
Mononuclear high-spin Fe(III) complexes: synthesis, crystal structures, magnetic and optical properties†
Olga
Danilescu
 *a,
Paulina
Bourosh
b,
Ion
Bulhac
a,
Olga
Kulikova
b,
Maria
Cocu
a and
Yurii
Chumakov
b
*a,
Paulina
Bourosh
b,
Ion
Bulhac
a,
Olga
Kulikova
b,
Maria
Cocu
a and
Yurii
Chumakov
b
aMoldova State University, Institute of Chemistry, 3 Academiei str., Chisinau MD2028, R. Moldova. E-mail: olga.danilescu@sti.usm.md; olgadanilescu@gmail.com
bMoldova State University, Institute of Applied Physics, 5 Academiei str., Chisinau MD2028, R. Moldova
First published on 18th March 2025
Abstract
The coordination agent H2L = 2,6-diacetylpyridine bis(picolinoylhydrazone) was used for the synthesis of two mononuclear iron(III) complexes [Fe(H2L)(H2O)2]X3·2.5H2O, where X = NO3− (1) and ClO4− (2). In the studied crystals of 1 and 2, characterized by IR spectroscopy and X-ray diffraction, the pentadentate Schiff base ligands are coordinated in mononuclear cationic complexes [Fe(H2L)(H2O)2]3+ by donor atoms N3O2 as double zwitterions. In 1, the cations are joined to centrosymmetric dimers via hydrogen bonds (HB), forming chains in the crystal, while in 2, the cations are assembled into tetramers through HB owing to the anions and water molecules. The crystal structures of the studied compounds were stabilized via hydrogen bonding that yielded supramolecular 3D networks via coordinated and crystallization water molecules, PyH+ groups of H2L and NO3− or ClO4− anions. The theoretical calculations, performed using the new DFT-TB2J approach revealed the magnetic exchange interactions between the iron ions, which were mediated through intermolecular hydrogen bonds. The UV-vis absorption spectra of 1 and 2 were not overlapped with their photoluminescence emission spectra because of the spin-gapless characteristic of the band structure of these compounds.
1. Introduction
Advances in coordination chemistry are to a large extent associated with the construction of new types of ligand systems. Schiff bases and their structural analogues are the most studied ligands in coordination chemistry.1 Schiff base derivates reveals catalytic,2 magnetochemical3 and biological4 properties. One class of intensively studied Schiff bases coordination compounds is represented by iron complexes with pentadentate ligands.5Understanding the relationship between the molecular and supramolecular structures is so necessary for molecular crystal engineering,6 bioinorganic chemistry (modeling of electron transfer and oxygen transfer enzymes)7 and catalysis.8,9
Over the past few years, the role of anions in supramolecular architecture has become more and more attractive due to its applicability in diverse areas of research, such as in molecular recognition, sensing, biomembrane transport, catalysis and designing novel selective anion-receptors. In supramolecular chemistry, especially coordination polymer chemistry, anions can promote the formation of complex compounds with different dimensionalities.10–12 Due to several major interactions of anions in the crystals such as electrostatic interactions, metal coordination bond, hydrogen and/or halogen bonding and anion-π interactions, the anions can act as a spectator (charge balance) or may have a structure-directing impact as a template, building unit, or secondary building unit.13,14 Anions binding has attracted continued interest because of the relevant role performed by this molecules in biological, chemical, and environmental processes. It is well known that anions can fulfil various roles during crystallization processes, and anion receptors have shown bright application prospects in trans-epithelial ion transport, chemical sensing, and simulation of enzyme catalysed organic reactions.15,16
Structural studies have shown that the nature and geometry of inorganic anions, such as linear (NCS−), spherical (Cl−, Br−, I−), trigonal planar (NO3−), tetrahedral (ClO4−, SO42−, H2PO4−, BF4−) can induce the formation of diverse supramolecular architectures, effectively influencing the final structure.17,18 Our recent study of iron(III) coordination compounds with isomeric Schiff base ligands19 also showed the contribution of different anions (ClO4−, SCN−, N3−) to the supramolecular assemblies. Anions were chosen to have the same charge with different coordination abilities: ClO4− < SCN− < N3−. Therefore, based on this and our previous studies, we are interested in finding an optimal synthetic method for obtaining of mononuclear Fe(III) coordination compounds with 2,6-diacetylpyridine bis(picolinoylhydrazone) (H2L). Analysis of the Cambridge Structural Database (CSD)20 has revealed five coordination compounds of transition metals (Co(II), Mn(II), Cd(II) and Cu(II)), where the ligand, 2,6-diacetylpyridine bis(picolinoylhydrazone) predominately acts as a pentadentate N3O2 ligand and surrounding metal in the equatorial plane, except for the Cu(II) complexes. The examination of published examples showed that H2L moiety forms neutral and ionic complexes, coordinating in the neutral and bi-deprotonated fashions (H2L and L2−), in consequence providing three mononuclear and two tetrameric coordination compounds. It is noteworthy that only mononuclear coordination compounds21–23 were prepared through one-pot template synthesis, while the tetrameric compounds24,25 – by direct reaction between H2L and copper(II) chloride/perchlorate salts. The search in the CSD has revealed also six coordination compounds of Fe(III), with ionic structures, where the ligands act as a pentadentate N3O2 ligands, surrounding metal in the equatorial plane. In the crystal packing of all compounds there are the noncovalent interactions between coordination cation, outer-sphere inorganic anions (NO3−,26,27 ClO4−19,28) and water molecules.
In the present work, we present the synthesis and crystal structures of two coordination compounds of Fe(III), namely, [Fe(H2L)(H2O)2](NO3)3·2.5H2O (compound 1) and [Fe(H2L)(H2O)2](ClO4)3·2.5H2O (compound 2), where H2L = 2,6-diacetylpyridine bis(picolinoylhydrazone). Both complexes contain the complex cation [Fe(H2L)(H2O)2]3+ and counter-anions with the same charge but various geometries: trigonal planar (NO3− in 1) and tetrahedral (ClO4− in 2). The crystals of compound 1 and 2 (Scheme 1), suitable for single crystal diffraction analysis, were obtained by the direct reaction between the pre-synthesized H2L ligand and iron(III) nitrate/perchlorate salt (Fig. S1†). Both complexes were characterized by single crystal X-ray diffraction method and IR spectroscopy.
 | ||
| Scheme 1 Schematic of the synthesis of the Schiff base ligand H2L and coordination compounds 1 and 2. | ||
2. Experimental
2.1. Materials and methods
All reagents (2,6-diacetylpyridine, picolinic acid hydrazide, Fe(ClO4)3·xH2O, Fe(NO3)3·9H2O) and methanol were obtained from commercial sources and were used without further purification. Schiff base H2L was synthesized by the condensation of 2,6-diacetylpyridine and picolinic acid hydrazide according to the previously reported method.29The IR spectra were obtained in Vaseline oil (400–4000 cm−1) and ATR (650–4000 cm−1) on an FT IR Spectrum-100 Perkin Elmer spectrometer.
