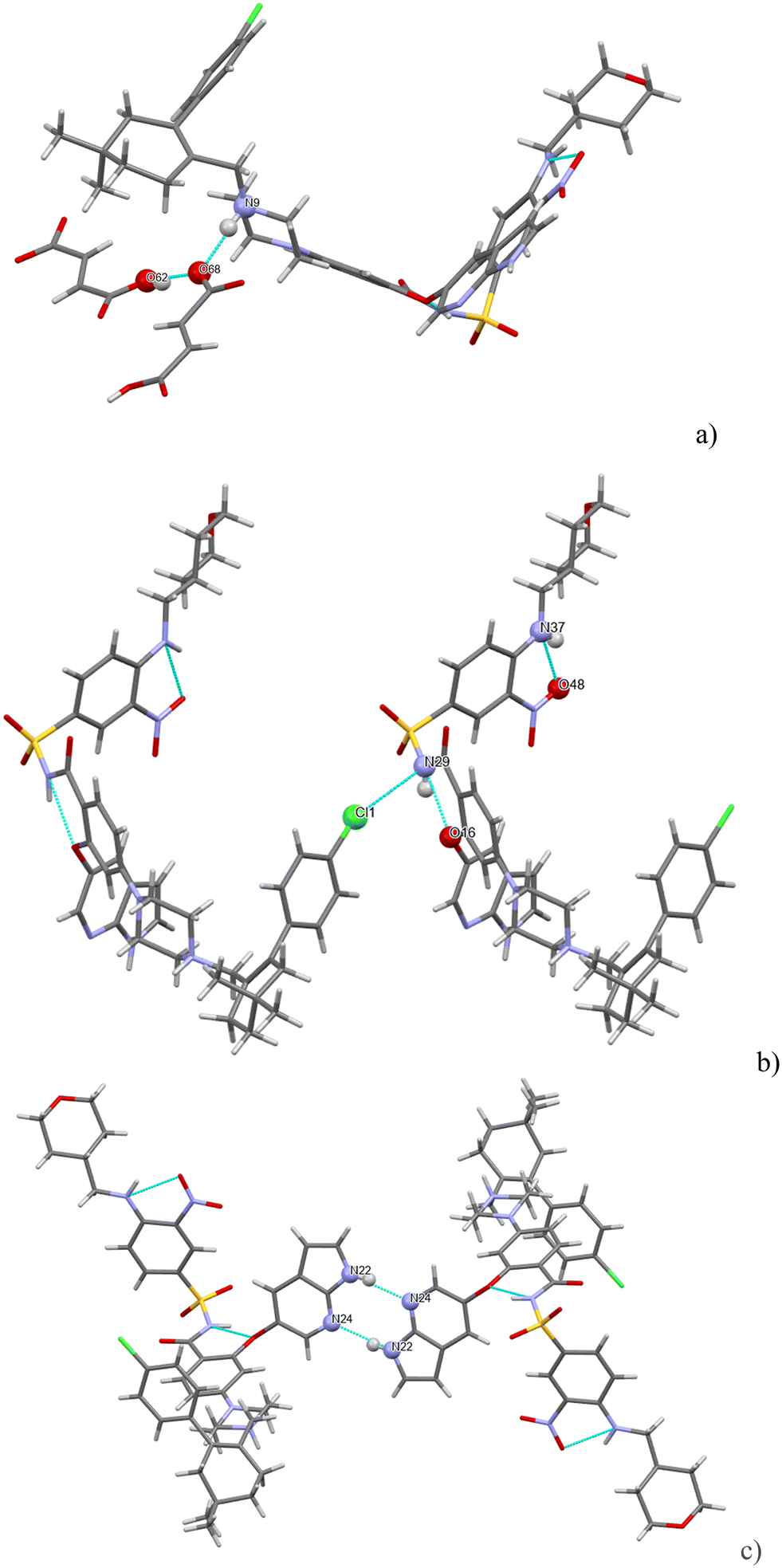 | ||
| Fig. 11 (a–c) The main H-bonding motifs in venetoclax fumarate acetonitrile solvate. | ||
Both fumarate and acetonitrile occupy flat channels in the structure (Fig. 12). Although the –A–B–A–B– aromatic interaction described in detail above for venetoclax acetonitrile solvate is present also in the structure of venetoclax fumarate acetonitrile solvate, the structures are very different, both from the view of molecular packing and from the conformations of the API.
