
Reversible phase transformations upon water uptake/removal in crystalline material of a bipodal N-donor ligand and evaluation of the stability of the hydrates formed†
Simran
Chaudhary
 a,
Marek
Wiśniewski
a,
Anna
Hoser
b,
Renny
Maria Losus
a,
Zbigniew
Rafiński
a and
Liliana
Dobrzańska
*a
a,
Marek
Wiśniewski
a,
Anna
Hoser
b,
Renny
Maria Losus
a,
Zbigniew
Rafiński
a and
Liliana
Dobrzańska
*a
aFaculty of Chemistry, Nicolaus Copernicus University in Toruń, Gagarina 7, 87-100 Toruń, Poland. E-mail: lianger@umk.pl
bDepartment of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Żwirki i Wigury 101, Warsaw, Poland
First published on 30th April 2025
Abstract
Isolation of the anhydrous form of 2,7-bis(imidazol-1-ylmethyl)-naphthalene (L, 1) as well as three of its hydrates, in particular the hemihydrate (L·0.5H2O, 2), monohydrate (L·H2O, 3) and dihydrate (L·2H2O, 4), allowed to gain insight into the mechanism of the hydration process. SCXRD studies revealed that in the hemihydrate, the water molecules interact with only one N-donor site originating from the ligand and serve as a bridge between two ligand molecules, whereas in the monohydrate, the water molecules interact with both N-donor sites available in the bipodal ligand, leading to the formation of binary supramolecular 1D chains. In the dihydrate, the water molecules are forming 1D chains by themselves and simultaneously interact with the ligand to yield 2D supramolecular layers. The lattice energies as well as the stabilisation energies of the hydrates indicate a clear relationship between the stability of the structures and the hydration level. Supportive studies on bulk material, such as temperature dependent PXRD, as well as TGA were also performed. PXRD indicated a reversible transition between anhydrate formed upon heating and dihydrate formed after immersion of the sample in water. Water adsorption/desorption isotherms further confirmed that the crystalline material undergoes transformations induced by water molecules.
1. Introduction
Hydrates are a class of multi-component crystalline solids, incorporating water into their crystal structure.1 Their formation is largely driven by the presence of hydrogen bond donors and acceptors within an anhydrous compound. Desiraju reported that the formation of hydrates is particularly favourable when the host molecule has a low hydrogen bond donor-to-acceptor ratio, typically less than 0.5.2 Later on, it was suggested that the presence of polar or charged functional groups in the molecule increases the likelihood of hydrate formation, as these groups enhance water interactions.3Hydrates can be divided into three categories (i) channel hydrates, (ii) isolated site hydrates, and (iii) metal ion-associated hydrates, based on structural features.4,5 Furthermore, there are two distinct types, namely stoichiometric and non-stoichiometric hydrates.6 However, this division can lead to confusion, as some authors include hydrates with a definite but non-integral ratio of water molecules in the group of non-stoichiometric hydrates7 and others restrict this subgroup to hydrates with no definite ratio of water molecules8 in relation to the host. The study of hydrates and their impact on the stability and structural diversity of molecular compounds has garnered substantial attention from both an experimental and theoretical point of view9–15 due to the pivotal roles that organic hydrates play in pharmaceutical formulations,16–19 and materials science.20 Their significance in the pharmaceutical industry is connected with the fact that their occurrence can alter the physicochemical properties of a drug substance. However, while hydrate formation can be beneficial in pharmaceuticals, the presence of water poses substantial challenges in materials science, such as stability issues in metal–organic materials (MOMs) and decreased gas sorption performance in physisorbents.21,22
Recently, computational and statistical tools have been employed to better understand hydrate formation23,24 and their properties, especially stability, complementing systematic studies on hydration and dehydration processes.25–28 Despite these advances, hydrate formation and their stability remain difficult to predict, posing an ongoing challenge in the fields mentioned above. Not without reason, Zaworotko and co-workers referred to hydrates as ‘the nemesis of crystal engineering’.29