The photoluminescence (PL) was measured using a pulsed N2 laser excited with λexc = 337 nm in the interval of 350–750 nm at room temperature. The excitation pulse duration was 15 ns, the repetition rate was 50 Hz, and the pulse energy was 0.2 mJ. The emission was detected using an FEU-79 instrument (multi-alkaline photocathode Sb(Na2K) with the adsorbed cesium layer on the photocathode surface, characteristic of the S20 type). The registration system self-time was 20 ns. The equipment used for recording was a boxcar integrator SR-250 (Stanford Research Systems).
The ultraviolet-visible (UV-vis) absorption spectrum was recorded on a Perkin Elmer Lambda 25 spectrophotometer.
Single crystal X-ray diffraction data of 1 and 2 were collected at room temperature on an Xcalibur E CCD diffractometer equipped with a CCD area detector and a graphite monochromator using MoKα radiation. Determination of the unit cell parameters and processing of experimental data were performed using the CrysAlis Oxford Diffraction Ltd software.30 The structures were solved by a direct method and refined by the least square method in the anisotropic approximation for non-hydrogen atoms using the SHELX-97 (ref. 31) and SHELXL2014 (ref. 32) program packages. The hydrogen atoms were included in the refinement in geometrically calculated positions, and their thermal factors UH were taken to be 1.2 times larger than those of carbon and nitrogen atoms bonded to them. The H-atoms in water molecules were determined from differential Fourier maps at the intermediate stages of refinement. The outer-sphere water molecules in the studied compounds were refined with full and partial occupancies. In 1, one of the three crystallization water molecules in the asymmetric unit cell has an occupancy factor of 0.5 while in 2, the site occupancy factors of the eight observed crystallization water molecules range from 0.4 to 0.85 and metal: crystallization water molecules ratio is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5. One of the oxygen atoms in the two ClO4− anions in compound 2 is statically disordered over two positions with the site occupation factors of 0.55 and 0.45 probabilities. The X-ray data and the details of the refinement for all compounds are summarized in Table 1. The figures were obtained using the Mercury program33 and some of them are presented in ESI.† The hydrogen atoms that are not involved in the hydrogen bonding were omitted from the generation of the main packing diagrams. The geometrical analysis was performed using the PLATON program.34 The Y–X⋯Cg (π-cycle) interactions were revealed in the studied compounds (X⋯Cg < 4.0 Å, γ < 30.0°, with γ being the angle between the XCg vector and the normal to the aromatic cycle Cg); for 1 and 2, X = O, Y = N, Cl and Cg is the centroid of the pyridine ring. Selected interatomic distances and bond angles, as well as all hydrogen bonds, are given in Tables S1 and S2;† coordinates of the basis atoms of the studied structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2400697 and 2400698).
:2.5. One of the oxygen atoms in the two ClO4− anions in compound 2 is statically disordered over two positions with the site occupation factors of 0.55 and 0.45 probabilities. The X-ray data and the details of the refinement for all compounds are summarized in Table 1. The figures were obtained using the Mercury program33 and some of them are presented in ESI.† The hydrogen atoms that are not involved in the hydrogen bonding were omitted from the generation of the main packing diagrams. The geometrical analysis was performed using the PLATON program.34 The Y–X⋯Cg (π-cycle) interactions were revealed in the studied compounds (X⋯Cg < 4.0 Å, γ < 30.0°, with γ being the angle between the XCg vector and the normal to the aromatic cycle Cg); for 1 and 2, X = O, Y = N, Cl and Cg is the centroid of the pyridine ring. Selected interatomic distances and bond angles, as well as all hydrogen bonds, are given in Tables S1 and S2;† coordinates of the basis atoms of the studied structures have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2400697 and 2400698).
| 1 | 2 | |
|---|---|---|
| CCDC | 2400697 | 2400698 |
| Empirical formula | C21H28FeN10O15.5 | C21H28Cl3FeN7O18.5 |
| Formula weight | 724.38 | 836.70 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| Z | 2 | 4 |
| a (Å) | 10.2159(6) | 13.4972(5) |
| b (Å) | 11.9887(4) | 16.7467(7) |
| c (Å) | 12.2929(5) | 17.1179(6) |
| α (deg) | 86.478(3) | 89.179(3) |
| β (deg) | 89.905(4) | 71.646(3) |
| γ (deg) | 83.462(4) | 70.421(4) |
| V (Å3) | 1492.92(12) | 3442.0(2) |
| D c (g cm3) | 1.611 | 1.615 |
| μ (mm−1) | 0.596 | 0.758 |
| F(000) | 748 | 1712 |
| Crystal size (mm3) | 0.50 × 0.25 × 0.18 | 0.45 × 0.40 × 0.08 |
| Reflections collected/unique | 9737/5553 | 21523/12139 |
| [R(int) = 0.0220] | [R(int) = 0.0248] | |
| Reflections with [I > 2σ(I)] | 4535 | 7619 |
| Parameters | 5553/3/455 | 12139/1/959 |
| GOF on F2 | 1.003 | 1.005 |
| R 1, wR2 [I > 2σ(I)] | 0.0463, 0.1217 | 0.0902, 0.2655 |
| R 1, wR2 (all data) | 0.0587, 0.1320 | 0.1326, 0.3042 |
2.2. Synthesis
3. Results and discussion
3.1. IR spectroscopy
The IR spectra of compounds 1 and 2 confirm the coordination of the organic ligand H2L to Fe(III) ions (Fig. S2†). Although the H2L ligand is neutral, in the spectra of complexes 1 and 2, the band ν(NH), observed in the free ligand at 3324 cm−1, is absent. The ν(C=O) stretching vibration band at 1698 cm−1 in the H2L spectrum represents the most intense and significant band and a characteristic of the Schiff base ligand. In the IR spectrum of H2L, the strong characteristic absorption in the 3650–3400 cm−1 region, represented by peaks at 3609 and 3483 cm−1, indicates the stretching vibrations ν(OH).The crystal structures of compounds 1 and 2 are stabilized by hydrogen bonds formed by the coordinated and crystallization water molecules, NO3− and ClO4− anions and NH+ (PyH+) (Table S2†). In the spectra of compounds 1 and 2 in the region 3650–2600 cm−1, a broad absorption is observed with the clear manifestation of four peaks at 3161, 3081, 2988 and 2952 cm−1 in the spectrum of compound 1 and seven peaks at 3516, 3369, 3219, 3158, 3100, 2988 and 2888 cm−1 in the spectrum of compound 2. The absorption bands at 3081, 2988, and 2952 cm−1 in the spectrum of 1 and 3100, 2988, and 2888 cm−1 in the spectrum of compound 2 are attributed to the ν(CH)arom. and ν(CH3) oscillations.