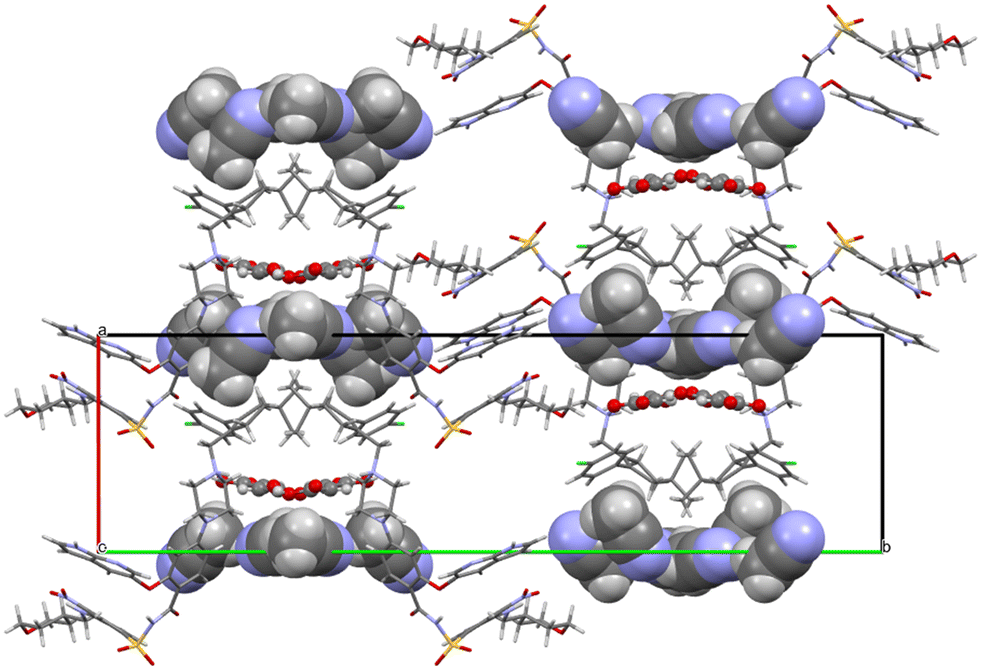 | ||
| Fig. 12 Molecular packing of venetoclax fumarate acetonitrile solvate. The solvent is shown in the spacefill mode, and the counterion is shown in ball-and-stick mode. | ||
The structure of venetoclax fumarate acetonitrile solvate contains three equivalents of solvent. However, in the bulk sample, no solvent was detected by 1H NMR. This suggests a full desolvation under our drying conditions. Comparison between the calculated and the bulk powder XRPD does not yield any similarities (Fig. S11†). However, this is to be expected as both 1H NMR and TGA confirm full desolvation of the bulk powder.
Venetoclax napsylate 1:2 acetonitrile hemisolvate crystallises in the triclinic system in the space group P1. There is one molecule of API and two molecules of the napsylate anion together with a molecule of solvent (with 0.5 occupancy) in the asymmetric unit (and also in the unit cell). Fig. S12† shows the asymmetric unit with the numbering of non C/H atoms.
The comparison between the calculated and the experimental XRPD looks somewhat similar but not identical (see Fig. S13†). As 1H NMR indicates that there was no residual solvent in the bulk sample, we can assume that the crystal structure from a single crystal would change slightly when all of the solvent is removed from the lattice. The molar ratio between the API and the counterion is 1:2 from both 1H NMR and the crystal structure.
H-bonding of venetoclax napsylate 1:2 acetonitrile hemisolvate is shown in Fig. 13.
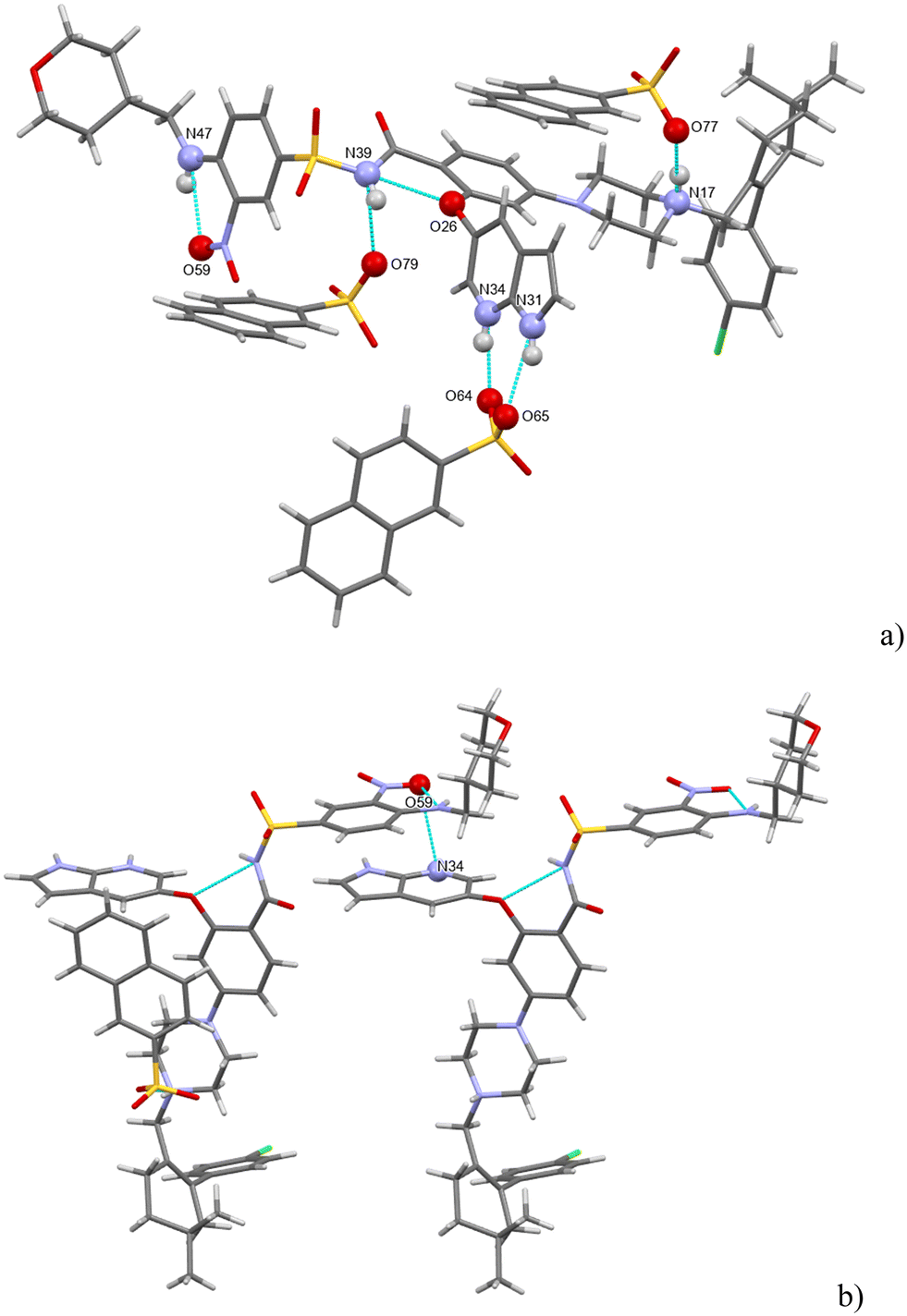 | ||
| Fig. 13 (a and b) The main H-bonding motifs in venetoclax napsylate 1:2 acetonitrile hemisolvate. | ||
Seven strong interactions can be identified. The molecule of venetoclax is protonated at two sites, on the aza-indole N34 and on the piperazine N17. Both of these NH form H-bonds to napsylates, N(17)H⋯O(77) and N(34)H⋯O(64). The other azaindole nitrogen, N(31)–H, also bonds to the same napsylate, O(65).