In here, we present a series of hydrates of a bipodal ligand containing a naphthalene core (Scheme 1), which were isolated by screening the crystallisation products from a range of solvents. Their structures were revealed by SCXRD. It is worth pointing out that based on a recent (2020) report, the number of hydrate–anhydrate pairs characterised via SCXRD studies is very limited. For example, only 167 pairs containing both the anhydrate and hemihydrate forms were deposited at CSD at that time.7 Recently (2024), the formation of seven hydrates was reported; however, five out of these show similar structural features and differ merely by the degree of hydration.30 Furthermore, it was revealed that the hemi- and monohydrate which were isolated, are metastable phases, formed upon dehydration of the dihydrate. As the latter is a channel hydrate, such behaviour is rather uncommon, as usually such transitions take place between anhydrate and channel hydrate in one go and without significant structural rearrangements, resulting in similar unit cell parameters.31
 | ||
| Scheme 1 Representation of 2,7-bis(imidazol-1-ylmethyl)-naphthalene (L, 1). | ||
2. Experimental
2.1. Reagents and materials
All commercially available chemicals were of reagent grade and were used without further purification. Anhydrous solvents were prepared using an INERT PureSolv solvent purification system.2.2. Measurements
1H and 13C NMR spectra were recorded in CDCl3 on a Bruker Avance 700 MHz and 400 MHz instrument, respectively and referenced to residual solvent peaks. Melting points were collected with a Stuart SMP50 automated melting point apparatus, starting at a temperature of 30 °C and ramping up at 2 °C min−1.Thermal analysis (TGA, DTA) was performed on a TA Instruments SDT 650 analyser at a heating rate of 2 °C min−1 under dry nitrogen with a flow rate of 100 ml min−1 covering the temperature range: 25–600 °C.
PXRD patterns were obtained on a Philips X'Pert X-ray diffractometer using CuKα radiation. The samples were measured at the 2θ range of 4–45° with a scan speed of 4° min−1. The PXRD data were analysed using the Powder Cell,32 Profex33 and Match34 software packages.
Isotherms were determined at a temperature of 25 °C using a gravimetric adsorption apparatus equipped with Baratron pressure transducers (MKS Instruments, Germany). Each sample was thermally desorbed before the measurement under high vacuum until a constant mass was obtained (after ca. 2 days).
2.3. Computational studies
2D fingerprint plots were generated with Crystal Explorer 17.0 by using a standard 0.6–2.4 Å range including reciprocal contacts.35 The lattice energy calculations were performed at the (B3LYP/6-31G(d, p)) level of theory. The results were corrected for dispersion (Grimme D3 correction) and the basis set superposition error (BSSE). Ghost atoms used for the BSSE estimation were selected up to 5 Å distance from the considered molecule in the crystal lattice. The general methodology was analogous to that described in previous reports.36 Before starting DFT calculations, O–H bonds positions were elongated to standard neutron mean values for all, determined by SCXRD structures of the hydrates.2.4. Synthesis
The synthesis of L was reported earlier as it was used during the preparation of naphthalenophane ligands.37 However, we used an alternative procedure described below.Imidazole (1.3 g, 19.08 mmol), dissolved in 20 ml dry THF, was slowly added to sodium hydride (60% in oil, 0.763 g, 19.08 mmol) in dry THF (20 mL) at 0 °C, and the resulting suspension was stirred at rt for 2 h (until the hydrogen gas evolution ceased). 2,7-Bis(bromomethyl)naphthalene (1 g, 3.18 mmol) solution in 25 ml dry THF was then slowly added and the resulting mixture was refluxed for another 12 h. The obtained mixture was dissolved in water and extracted with DCM (3 × 50 mL). The combined organic layers were washed with water (100 mL), dried over anhydrous magnesium sulphate and filtered. After removal of the solvent under reduced pressure, the product was re-dissolved in ethyl acetate and hexane was added dropwise until the solution became cloudy, and this was left at rt to precipitate the pure product as a yellowish solid (mass – 0.75 g, yield – 52%). Melting point – 138–141 °C.
1H NMR (700 MHz, CDCl3): δ 7.83–7.84 (2H, d), 7.61 (2H, s), 7.54 (2H, s), 7.28–7.30 (2H, d), 7.13 (2H, s), 6.95 (2H, s), 5.28 (4H, s).
13C NMR (100 MHz, CDCl3): δ 137.52, 134.57, 133.19, 132.57, 129.97, 128.84, 126.16, 125.46, 119.37, 50.86.