Absorption in the region of frequencies higher than 3100 cm−1 is attributed to the ν(OH) oscillations with different degrees of association35 involved in the formation of hydrogen bonds. In the spectral region of 3650–2600 cm−1 of compound 1, no clear peaks are highlighted, unlike those attributed to ν(CH)arom. and ν(CH3), which is evidence that the degree of association of the oscillations ν(OH) of the coordinated H2O, ν(OH) of the outer-sphere water molecules, and ν(NH+) with the participation of NO3− anions is pronounced.
The clearly visible absorption bands at 3516, 3369, 3219, and 3158 cm−1 in the spectrum of compound 2 can be explained by the grouping of hydrogen bonds according to their length into several groups, and the broad bands of weak intensity can be the PyH+ oscillations.36
In the spectra of compounds 1 and 2, the absorption bands of medium intensity at 3325 cm−1 and very high intensity at 1698 cm−1 disappear; however, they are observed in the spectrum of the non-coordinated Schiff base attributed to ν(NH) and ν(C=O), respectively. This is explained by the formation of metallocycles upon the complexation of the Schiff base, in which the redistribution of electron density occurs between the chemical bonds in the metallocycle.35
The absorption bands at 1521 cm−1 in the spectrum of compound 1 and 1529 cm−1 in the spectrum of compound 2 are attributed to the ν(C![[horiz bar, triple dot above]](https://www.rsc.org/images/entities/char_e0f1.gif) O) + ν(CC) oscillations.35 The highest intensity absorption band at 1382 cm−1 and the band at 825 cm−1 in spectrum 1 are attributed to the NO3− anions oscillations. In the spectrum of complex 2, a very intensive band at 1059 cm−1 is attributed to the ClO4− anion oscillations.35
O) + ν(CC) oscillations.35 The highest intensity absorption band at 1382 cm−1 and the band at 825 cm−1 in spectrum 1 are attributed to the NO3− anions oscillations. In the spectrum of complex 2, a very intensive band at 1059 cm−1 is attributed to the ClO4− anion oscillations.35
The type of substitutions in aromatic rings are distinguished by absorption bands that are manifested by the δ(CH)planar oscillations (in the region 1275–1000 cm−1 for 1,2- and 1175–1000 cm−1 for 1,2,3-substitution type) and δ(CH)noplanar (770–735 cm−1 for 1,2- and 810–750 cm−1 for the 1,2,3-substitution type).35,36 As seen in the spectra of the ligand and the complexes, the absorption bands of the δ(CH)nonplanar oscillations from the benzene rings are more pronounced and are manifested at 814 and 697 cm−1 in H2L, at 812 and 690 cm−1 in 1, and 808 and 679 cm−1 in 2 for four neighbouring hydrogen atoms and at 710 cm−1 in H2L, at 746 cm−1 in 1 and 744 cm−1 in 2 for the 1,2,3-substitution type.35
3.2. Crystal structures
The X-ray analysis indicated that 1 and 2 are ionic structures containing the mononuclear cationic complex [Fe(H2L)(H2O)2]3+, and NO3−/ClO4− anions, and the Schiff base ligand is coordinated in the neutral mode. Both salts crystallize in a triclinic space group P; however, an ideal asymmetric unit of this space group of 1 comprises one [Fe(H2L)(H2O)2]3+ cation, three non-coordinated nitrate anions and three outer-sphere water molecules, while in 2, there are two independent [Fe(H2L)(H2O)2]3+ cations A and B, three non-coordinated perchlorate anions and eight outer-sphere water molecules. In the studied structures, the proton transfers are observed from the nitrogen atoms of the amide groups to the N atoms of the terminal pyridine rings (PyH+); thus, the Schiff base ligands in 1 and 2 are coordinated in the form of double zwitterions. In these complexes, the Fe(III) coordination polyhedron is a pentagonal bipyramid. In each compound, the metal atoms are seven-coordinated (N3O4) with a practically symmetrical pentagonal bipyramidal environment. The equatorial plane contains one central pyridine nitrogen N4, two azomethine nitrogen atoms N3 and N5, as well as two carbonyl oxygen atoms O1 and O2. The Fe–O and Fe–N distances are within the range of 2.061(4)–2.085(2) and 2.185(5)–2.200(4) Å, respectively. The apical vertices of the pentagonal bipyramid are occupied by water molecules with Fe–Ow distances in the range of 1.995(2)–2.040(2) Å (Table S1, Fig. 1 and S3†).
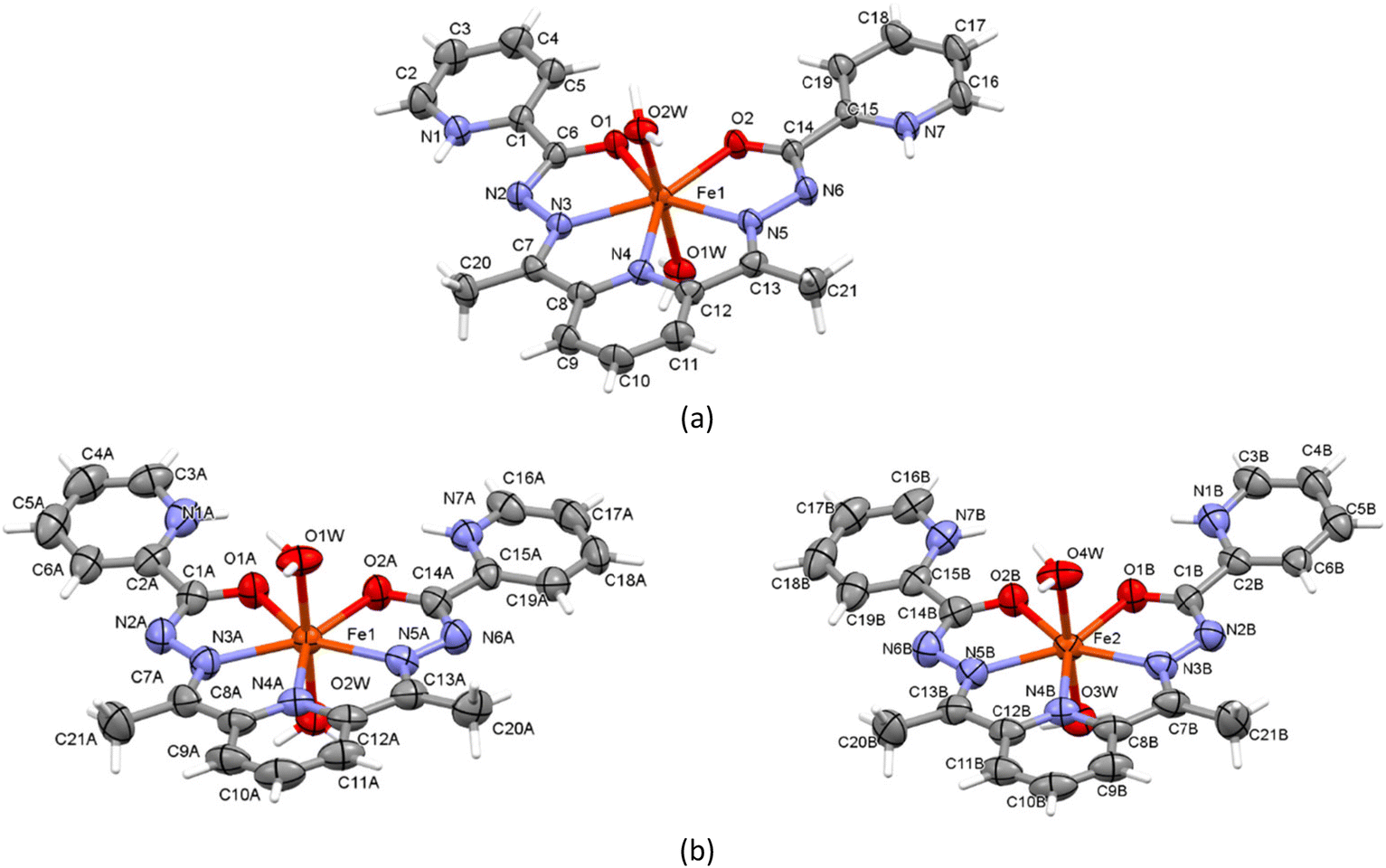 | ||
| Fig. 1 Atom labelling of complexes in 1 (a) and 2 (b). Thermal ellipsoids are drawn at 50% probability level. | ||