There are two intramolecular H-bonds within the API: the amino N(47)–H connects to the adjacent nitro O(59) and the amidic N(39)–H connects to the ether O(26). N(39)–H actually forms a 3-center H-bond with the other acceptor being O(79) from a napsylate.
Acetonitrile occupies discrete cavities in the structure. It is held together only by multiple weaker interactions such as C–H⋯O(42) and C–H⋯O(38) or C–H⋯Cl(1). The crystal packing is heavily influenced by aromatic interactions. This is understandable because in addition to the two aromatic regions of venetoclax, there are two naphthalene rings present belonging to the counterions. Some examples of these include a close π–π aromatic interaction between the A and B aromatic regions of venetoclax (Fig. 14) or the edge-to-face interaction azaindole (region B)–napsylate.
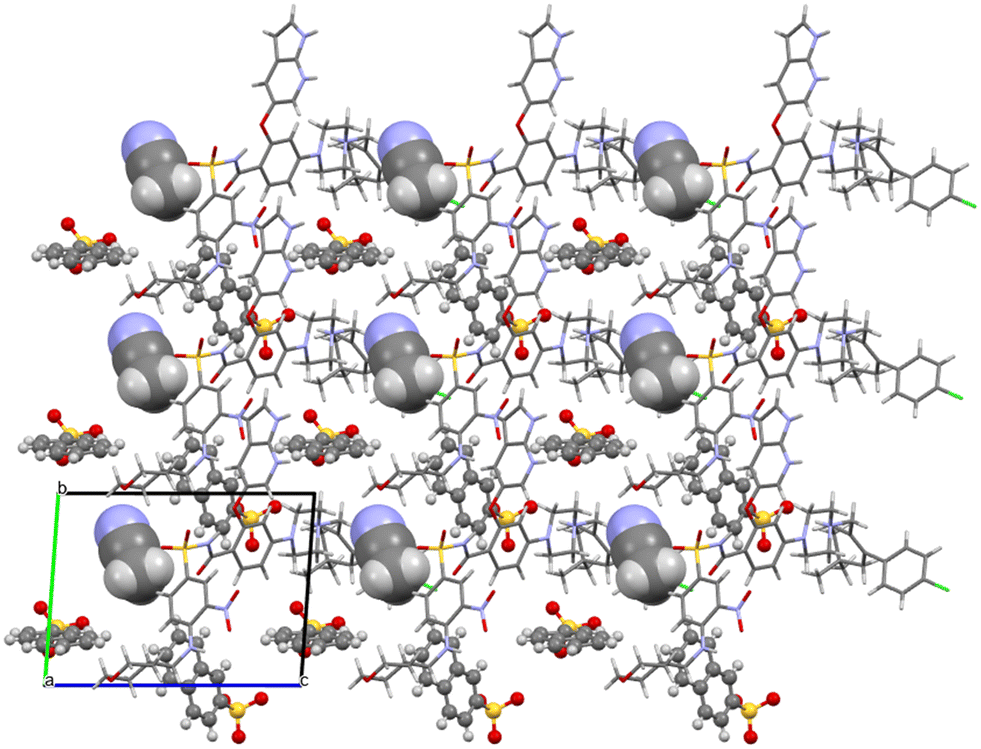 | ||
| Fig. 14 Molecular packing of venetoclax napsylate 1:2 acetonitrile hemisolvate. The solvent is shown in the spacefill mode, and the counterion is shown in ball-and-stick mode. | ||
The H-bonding pattern is very different from the above-mentioned structures. The only exceptions are a common intramolecular amidic N(H)⋯ ether (O) H-bond and that in both structures, the solvent is held only by weak interactions.
Venetoclax tosylate 1:2 crystallizes in the triclinic system in the space group P. There is one molecule of API and two molecules of the toluene sulfonate (tosylate) anion in the asymmetric unit. Fig. S14† shows the asymmetric unit with the numbering of non C/H atoms.
The comparison between the calculated and experimental XRPD confirms that the single-crystal is representative of the bulk sample (see Fig. S15†). Together with the 1H NMR results, this confirms the molar ratio between the API and counterion as 1:2.