The obtained pure compound was crystallized from solvents of different geometry and polarity, such as acetone, acetonitrile, DCM, MeOH, and THF, by dissolving 10 mg of the compound in 10 ml of each solvent. Vials covered with parafilm were left to undergo slow evaporation, which resulted in crystals of good quality for SCXRD in all vials.
2.5. Structure determination
X-ray crystallographic data for 1–4 were collected on a Rigaku XtaLab Synergy-S diffractometer equipped with graphite-monochromated CuKα radiation (λ = 1.5406 Å). The crystals were mounted on a loop upon coating with paratone-N oil. Data collection was carried out at 100(2) K to minimize solvent loss, possible structural disorder and thermal motion effects. Data frames were processed (unit cell determination, intensity data integration, correction for Lorentz and polarisation effects, and empirical absorption correction) by using the corresponding diffractometer's software package, CrysAlisPro.38 The structures were solved by using direct methods with SHELXS-2019/3 (ref. 39) and refined by using full-matrix least-squares methods based on F2 by using SHELXL-2019/3 (ref. 40) using the Olex41 interface. The programs Mercury42 and POV-Ray43 were both used to prepare molecular graphics. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were positioned geometrically with C–H = 0.95 Å (aromatic) and 0.99 Å (methylene) and refined as riding, with Uiso(H) = 1.2Ueq(C), except the water hydrogen atoms which were located in a difference map and refined freely in case of 2 and 3 and with restrained O–H bond lengths in case of 4. A summary of the data collection and structure refinement parameters is provided in Table 1.| Compound reference | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| a R 1 = ∑‖Fo∣ − ∣Fc‖/∑∣Fo∣. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | ||||
| Chemical formula | C18H16N4 | C18H17N4O0.5 | C18H18N4O | C18H20N4O2 |
| Formula mass | 288.35 | 297.35 | 306.36 | 324.38 |
| Crystal system | Triclinic | Monoclinic | Orthorhombic | Monoclinic |
| a/Å | 9.0492(3) | 20.5410(4) | 9.72620(10) | 18.149(4) |
| b/Å | 9.5052(4) | 5.46830(10) | 10.12810(10) | 9.6466(19) |
| c/Å | 9.9724(3) | 26.6872(5) | 31.3817(5) | 9.3799(19) |
| α/° | 62.525(4) | 90 | 90 | 90 |
| β/° | 75.236(3) | 102.478(2) | 90 | 93.06(3) |
| γ/° | 80.607(3) | 90 | 90 | 90 |
| Unit cell volume/Å3 | 734.86(5) | 2926.82(10) | 3091.35(7) | 1639.9(6) |
| Temperature/K | 100(2) | 100(2) | 100(2) | 100(2) |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c | Pbca | P21/c |
| No. of formula units per unit cell, Z | 2 | 8 | 8 | 4 |
| Radiation type | CuKα | CuKα | CuKα | CuKα |
| Absorption coefficient, μ/mm−1 | 0.632 | 0.676 | 0.680 | 0.089 |
| No. of reflections measured | 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 217 217 |
10042 |
26217 |
8358 |
| No. of independent reflections | 3044 | 2997 | 3187 | 2892 |
| R int | 0.0441 | 0.0307 | 0.0644 | 0.0470 |
| Final R1a values (I > 2σ(I)) | 0.0391 | 0.0371 | 0.0383 | 0.0581 |
| Final wR2b values (I > 2σ(I))b | 0.1008 | 0.0936 | 0.1030 | 0.1174 |
| Final R1a values (all data) | 0.0444 | 0.0421 | 0.0428 | 0.0880 |