We have recently studied six Fe(III) coordination compounds with isomeric Schiff base ligands and these compounds differ from each other in the charge of the ligands, the anions and in the axial vertices of the iron(III) coordination polyhedra.23 In all complexes the metal's coordination polyhedron is the pentagonal bipyramid, formed by the coordination cores N3O4, N5O2, N4O3 and found Fe–O and Fe–N distances are consistent with similar bond lengths in 1 and 2. In the unit cell of 1, the mononuclear cations are joined into centrosymmetric dimers by the O(1w)–H⋯O(2) hydrogen bonds (HB), forming the R22(8) synthons (Fig. 2a and S3a†) and these dimers are linked by water molecules O(2w) and one of the three NO3− groups into chains along the [011] direction through the hydrogen bonds O(2w)–H⋯O(7), N(1)–H⋯O(8), C(20)–H⋯O(8), C(2)–H⋯O(6), and C(16)–H⋯O(7) (Table S2,†Fig. 2b). Finally, these chains are further extended into a 3D supramolecular architecture via hydrogen bonding involving the remaining NO3− anions and outer-sphere water molecules (Table S2, Fig. 2 and S4b and c†). Additionally, in the crystal packing of 1, the N–O⋯π interactions are present between two nitrate groups with the N4-containing pyridine ring of the cation with the O⋯Cg distances of 3.537 and 3.268 Å (Fig. S4d†).
 | ||
| Fig. 2 Two types of dimers in chains along the [011] direction of 1 (a and b). Fragment of crystal packing of 1 without the hanging contacts (c). | ||
In the crystal of compound 2, the independent cations A and B are linked by an extensive system of hydrogen bonds, which includes bifurcated HB involving two outer-sphere water molecules O8(w) and O(10w) and two tetrahedral ClO4− anions: O(2w)–H⋯O(8w), O(8w)–H⋯O(5), O(4w)–H⋯O(5), O(4w)–H⋯O(6), O(8w)–H⋯O(12), O(10w)–H⋯O(12), and O(4w)–H⋯O(10) (Table S2,†Fig. 3a). In turn, these hydrogen bonded cations A and B in the unit cell form the tetramer due to the centre of symmetry (Fig. 3b). In the tetramer, the centrosymmetric cations A are joined by the water molecule O8w and one of the ClO4− anions via the O(2w)–H⋯O(8w), O(8w)–H⋯O(12), and C(9A)–H⋯O(11) hydrogen bonds (Fig. 3d). In turn, the cations A and B′ are related by the water molecules O(10w) and anion through C(9A)–H⋯O(11)′, O(10w)′–H⋯O(12)′, O(4w)′–H⋯O(10w)′, as well as the C(20B)–H⋯N(2A) HB (Fig. 3d and e). However, the centrosymmetric complexes B⋯B are not linked to each other directly by hydrogen bonds. As in the case of compound 1, the Cl–O⋯π interactions are observed in 2. All ClO4− anions form the anion⋯π interactions between Cl–O and the coordinated pyridine rings (Cg(N4A/C8A–C12A) and Cg((N4B/C8B–C12B)) and terminal pyridines rings (Cg(N1A/C2A–C6A), Cg(N7A/C15A–C19A) Cg(N1B/C2B–C6B), and Cg(N7B/C15B–C19B)) (Table S3,†Fig. 3a). The hydrogen bonding in 2, involving the perchlorate anions, water molecules and Cl–O⋯π interactions lead to a 3D supramolecular structure of this compound.
 | ||
| Fig. 3 View of two independent complexes of 2 (a). Formation of a tetramer in the crystal structure of 2 (b). Fragment of crystal packing of 2 without hanging contacts (c). Hydrogen bonding and magnetic coupling between complexes A⋯A′ and A⋯B′ (d and e). | ||
3.3. Magnetic exchange
The iron(III) Schiff base complexes are interesting because of their structural diversities and applications in molecular magnetism. The iron(III) complexes with the N3O2-donor Schiff base ligands belong to a family of coordination compounds in which their structural features affect the resulting magnetic behaviour, spin states and spin crossover properties.37,38 Moreover, it was found that the magnetic properties of the high spin Fe(III) complexes with two different Schiff base ligands and different pseudohalides as counterions can be very well reproduced with a weak intermolecular antiferromagnetic interaction mediated through the inter-molecular hydrogen bonds.39 Therefore, the TB2J python package40,41 was used to study the magnetic interactions in coordination compounds 1 and 2. It was developed in the framework of density functional theory (DFT) and successively applied for extended systems, such as bulk or 2D materials.42–47The converged Hamiltonian and overlap matrix, obtained by Siesta software (v. 1.4.5),48,49 were used by the TB2J script to calculate the exchange interactions. TB2J python script, based on Green's function method, uses the Liechtenstein–Katsnelson–Antropov–Gubanov formula50 implemented in this package. This approach treats the local spin rotation of the numerical atomic orbitals for the magnetic atoms as a perturbation, avoiding the problems related to the total energy mapping. The calculations of the electronic structures of 1 and 2, based on the geometry determined by X-ray measurements, were done by DFT in generalized gradient approximation with Perdew–Burke–Ernzerhof exchange-correlation functional.51 The norm-conserving pseudopotentials of Troullier–Martins52,53 and atomic orbital basis sets were used, as implemented in the Siesta code. A linear combination of numerical atomic orbitals with double-polarized and single-polarized basis sets was applied for Fe and oxygen and nitrogen atoms, respectively, and a real-space grid cut off 200 Ry was used for the calculation of the electronic crystal structures. The first-principles calculations were performed with the 4 × 3 × 3 k-points sampling of the Brillouin zone. Owing to the confined Fe-3d orbitals, the DFT+U approach was used to simulate the magnetic and electronic properties of the studied compounds, as implemented in the Siesta program.