H-bonding of venetoclax tosylate is shown in Fig. 15. Seven strong interactions can be identified. The molecule of venetoclax is protonated at two sites, on the azaindole N26 and on the piperazine N11. Both of these NH form H-bonds to tosylates, N(26)H⋯O(75) and N(11)H⋯O(63). N(11)–H actually forms a 3-center H-bond with the other acceptor being O(65) from the same tosylate. The other azaindole nitrogen, N(23)–H also bonds to the same tosylate as N(26), O(76). There are two intramolecular H-bonds within the API: the amino N(42)–H connects to the adjacent nitro O(39) and the amidic N(31)–H connects to the ether O(18). N(42)–H actually forms a 3-center H-bond with the other acceptor being amidic O(30).
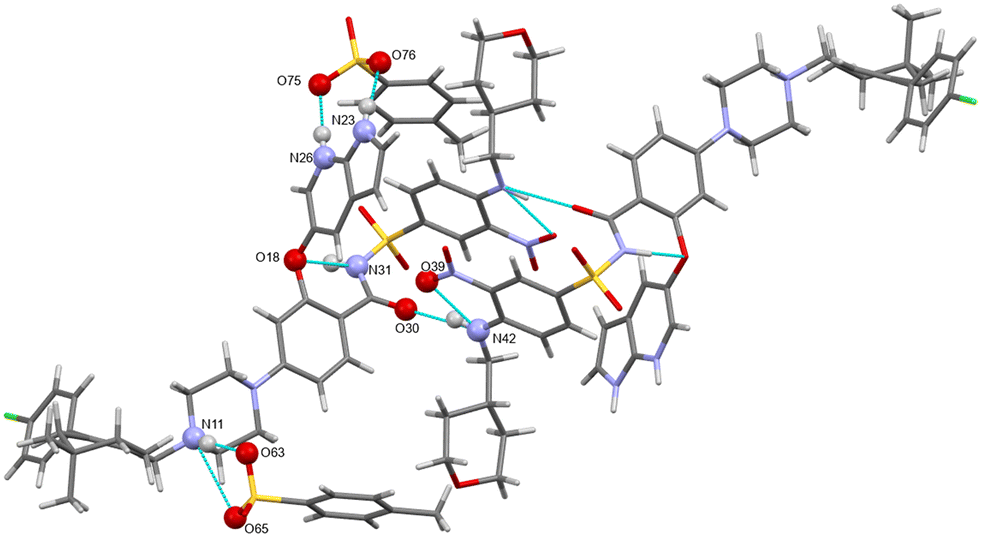 | ||
| Fig. 15 The main H-bonding motifs in venetoclax tosylate 1:2. | ||
The H-bonding pattern is partially similar to the structure of venetoclax napsylate acetonitrile hemisolvate. For example, the interactions between the protonated sites of venetoclax with the sulphonic groups of the counterions is very similar (Fig. 16).
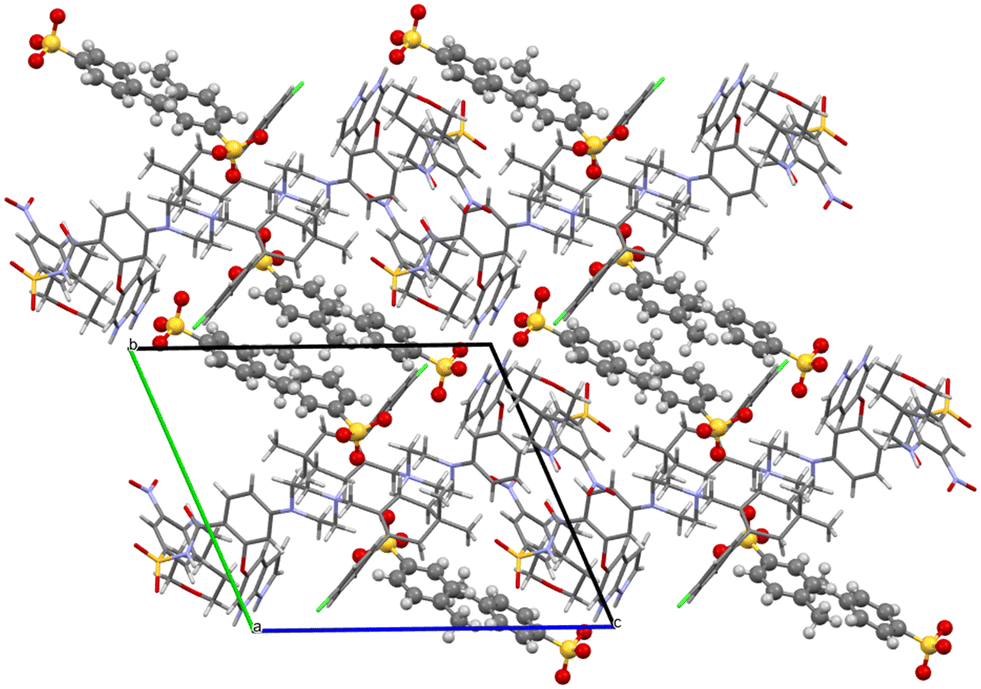 | ||
| Fig. 16 Molecular packing of venetoclax tosylate 1:2. The solvent is shown in the spacefill mode, and the counterion is shown in ball-and-stick mode. | ||
Comparison of venetoclax structures
All the available crystal structures of venetoclax were compared in regard to the conformation of the venetoclax molecule and its molecular crystal packing. The set included venetoclax acetonitrile solvate, venetoclax fumarate acetonitrile solvate, venetoclax oxalate acetonitrile solvate, venetoclax tosylate, venetoclax napsylate