| Final wR2b values (all data) | 0.1042 | 0.0973 | 0.1061 | 0.1293 |
| Goodness of fit on F2 | 1.056 | 1.048 | 1.039 | 1.057 |
3. Results and discussion
Crystallization of L from a range of solvents allowed us to isolate a series of hydrates: hemi- (2), mono- (3) and dihydrate (4). Interestingly, none any polymorph of the ligand was isolated during these screenings, which was a case for the related ligands containing more flexible biphenyl core.443.1. Crystal structures
1 crystallizes in the centrosymmetric space group of the triclinic system, with one molecule in the asymmetric unit. Its molecular structure is shown in Fig. 1. The angle between the mean planes of the imidazole rings is 52.6° and the angles between the mean planes of the imidazole rings and the naphthalene core are 84.13° and 89.04°, respectively. The molecules form rows extending along the a axis in which they adopt orientations alternating by an inversion centre. There are numerous C–H⋯N hydrogen bonds, resulting in the formation of a 3D supramolecular assembly, which is further stabilized by C–H⋯π interactions involving the methylene groups, naphthalene core and imidazole ring as donors and the naphthalene core, and imidazole rings as acceptors of these hydrogen bonds (Table 2). | ||
| Fig. 1 Molecular structure of 1 with atomic displacement plot shown at 50% probability. | ||
| Compound | D–H⋯A | H⋯A (Å) | D⋯A (Å) | D–H–A (°) | Compound | D–H⋯A | H⋯A (Å) | D⋯A (Å) | D–H–A (°) |
|---|---|---|---|---|---|---|---|---|---|
| (1) Cg1 is the centroid of the imidazole ring N1–C5, Cg3 is the centroid of the benzene ring C7–C12, Cg4 is the centroid of the benzene ring C10–C16, (2) Cg2 is the centroid of the imidazole ring N18–C22, Cg4 is the centroid of the benzene ring C10–C16, (3) Cg1 is the centroid of the imidazole ring N1–C5, Cg2 is the centroid of the imidazole ring N18–C22, (4) Cg1 is the centroid of the imidazole ring N1–C5, Cg3 is the centroid of the benzene ring C7–C12, Cg4 is the centroid of the benzene ring C10–C16, Symmetry codes (1): (i) 1 − x, 2 − y, 1 − z, (ii) x, y, z − 1, (iii) x, y − 1, z − 1, (iv) 1 − x, 1 − y, 1 − z, (v) x, 1 + y, 1 + z, (vi) 2 − x, 1 − y, −z, (vii) x, y, 1 + z, (viii) 2 − x, −y, −z; (2): (i) x, y, z, (ii) −x, y, 1/2 − z, (iii) 1/2 + x, 1/2 + y, z, (iv) x − 1/2, y − 3/2, z, (v) 1 − x, 2 − y, 1 − z, (vi) 3/2 − x, 5/2 − y, 1 − z, (vii) x, y − 1, z, (viii) 1/2 + x, 3/2 + y, z, (ix) x, 1 + y, z; (3): (i) x, y, z, (ii) 3/2 − x, 1 − y, z − 1/2, (iii) x − 1/2, 3/2 − y, 1 − z, (iv) 1/2 + x, 3/2 − y, 1 − z, (v) 1 − x, 1/2 + y, 3/2 − z, (vi) x − 1/2, y, 3/2 − z, (vii) 3/2 − x, y − 1/2, z, (viii) x, 3/2 − y, 1/2 + z; (4): (i) x, y, z, (ii) x − 1, y, z − 1, (iii) x, 3/2 − y, z − 1/2, (iv) 2 − x, 1/2 + y, 3/2 − z, (v) 1 − x, 1/2 + y, 1/2 − z, (vi) 1 − x, 2 − y, 1 − z, (vii) 1 − x, y − 1/2, 1/2 − z, (viii) 1 + x, 3/2 − y, 1/2 + z. | |||||||||
| 1 | C8–H8⋯N3i | 2.61 | 3.532(2) | 164 | 3 | O23–H23B⋯N3i | 2.01 | 2.887(1) | 170 |
| C13–H13⋯N3ii | 2.76 | 3.708(1) | 172 | O23–H23A⋯N20ii | 1.99 | 2.873(1) | 178 | ||
| C21–H21⋯N3iii | 2.77 | 3.605(2) | 148 | C6–H6A⋯N3iii | 2.67 | 3.649(1) | 170 | ||