This method requires the knowledge of the Hubbard U parameter that describes the effective Coulomb on-site electron–electron interaction as well as the Hund exchange interaction parameter J, and these parameters were taken from.54 The magnetic interaction pathways and calculated isotropic exchange parameters Jiso are presented in Table 2. The magnetic exchange interactions occur only between those iron ions in the complexes, which are related by hydrogen bonds. The antiferromagnetic (AFM) interactions are observed for symmetrically related atoms Fe⋯Fe′ in complexes 1, and A⋯A′ and A⋯B′ in 2 (Fig. 3a, d and e), where the isotropic exchange parameters are in the range of −0.196–0.285 cm.−1 However, the very weak ferromagnetic interactions Fe1⋯Fe2 exist for independent compounds A and B, which are joined by extensive hydrogen bonds involving two outer-sphere water molecules and two ClO4− anions (Fig. 3a) if it is compared with the hydrogen-bound complexes A⋯A′ and B⋯B′.
| Compound | Coupling | J iso | d |
|---|---|---|---|
| 1 | Fe1⋯Fe1′ | −0.227 | 8.192 |
| 2 | Fe1⋯Fe2 | 0.01 | 8.408 |
| Fe1⋯Fe2′ | −0.196 | 9.469 | |
| Fe1⋯Fe1′ | −0.285 | 10.949 |
The spin-polarized density of states (DOS) and projected DOS have been calculated for 1 and 2 to understand the magnetic coupling in these compounds (Fig. 4). In compounds 1 and 2, both the majority (spin-up) and minority (spin-down) spin channels are the spin-gapless channels that might lead to a minimal amount of energy required to excite electrons from the valence to the conduction band due to zero gap and the availability of both charge carriers, i.e., electrons and holes, which may be spin-polarized simultaneously. In each of the studied compounds, the majority and minority spin channels arise within ca. −9.9–2.7 and −11.9–2.3 eV, respectively. However, the spin-up and spin-down states of iron(III) ions in 1 and 2 are delocalized within the same aforementioned ranges of energy (Fig. 4a and b), and such hybridization between Fe3+ and ligands atoms could facilitate the magnetic exchange observed in these compounds.
 | ||
| Fig. 4 DOS (upper panel) and PDOS (lower panel) of 1 (a) and 2 (b). Spin-up (positive) and spin-down (negative) density of states is shown by black and red color respectively. | ||
3.4. Ultraviolet-visible (UV-vis) spectroscopic study
The UV-Vis absorption spectra of Schiff base ligand and coordination compounds 1, 2 were determined in ethanol solution of 5 × 10−5 mol L−1. It was found that the profiles of the electronic spectra of complexes 1 and 2, are very similar, which confirms the ionic nature of these compounds, having the same complex cation [Fe(H2L)(H2O)2]3+ (Fig. 5). The broad bands of these compounds are observed in the UV region of spectra and maxima are centered at 363 and 365 nm, respectively. Two broad absorption bands of Schiff base ligand, whose maxima are centered at 275 and 313 nm, are also observed in UV region of spectra and the corresponding energy transfers could be attributed to π → π* transitions. In the wavelength region 272–317 nm the shapes of ligand and complexes absorption curves are similar, however for 1 and 2 there is the intensity decreasing compared with that in spectrum of ligand. Because of coordination, the bands, related to electronic transitions in the complexes, are red shifted relatively to those observed at the free Schiff base ligand. | ||
| Fig. 5 Absorption spectra of the H2L ligand and Fe(III) coordination compounds 1 and 2. | ||
The UV-vis spectral titration experiments of the H2L ligand with Fe3+ were carried out by gradually increasing the amount of Fe3+ (10–100 μL) in the ethanol media of the ligand. The successive addition of Fe3+ to H2L is accompanied by the appearance of shoulder peaks in the 200–350 nm region and one isosbestic point at 360 nm (1) and at 365 nm (2) (Fig. S5†). The ratio of the intensities of the peaks forming the spectra changed with the increasing Fe3+ concentration for both complexes: the intensity of the peaks at 285 and 325 nm decreased, and the intensity of the peak at 210 nm increased. The gradual addition of Fe3+ to the Schiff base ligand showed a hypochromic shift at 285 and 325 nm and a hyperchromic shift at 210 and 380 nm. The results show that the addition of Fe3+ to an ethanol solution of H2L leads to the formation of stable coordination compounds.
The DFT calculations were performed at the B3LYP/6-311++G(2d,2p)/IEFPCM (ethanol) level of theory using the Gaussian16 software55,56 for a detailed description of the observed UV-vis spectra of the complex cation [Fe(H2L)(H2O)2]3+. The molecular structure of the cation, determined by the X-ray method, was fully optimized with the aforementioned basis set in ethanol solvent, and all calculated frequencies were positive, confirming that a definite absolute minimum in the potential energy surface was obtained. To inspect the ground state, a series of DFT calculations at different levels of theory were done for the doublet, quartet and sextet states for [Fe(H2L)(H2O)2]3+, and it was shown that the high spin (HS) state (S = 5/2) is the most stable (Table S4†).
The HS and low spin (LS) states are energetically separated by 0.79 eV, indicating that the transition from a ground HS state to an excited LS state is not very likely to be observed. The time-dependent DFT (TDDFT) formalism was employed to study the UV-vis spectra in the solvent, and the GausSum-3.0 software57 was used for analyzing this spectrum. The obtained results are reported in Tables 3 and S5 (ESI†).