acetonitrile solvate and the hydrate published previously. This was aided by the calculation of similarity dendrograms (see Fig. S16 and S17 in ESI†) in the software CrystalCMP. As can be seen from the dendrograms, the structures are generally all very different. Fig. 17a shows the overlay of the conformations. We can see that all of the torsion angles are varied, resulting in many different conformations. The only somewhat similar pair was the venetoclax fumarate acetonitrile solvate and venetoclax oxalate acetonitrile solvate, shown right in Fig. 17b.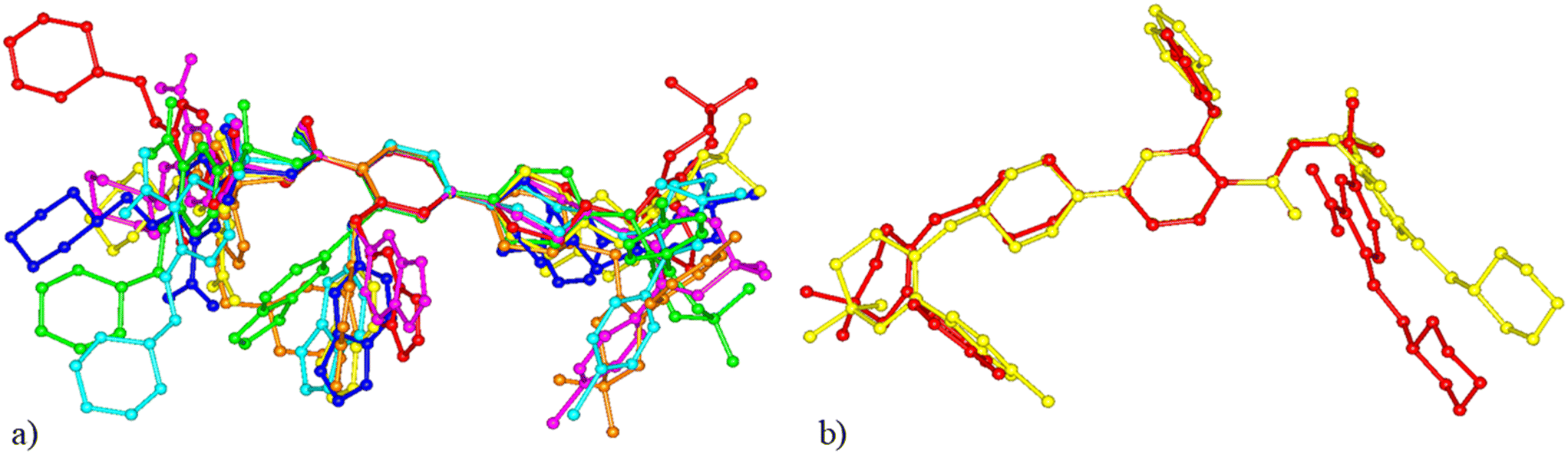 | ||
| Fig. 17 Comparison of venetoclax conformations. (a) All structures. (b) The most similar conformation in venetoclax fumarate acetonitrile solvate (yellow), venetoclax oxalate acetonitrile solvate (red). | ||
The molecular packing of venetoclax was unique in all of the examined structures.
This conformational and structural flexibility of venetoclax may be caused by its large molecular size. We suspect that it is one of the driving factors behind its observed ‘promiscuity’ in regard to forming many different phases and solvatomorphs.
Molecular electrostatic potential surfaces and pKa
The molecular electrostatic potential (MEP) and pKa values are crucial indicators of a molecule's ionization behavior and its capacity to form molecular salts. MEP maps visually represent charge distribution, while pKa values measure susceptibility to protonation and deprotonation. Fig. 18 in the Introduction presents the pKa values of different parts of VEN. The MEP maps (Fig. 19) highlight regions of high (red) and low (blue) electrostatic potential, showcasing VEN in its neutral state, +1 and +2 cation forms.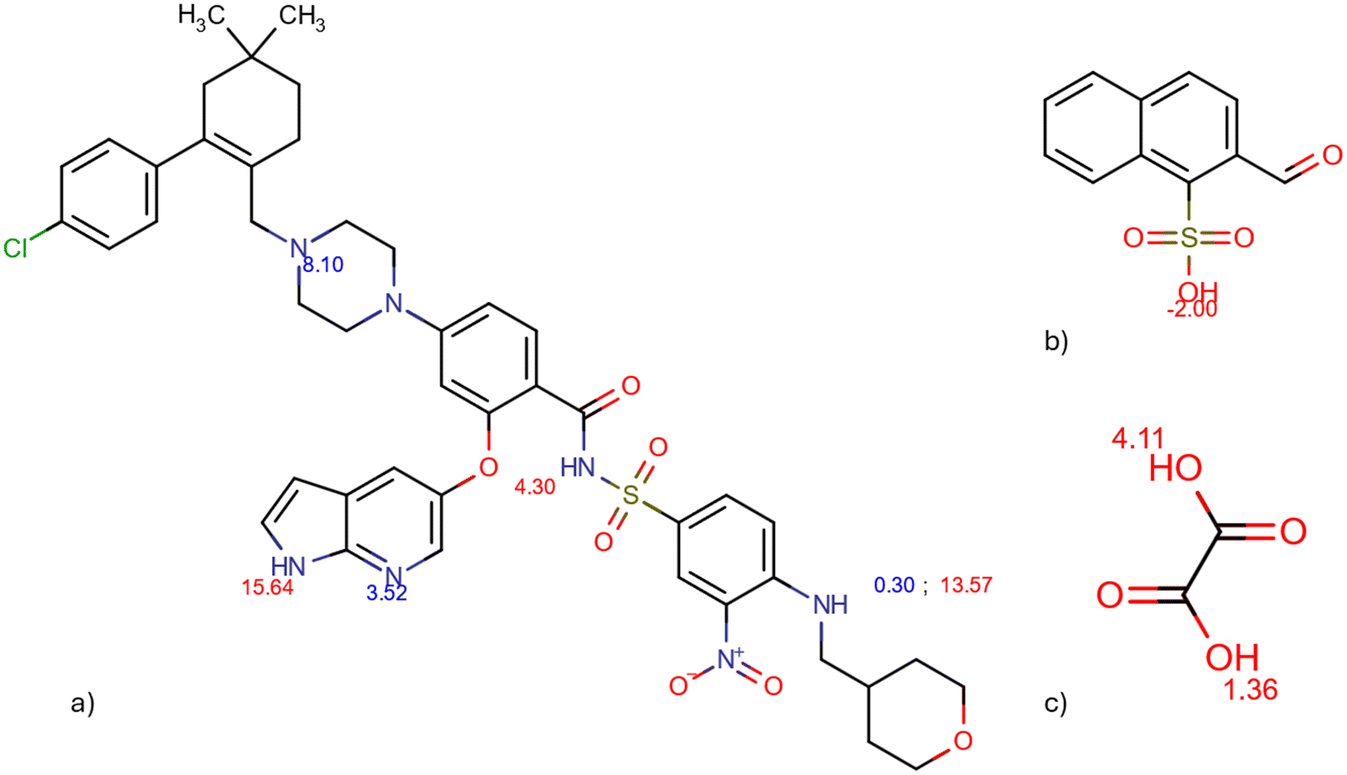 | ||