| Energy, eV | λ, nm | f | Major contributions | Assignment |
|---|---|---|---|---|
| 2.88 | 430.23 | 0.088 | HOMO(A) → LUMO(A) (38%), HOMO(B) → L + 2(B) (56%) | LLCT |
| 3.32 | 373.8 | 0.17 | HOMO(A) → L + 1(A) (25%), HOMO(B) → L + 4(B) (58%) | LLCT |
| 3.34 | 370.91 | 0.251 | H-1(A) → LUMO(A) (44%), H-1(B) → L + 2(B) (39%) | LLCT |
| 3.55 | 349.63 | 0.06 | H-1(A) → L + 1(A) (19%), HOMO(A) → L + 2(A) (10%), H-1(B) → L + 4(B) (53%) | LMLCT |
The calculated electronic transitions in the studied cation, which lead to UV and visible absorption, are in good agreement with the experimental data. The values of transition energies, whose oscillator strengths (f) are greater than 0.05, are in the same range 3.55–2.88 eV (350–430 nm), as shown in Fig. 5. The maximum of the absorption bands on the experimental curves are centred at 363 and 365 nm while the calculated wavelength with f = 0.25 is equal to 371 nm. As shown in Table 3, the occupied HOMO−1 alpha (A) and beta (B) spin molecular orbitals (MO) and antibonding LUMO(A) and LUMO+2(B) play important roles in this electron transition. The ligand 2,6-diacetylpyridine bis(picolinoylhydrazone) shows the main contribution in both the occupied and unoccupied MOs of the given transition, which is equal to 98% with the exception of HOMO−1(B) (96%) (Table S5†). The frontier MOs are π (π*) – orbitals (Fig. 6), and thus, the absorption maximum arises from the ligand-to-ligand π → π* charge transfer (LLCT). HOMO−1(A) and HOMO−1(B) ate centered mainly on pyridine nitrogen atom N4 and O–C–N–N–C fragments of metallocycles while LUMO(A) and LUMO+2(B) are centered practically on all non-hydrogen atoms of ligand. The same situation is observed for other electron transitions, listed in Table 3, with the exception of a transition with f = 0.06, where the corresponding metal and ligand contributions consist of 16 and 84%, respectively, which could be attributed to the metal–ligand-to-ligand charge transfer (LMLCT).
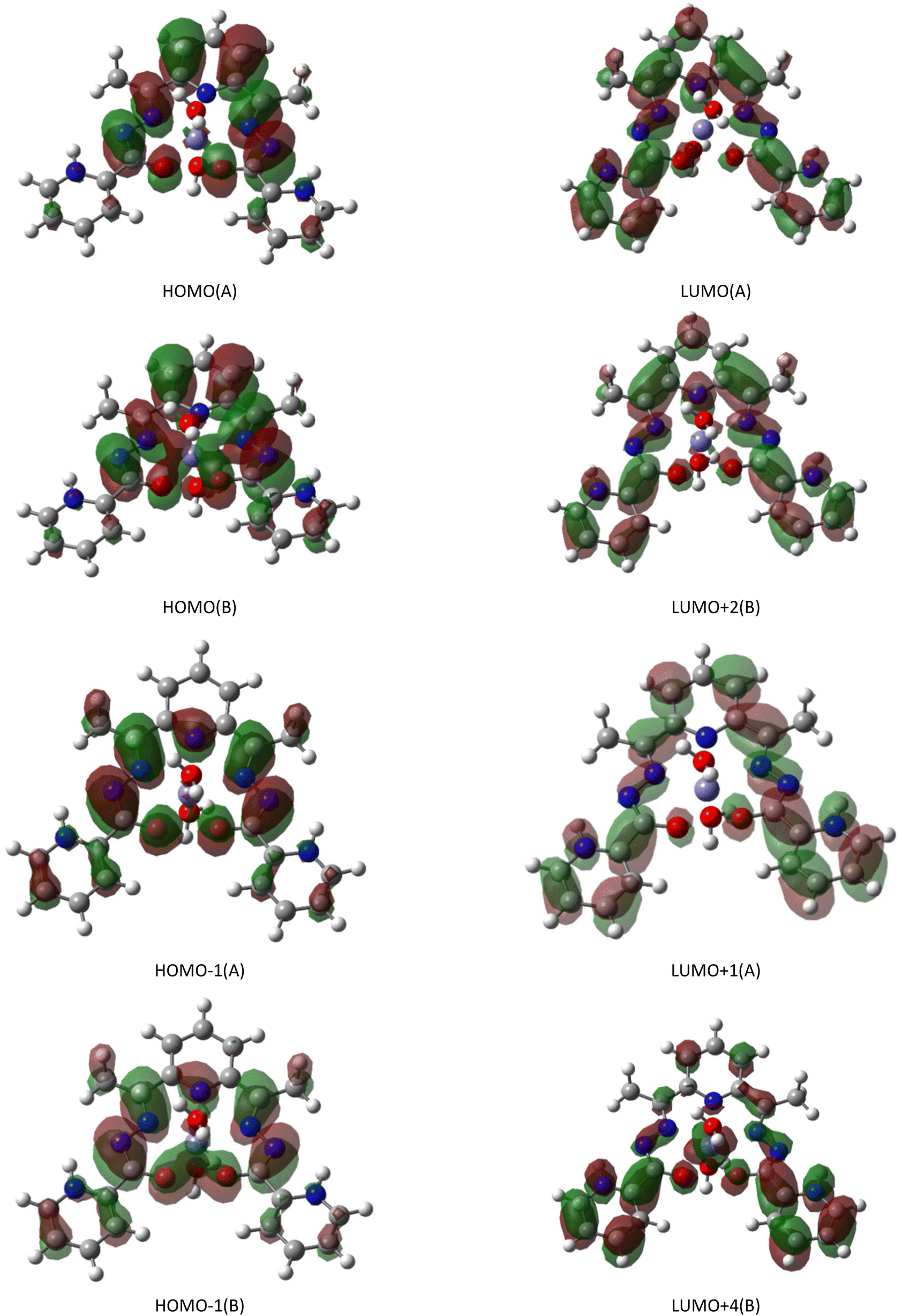 | ||
| Fig. 6 Selected frontier alpha (A) and beta (B) spin molecular orbitals for the calculated absorption transitions. | ||
3.5. Photoluminescent properties
The shape of the spectral dependencies of the luminescence spectra of all samples indicates the superposition of several radiative processes. For the deconvolution of the bands, the Gaussian function was used, which allowed the resolution of the spectra as superpositions of several radiative processes. The band of the PL spectrum for the studied complexes is a superposition of three peaks: in the red region of the spectrum, with 1.85 eV for 2 and 1.95 eV for 1; in the yellow region of the spectrum, with a maximum at 2.1 eV; and in the blue region of the spectrum with a maximum at 2.6 eV (Fig. 7). | ||
| Fig. 7 Solid-state PL emission spectra for complexes 1 and 2. | ||
The PL observed in the complexes is very weak compared to the ligand. The intensity of the PL peaks of the samples is more than 500 times weaker than the PL spectrum of the ligand H2L (Fig. S6†). The addition of iron to the ligand solution leads to changes in the photoluminescence spectra because of complexation. The maximum of the photoluminescence spectral band is shifted to the red region relative to the ligand.
The band structures of compounds 1 and 2 display metallic character because the majority and minority spin channels are spin-gapless. Therefore, the intensity reduction of the PL spectra in 1 and 2 in comparison with the ligand may be explained by the mechanism proposed in ref. 58 (Scheme S1†). In accordance with this mechanism, the PL is caused by emissions within the filled bands. However, in both the studied compounds, the number of electronic states is limited in the energy range of 2.2 eV above the Fermi level (Fig. 7).