| Fig. 18 pKa values of venetoclax (a), naphtalenesulfonic acid (b), and oxalic acid (c) calculated by Chemicalize. | ||
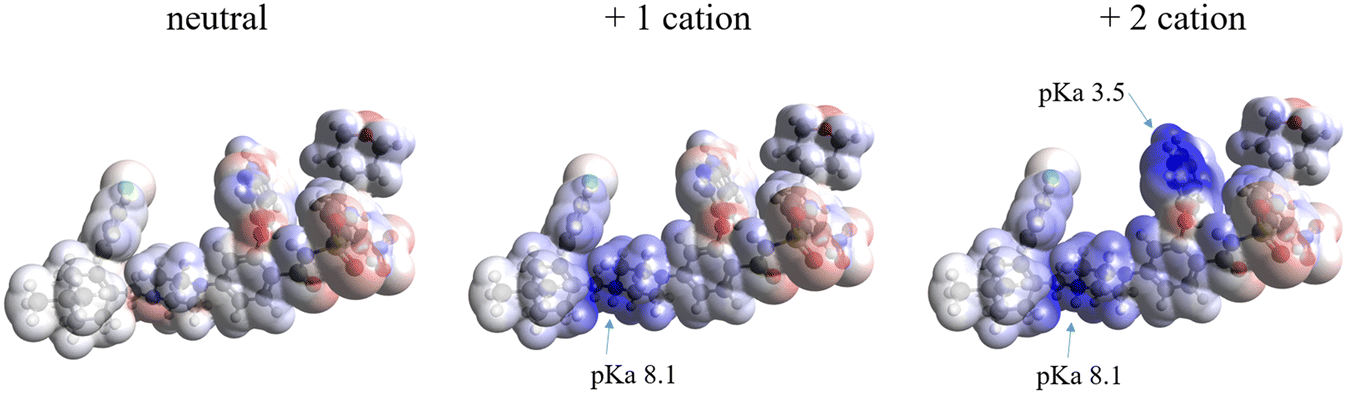 | ||
| Fig. 19 Molecular electrostatic potential surface of venetoclax as a neutral molecule (left) and as it exists in the herein reported salts, as a +1 cation (middle) and +2 cation (right). pKa indicated for the protonated regions. | ||
Key functional groups, based on their pKa values, are annotated: the piperazine nitrogen (N) and the 7-azaindole nitrogen (N), which indicate their susceptibility to ionization. Notice how VEN's basic functional groups transition from red (high electrostatic potential) when unprotonated to dark blue (low electrostatic potential) upon protonation. MEPs also suggest likely interaction sites for non-bonded interactions. Comparing MEPs with crystal structures reveals that the observed hydrogen bonding patterns stem from the molecules' electrostatic potentials.
We can understand the 1:1 and 1:2 character of salts formed with dicarboxylic and sulfonic acids, respectively. All salts with dicarboxylic acids consist of mono-protonated venetoclax crystallizing in a 1:1 ratio with the organic anion. Despite counterions having two deprotonable groups, only one is sufficiently acidic to protonate venetoclax. For instance, oxalic acid, the strongest dicarboxylic acid in this study, has two acidic groups with pKa values of 1.36 (stronger) and 4.11 (weaker). Venetoclax has two basic regions with pKa values of 8.10 (stronger) and 3.52 (weaker).
Considering the possible 1:1 and 1:2 ratios:
• 1:1 ratio: compare the strongest basic and strongest acidic sites, and the weaker basic and weaker acidic sites, to determine likely proton transfer. ΔpKa values are +6.74 (i.e., 8.1–1.36) and −0.59 (i.e., 3.52–4.11). Generally, a ΔpKa value greater than +3 suggests salt formation. Thus, in a 1:1 ratio with oxalic acid, only one site of venetoclax (the piperazine N) is protonated, as observed in the crystal structure of venetoclax oxalate acetonitrile solvate.
• 1:2 ratio: compare the strongest basic site of venetoclax with the strongest acidic site of oxalic acid, and the weaker basic site of venetoclax with the strongest acidic site of oxalic acid (a second acid molecule would be available). ΔpKa values are +6.74 (i.e., 8.1–1.36) and 2.16 (i.e., 3.52–1.36). It is unlikely for double protonation of venetoclax to occur with oxalic acid, supported by the experimentally observed 1:1 ratio in VEN oxalate. ΔpKa values for other dicarboxylic acids would be even lower.
In contrast, sulfonic acids are stronger and have only one deprotonable group. For example, naphthalene sulfonic acid, with a pKa of −2.00, yields ΔpKa values of 10.10 (i.e., 10.1–(−2)) and 5.52 (i.e., 3.52–(−2)) against venetoclax, indicating that it is strong enough to protonate venetoclax into a +2 cation and form a 1:2 salt, as experimentally observed.
4. Conclusion
This study successfully identified and characterized ten new crystalline salts of venetoclax using dicarboxylic and sulfonic acids as counterions. The salt screening process demonstrated venetoclax's high propensity for salt formation, with 10 out of 13 counterions resulting in positive hits. The formation of these salts was confirmed through a combination of Raman spectroscopy, 1H NMR, and XRPD, with the stoichiometric ratios of the components determined.Thermal analysis revealed that all prepared salts, except dodecylbesylate, exhibited significantly higher melting points compared to that of pure API, indicating enhanced thermal stability. All salts exhibit improved dissolution rates ranging from a 0.07-fold increase for venetoclax malonate to a 2.9-fold increase for venetoclax napsylate, which appears to be the most promising candidate for further research. Despite attempts to correlate dissolution behaviour with melting points, no clear trend was observed, suggesting that factors beyond the melting point influence dissolution rates. The lack of strong correlation aligns with previous findings that crystal structures ultimately govern solid-state properties. The crystal structures of venetoclax, venetoclax fumarate, venetoclax oxalate, and venetoclax napsylate (all solvated with acetonitrile) as well as unsolvated venetoclax tosylate were solved from single-crystal data. Although all structures are unique, some similarities were found. The salt solvates are more similar to each other than to the API solvate. Intramolecular amidic N(H) and ether (O) H-bonds were found in all structures. Also, in all structures, the solvent was held by only weak interactions. The API conformations were different in all structures. The overall molecular packing was somewhat similar between the salt solvates but completely different in the API solvate. The distinction between 1:1 and 1:2 salt formation, as elucidated by pKa values and molecular electrostatic potential (MEP) maps, underscores the selective protonation of venetoclax in the presence of different counterions. Specifically, our findings demonstrate that while dicarboxylic acids predominantly form 1:1 salts, the MEP highlights regions of high electrostatic potential facilitating this interaction. Conversely, stronger sulfonic acids facilitate the formation of 1:2 salts, as evidenced by the MEP showcasing regions conducive to further protonation. This nuanced understanding is crucial for optimizing the physicochemical properties of venetoclax salts for therapeutic applications.
Data availability
Crystallographic data for venetoclax acetonitrile solvate, venetoclax oxalate acetonitrile solvate, venetoclax fumurate acetonitrile solvate, venetoclax napsylate acetonitrile solvate and venetoclax tosylate have been deposited at the CCDC under 2377711, 2377712, 2377714, 2393684 and 2393682.The data supporting this article have been included as part of the ESI.† This contains full crystallographic information about the prepared new solid forms, Raman spectra; solution 1H NMR spectra; comparison of XRPD patterns of salts to starting materials; UV-vis spectra of venetoclax and aromatic acids, IDR curves and their comparison as well as measured and calculated XRPD patterns for solved structures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to acknowledge the Research and Development department of Zentiva k.s. for the materials and all the help provided. The work was supported by the project New Technologies for Translational Research in Pharmaceutical Sciences/NETPHARM, project ID CZ.02.01.01/00/22_008/0004607, co-funded by the European Union and by the Czech Science Foundation project 24-10558S (EZ). The crystallographic part was supported by the CzechNanoLab Research Infrastructure supported by MEYS CR (LM2023051).References
- EMA, EPAR, 2016 Search PubMed.
- A. Alaarg, R. Menon, D. Rizzo, Y. Liu, J. Bien, T. Elkinton, T. Grieme, L. R. Asmus and A. H. Salem, Clin. Transl. Sci., 2022, 15, 244–254 CrossRef CAS PubMed.
- D. P. Elder, R. Holm and H. L. D. Diego, Int. J. Pharm., 2013, 453, 88–100 CrossRef CAS PubMed.
- P. C. Vioglio, M. R. Chierotti and R. Gobetto, Adv. Drug Delivery Rev., 2017, 117, 86–110 CrossRef PubMed.
- N. Biswas, in Pharmaceutical Salts and Co-crystals, ed. J. Wouters and L. Quéré, The Royal Society of Chemistry, 2011 Search PubMed.