Moreover, within this interval, related to the observed maxima of the PL peaks of the studied compounds, no spin-up electron states of iron ions are present. Thus, the number of photoexcited carrier relaxations in 1 and 2 could be less than in the ligand, which might lead to a decrease in the PL intensity of these compounds.
Conclusion
The new mononuclear iron(III) complexes, [Fe(H2L)(H2O)2](NO3)3·2.5H2O (1) and [Fe(H2L)(H2O)2](ClO4)3·2.5H2O (2), were synthesized and characterized by IR spectroscopy and X-ray diffraction. It was found that in the crystalline state, compounds 1 and 2 form the supramolecular dimers and tetramers, respectively, which are extended into a 3D supramolecular architecture via hydrogen bonding involving the NO3−/ClO4− counter anions and water molecules. The intermolecular magnetic couplings in the mononuclear high spin iron(III) Schiff base complexes were studied by the TB2J python package used for calculating the magnetic interaction parameters in Heisenberg models from density functional theory. The DFT-TB2J approach revealed the antiferromagnetic interactions and very weak ferromagnetic coupling between iron ions, mediated through the intermolecular hydrogen bonds. The UV-vis absorption spectra of the Schiff base ligand and coordination compounds 1 and 2 confirmed the presence of a complex cation [Fe(H2L)(H2O)2]3+ in ethanol solution. The TDDFT calculations were used to study the UV-vis spectra of the studied complexes, and it was shown that the absorption maxima arise from the ligand-to-ligand π → π* charge transfer. The measured photoluminescence spectra revealed that the PL peaks in 1 and 2 are considerably weaker than those in the ligand, and it is related to the spin-gapless character of the band structure of these complexes.Data availability
The data supporting this article have been included as part of the ESI.†The crystallographic data for compounds 1 and 2 have been deposited at the Cambridge Crystallographic Data Centre (CCDC 2400697 and 2400698) and can be obtained from http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: E-mail: deposit@ccdc.cam.ac.uk.
Author contributions
OD: conceptualization, methodology, software, investigation, validation, data curation, writing – original draft preparation, writing – review and editing, supervision. PB: methodology, software, investigation, validation, writing – review and editing. IB: formal analysis, investigation, writing – review and editing. OK: methodology, software, formal analysis, visualization, writing – original draft preparation. MC: formal analysis, investigation, writing – review and editing. YC: conceptualization, methodology, software, investigation, validation, data curation, writing – original draft preparation, writing – review and editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful for the sub-programs financed by the Ministry of Education and Research of the Republic of Moldova 011201, 011202 and 010602. Yu. Chumakov thanks the National Initiatives for Open Science in Europe for granting access to the e-infrastructure purchased under the National Roadmap for RI, financially coordinated by the MES of the Republic of Bulgaria (grant no. D01-325/01.12.2023).References
- M. Gruzdev, U. Chervonova and V. Vorobeva, Symmetry, 2022, 14, 1140 CrossRef CAS.
- X. Liu, C. Manzur, N. Novoa, S. Celedón, D. Carrillo and J.-R. Hamon, Coord. Chem. Rev., 2018, 357, 144–172 CrossRef CAS.
- I. Nemec, R. Herchel and Z. Trávníček, Dalton Trans., 2015, 44, 4474–4484 RSC.
- E. Coropceanu, A. Ciloci, A. Ştefîrţă and I. Bulhac, Study of useful properties of some coordination compounds containing oxime ligands, Academica Greifswald, 2020, p. 266 Search PubMed.
- M. Dolai, S. Pakrashy, A. K. Ghosh, S. Biswas, S. Konar, F. A. Alasmary, A. S. Almalki and M. A. Islam, J. Mol. Struct., 2023, 1274, 134584 CrossRef CAS.
- H. Toftlund, Coord. Chem. Rev., 1989, 94, 67–104 CrossRef CAS.
- R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed.
- B. Meunier, Biomimetic Oxidations, Catalyzed by Transition Metal Complexes, Imperial College Press, London, 2000 Search PubMed.
- G. D. Hosken and R. D. Hancock, J. Chem. Soc., Chem. Commun., 1994, 11, 1363 RSC.
- A. A. Khandar, F. A. Afkhami, S. A. Hosseini-Yazdi, J. M. White, S. Kassel, W. G. Dougherty, J. Lipkowski, D. Van Derveer, G. Giester and F. Costantino, Inorg. Chim. Acta, 2015, 427, 87–96 CrossRef CAS.
- T. Balić, F. Perdih, T. Mršo and I. Balić, Polyhedron, 2020, 190, 114774 CrossRef.
- N. Kaeosamut, Y. Chimupala and S. Yimklan, Cryst. Growth Des., 2021, 21, 2942–2953 CrossRef CAS.
- R. Custelcean, Chem. Soc. Rev., 2010, 39, 3675–3685 RSC.
- J. Zhao, D. Yang, X.-J. Yang and B. Wu, Coord. Chem. Rev., 2018, 378, 415–444 CrossRef.
- L. Yong-Jun, L. Hui-Biao and L. Yu-Liang, CCS chem., 2015, 31, 1687–1704 Search PubMed.
- Y. Marunaka, J. Physiol. Sci., 2023, 73, 31 CrossRef CAS PubMed.
- P. Gamez, Inorg. Chem. Front., 2014, 1, 35–43 RSC.
- O. Danilescu, P. Bourosh, I. Bulhac, S. Shova, V. Ch. Kravtsov, M. N. Caraba, I. V. Caraba, R. Popescu, M. Crisan, D. Haidu, O. Kulikova, N. V. Costriucova, O. Petuhov and L. Croitor, Polyhedron, 2024, 258, 117039 CrossRef CAS.
- O. Danilescu, I. Bulhac, P. N. Bourosh and L. Croitor, Polyhedron, 2022, 215, 115679 CrossRef CAS.
- CrysAlis RED, O.D.L. Version 1.171.34.76, 2003 Search PubMed.
- C. Pelizzi, G. Pelizzi and F. Vitali, Transition Met. Chem., 1986, 11, 401 CrossRef CAS.
- C. Pelizzi, G. Pelizzi, G. Predieri and S. Resola, J. Chem. Soc., Dalton Trans., 1982, 1349 RSC.
- O. Danilescu, P. Bourosh, O. V. Kulikova, Y. M. Chumakov, I. Bulhac and L. Croitor, Inorg. Chem. Commun., 2022, 146, 110199 CrossRef CAS.
- A. Mangia, C. Pelizzi and G. Pelizzi, Acta Crystallogr., 1974, 30, 2146 CrossRef.
- A. Bonardi, S. Ianelli, C. Pelizzi, G. Pelizzi and C. Solinas, Inorg. Chim. Acta, 1991, 187, 167–174 CrossRef CAS.
- M. U. Anwar, K. V. Shuvaev, L. N. Dawe and L. K. Thompson, Inorg. Chem., 2011, 50, 12141 CrossRef CAS PubMed.
- I. Bulhac, A. Deseatnic-Ciloci, P. Bourosh, J. Tiurina, O. Bologa, C. Bivol, S. Clapco, A. Verejan, S. Labliuc and O. Danilescu, Chem. J. Mold., 2016, 11, 39 CAS.
- K. Andjelkovic, A. Bacchi, G. Pelizzi, D. Jeremic and I. Ivanovic-Burmazovic, J. Coord. Chem., 2002, 55, 1385–1392 CrossRef CAS.
- C. Pelizzi and G. Pelizzi, Acta Crystallogr., Sect. B, 1979, 35, 126–128 CrossRef.
- CrysAlis RED, O.D.L. Version 1.171.34.76, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- K. Nakamoto, Infrared Spectra of Inorganic and Coordination Compounds, John Willey & Sons, New York, 5th edn, 1997 Search PubMed.
- L. Bellamy, IR spectra of complex molecules, Foreighn Liter, Leningrad, 1963 Search PubMed.
- C. Faulmann, J. Chahine, L. Valade, G. Chastanet, J.-F. Létard and D. de Caro, Eur. J. Inorg. Chem., 2013, 5–6, 1058–1067 CrossRef.
- V. Vorobeva, D. Starichenko, M. Gruzdev, U. Chervonova, R. Zaripov and A. Korolev, Appl. Organomet. Chem., 2022, 36, e6748 CrossRef CAS.
- T. Basak, C. J. Gomez-Garcıa, R. M. Gomila, A. Frontera and S. Chattopadhyay, RSC Adv., 2021, 11, 3315 RSC.
- X. He, N. Helbig, M. J. Verstraete and E. Bousquet, Comput. Phys. Commun., 2021, 264, 107938–107954 CrossRef CAS.
- X. Li, H. Yu, F. Lou, J. Feng, M.-H. Whangbo and H. Xiang, Molecules, 2021, 26, 803 CrossRef CAS PubMed.
- D. L. Esteras and J. J. Baldoví, Condensed Matter > Materials Science, arXiv, 2021, preprint, arXiv:2104.03023 [cond-mat.mtrl-sci], DOI:10.48550/arXiv.2104.03023.
- I. J. Onuorah, M. M. Isah, R. Renzi and P. Bonfà, Phys. Rev. Mater., 2021, 5, 124407 CrossRef CAS.
- S. Achilli, C. Besson, X. He, P. Ordejón, C. Meyer and Z. Zanolli, Phys. Chem. Chem. Phys., 2022, 24, 3780–3787 RSC.
- F. Ibrahim, A. Hallal, A. Kalitsov, D. Stewart, B. Dieny and M. Chshiev, Phys. Rev. Appl., 2022, 17, 054041 CrossRef.
- S. Teh and H.-T. Jeng, Nanomaterials, 2023, 13, 2644–2660 CrossRef CAS PubMed.
- M. Darii, J. Van Leusen, V. Ch. Kravtsov, Y. Chumakov, K. Krämer, S. Decurtins, Sh.-X. Liu, P. Kögerler and S. G. Baca, Cryst. Growth Des., 2023, 23, 6944 CrossRef CAS.
- J. M. Soler, E. Artacho, J. D. Gale, A. Garca, J. Junquera, P. Ordejon and D. Sanchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2767 CrossRef CAS.
- A. García, N. Papior, A. Akhtar, E. Artacho, V. Blum, E. Bosoni, P. Brandimarte, M. Brandbyge, J. I. Cerdá, F. Corsetti, R. Cuadrado, V. Dikan, J. Ferrer, J. Gale, P. García-Fernández, V. M. García-Suárez, S. García, G. Huhs, S. Illera, R. Korytár, P. Koval, I. Lebedeva, L. Lin, P. López-Tarifa, S. G. Mayo, S. Mohr, P. Ordejón, A. Postnikov, Y. Pouillon, M. Pruneda, R. Robles, D. Sánchez-Portal, J. M. Soler, R. Ullah, V. W.-Z. Yu and J. Junquera, J. Chem. Phys., 2020, 152, 204108 CrossRef PubMed.
- A. I. Liechtenstein, M. I. Katsnelson, V. P. Antropov and V. A. Gubanov, Local spin density functional approach to the theory of exchange interactions in ferromagnetic metals and alloys, J. Magn. Magn. Mater., 1987, 67, 65–74 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- N. Troullier and J. L. Martins, Phys. Rev. B:Condens. Matter Mater. Phys., 1991, 43, 8861–8869 CrossRef PubMed.
- A. Garcia, ATOM User Manual, Version 4.2.0, 2017, pp. 1–17 Search PubMed.
- E. B. Linscott, D. J. Cole, M. C. Payne and D. D. O'Regan, Phys. Rev. B, 2018, 98, 235157 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- D. M. Chipman, Theor. Chem. Acc., 2002, 107, 80–89 Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- P. Apell, R. Monreal and S. Lundqvist, Phys. Scr., 1988, 38, 174–179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photographs of crystals' compound 1 and 2 selected for the single crystal X-ray experiment (Fig. S1); the IR spectra of compounds 1, 2 and H2L Schiff base ligand (Fig. S2); selected bond lengths (Å) and angles (°) in coordination metal environment in 1 and 2 (Table S1); hydrogen bond distances (Å) and angles (°) in 1 and 2 (Table S2); geometrical parameters for Cl–O⋯π interactions (Table S3); coordination geometry of Fe(III) ion in 1 and 2 with partial atom labelling (Fig. S3); formation dimers in 1 (a); fragments of crystal packing of 1 (b, c); anion⋯π interactions in 1. Due to center of symmetry in the crystal the outer-sphere water molecules O4W and O5W form the four-membered rings (d) (Fig. S4); UV-visible study of H2L ligand titration profiles with varying concentration of Fe3+ ions from 10 to 100 μL: Fe(NO3)3 (a) and Fe(ClO4)3 (b) solutions (Fig. S5); the total energies (A.U.) for different spin multiplicities calculated at different levels of theory within the DFT methodology (Table S4); frontier orbital energies and percentage composition of molecular orbitals (MO) computed for studied cation. The MOs consist of the following fragments: iron ion, 2,6-diacetylpyridine (A) and two picolinoylhydrazones (B1 and B2) (Table S5); comparison of solid-state PL emission spectra for complexes 1, 2 and coordination agent (Fig. S6); schematic picture of the luminescence taken from ref. 58. A photon of energy ℏω1, promotes an electron from the filled (narrow) d-band to above the Fermi level EF (sp-band). The hole relaxes in momentum space by e.g., absorbing (E > E′) or emitting (E < E′) phonon and is eventually filled by an electron from the filled sp-band emitting photon of energy ℏω2, which is the luminescence photon (Scheme S1). CCDC 2400697 and 2400698. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00125k |
| This journal is © The Royal Society of Chemistry 2025 |