- A. T. M. Serajuddin, Adv. Drug Delivery Rev., 2007, 59, 603–616 CrossRef CAS PubMed.
- K. Kawakami, Adv. Drug Delivery Rev., 2012, 64, 480–495 CrossRef CAS PubMed.
- E. Pindelska, A. Sokal and W. Kolodziejski, Adv. Drug Delivery Rev., 2017, 117, 111–146 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, M. Lusi, H. P. Wheatcroft and A. D. Bond, Faraday Discuss., 2022, 235, 446–466 RSC.
- A. J. Cruz-Cabeza, CrystEngComm, 2012, 14, 6362–6365 RSC.
- C. Saal and A. Becker, Eur. J. Pharm. Sci., 2013, 49, 614–623 CrossRef CAS PubMed.
- S. S. Bharate, Drug Discovery Today, 2021, 26, 384–398 CrossRef CAS PubMed.
- G. S. Paulekuhn, J. B. Dressman and C. Saal, J. Med. Chem., 2007, 50, 6665–6672 CrossRef CAS PubMed.
- H. Qadri, A. A. Malik, A. A. Ahangar, M. A. Mir, A. A. Dar and A. H. Shah, RSC Pharm., 2024, 1, 705–715 RSC.
- R. Thakuria and A. Nangia, Cryst. Growth Des., 2013, 13, 3672–3680 CrossRef CAS.
- K. K. Sarmah, A. Sarma, K. Roy, D. R. Rao and R. Thakuria, Cryst. Growth Des., 2016, 16, 1047–1055 CrossRef CAS.
- W. Gong, P. K. Mondal, S. Ahmadi, Y. Wu and S. Rohani, Int. J. Pharm., 2021, 608, 121063 CrossRef CAS PubMed.
- R. Thakuria and A. Nangia, CrystEngComm, 2011, 13, 1759–1764 RSC.
- A. O. Surov, A. N. Manin, A. P. Voronin, K. V. Drozd, A. A. Simagina, A. V. Churakov and G. L. Perlovich, Eur. J. Pharm. Sci., 2015, 77, 112–121 CrossRef CAS PubMed.
- T. Hibbard, B. Nyambura, P. Scholes, M. Totolici, K. Shankland and H. Al-Obaidi, J. Pharm. Sci., 2023, 112, 195–203 CrossRef CAS PubMed.
- S. S. Bharate, Pharm. Res., 2021, 38, 1307–1326 CrossRef CAS PubMed.
- G. Bolla and A. Nangia, Cryst. Growth Des., 2012, 12, 6250–6259 CrossRef CAS.
- L. Rajput, Cryst. Growth Des., 2014, 14, 5196–5205 CrossRef CAS.
- E. Skořepová, D. Bím, M. Hušák, J. Klimeš, A. Chatziadi, L. Ridvan, T. Boleslavská, J. Beránek, P. Šebek and L. Rulíšek, Cryst. Growth Des., 2017, 17, 5283–5294 CrossRef.
- J. H. Seo, J. B. Park, W.-K. Choi, S. Park, Y. J. Sung, E. Oh and S. K. Bae, Drug Des., Dev. Ther., 2015, 9, 3961–3968 CAS.
- A. O. Surov, N. A. Vasilev, M. V. Vener, O. D. Parashchuk, A. V. Churakov, O. V. Magdysyuk and G. L. Perlovich, Cryst. Growth Des., 2021, 21, 4516–4530 CrossRef CAS.
- N. J. Koehl, L. J. Henze, R. Holm, M. Kuentz, J. J. Keating, T. De Vijlder, A. Marx and B. T. Griffin, J. Pharm. Sci., 2022, 111, 164–174 CrossRef CAS PubMed.
- F. Perdih, N. Žigart and Z. Časar, Crystals, 2021, 11, 261 CrossRef CAS.
- D. Baker, CrysAlisPro User Manual, 2023 Search PubMed.
- V. Petříček, L. Palatinus, J. Plášil and M. Dušek, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- V. Petříček, L. Palatinus, J. Plášil and M. Dušek, Z. Kristallogr. - Cryst. Mater., 2023, 238, 271–282 CrossRef.
- J. Rohlicek and M. Husak, J. Appl. Crystallogr., 2007, 40, 600–601 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- Committee for Human Medicinal Products, ICH guideline, 2019 Search PubMed.
- N. J. Koehl, L. J. Henze, M. Kuentz, R. Holm and B. T. Griffin, Pharmaceutics, 2020, 12, 564 CrossRef CAS PubMed.
- M. Okezue, S. Bogdanowich-Knipp, D. Smith, M. Zeller, S. Byrn, P. Smith, D. K. Purcell and K. Clase, AAPS PharmSciTech, 2021, 22, 228 CrossRef CAS PubMed.
- R. K. Khankari, D. Law and D. J. W. Grant, Int. J. Pharm., 1992, 82, 117–127 CrossRef CAS.
- L. S. de Moraes, D. Edwards, A. J. Florence, A. Johnston, B. F. Johnston, C. A. Morrison and A. R. Kennedy, Cryst. Growth Des., 2017, 17, 3277–3286 CrossRef.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CrossRef CAS.
- A. D. Bond, in Pharmaceutical Salts and Co-crystals, ed. J. Wouters and L. Quéré, The Royal Society of Chemistry, 2011, 10.1039/9781849733502-00009.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2377711, 2377712, 2377714, 2393682 and 2393684. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ce01121j |
| This journal is © The Royal Society of Chemistry 2025 |