| C22–H22⋯N3iv | 2.99 | 3.915(2) | 165 | C9–H9⋯N3iv | 2.88 | 3.741(1) | 152 | ||
| C4–H4⋯N20v | 2.90 | 3.675(2) | 139 | C16–H16⋯N20v | 2.90 | 3.538(1) | 125 | ||
| C5–H5⋯N20vi | 2.69 | 3.557(2) | 156 | C17–H17A⋯N20vi | 2.75 | 3.503(2) | 133 | ||
| C12–H12⋯N20vii | 2.53 | 3.471(1) | 169 | C4–H4⋯O23vii | 2.55 | 3.371(2) | 145 | ||
| C17–H17B⋯N20viii | 2.76 | 3.446(1) | 127 | C9–H9⋯O23iv | 2.85 | 3.666(1) | 145 | ||
| C13–H13⋯Cg1ii | 2.76 | 3.599(1) | 148 | C13–H13⋯O23iv | 2.52 | 3.421(1) | 158 | ||
| C6–H6A⋯Cg3iv | 2.62 | 3.524(2) | 151 | C16–H16⋯O23iii | 2.92 | 3.802(1) | 155 | ||
| C19–H19⋯Cg3vi | 2.59 | 3.517(1) | 166 | C21–H21⋯O23viii | 2.33 | 3.143(2) | 143 | ||
| C2–H2⋯Cg4iv | 2.89 | 3.780(1) | 156 | ||||||
| 2 | O23–H23A⋯N3i | 1.99 | 2.896(1) | 156 | 4 | O23–H23A⋯N3i | 1.93 | 2.856(3) | 165 |
| O23–H23B⋯N3ii | 1.94 | 2.896(1) | 168 | O23–H23B⋯O24i | 1.82 | 2.754(3) | 174 | ||
| C17–H17A⋯N3iii | 2.76 | 3.723(2) | 164 | O24–H24B⋯N20ii | 1.82 | 2.776(2) | 168 | ||
| C2–H2⋯N18iv | 2.67 | 3.399(2) | 134 | O24–H24A⋯O23iii | 1.86 | 2.791(3) | 167 | ||
| C9–H9⋯N20v | 2.82 | 3.632(2) | 144 | C17–H17A⋯N20iv | 2.71 | 3.693(4) | 170 | ||
| C21–H21⋯N20vi | 2.81 | 3.571(1) | 138 | C4–H4⋯O23iii | 2.93 | 3.784(3) | 150 | ||
| C22–H22⋯N20vii | 2.64 | 3.439(2) | 142 | C6–H6A⋯O23v | 2.71 | 3.515(3) | 138 | ||
| C16–H16⋯O23iii | 2.94 | 3.787(1) | 149 | C12–H12⋯O23v | 2.91 | 3.712(3) | 143 | ||
| C17–H17B⋯O23viii | 2.53 | 3.273(1) | 132 | C16–H16⋯O23vi | 2.85 | 3.412(3) | 118 | ||
| C2–H2⋯Cg2iv | 2.71 | 3.372(1) | 128 | C9–H9⋯O24vii | 2.72 | 3.585(3) | 153 | ||
| C19–H19⋯Cg4ix | 2.85 | 3.753(1) | 160 | C12–H12⋯O24v | 2.81 | 3.686(3) | 153 | ||
| C13–H13⋯O24vii | 2.77 | 3.624(3) | 150 | ||||||
| C16–H16⋯O24v | 2.82 | 3.692(3) | 153 | ||||||
| C21–H21⋯O24viii | 2.74 | 3.548(3) | 144 | ||||||
2 crystallizes as a hemihydrate in the C2/c space group of the monoclinic system, wherein the asymmetric unit contains one ligand molecule and half a water molecule, as the O atom is located on an inversion centre (Fig. 2). The angle between the mean planes of the imidazole rings is 46.42° and the angles between the mean planes of the imidazole rings and the naphthalene core are 77.35° and 84.71°, respectively, revealing certain conformational differences in comparison with the anhydrate form (Fig. S1†). A water molecule is interconnecting two ligands via strong O–H⋯N hydrogen bonds, namely O23–H23⋯N3 with an O⋯N distance of 2.897(1) Å and an O–H–N angle of 170°. Furthermore, it also acts as a hydrogen bond acceptor, participating in C–H⋯O interactions and yielding 2D supramolecular layers expanding along the a–b plane, which are further stabilised via C–H⋯π interactions. These are further extended in a third direction via weak C–H⋯N hydrogen bonds (see Table 2).
 | ||
| Fig. 2 On the left: representation of the asymmetric unit of 2 with atomic displacement plot shown at 50% probability; on the right: the supramolecular discrete motive stabilised by a water molecule. | ||
3 crystallizes as a monohydrate in the Pbca space group of the orthorhombic system with one ligand molecule and one water molecule in the asymmetric unit (Fig. 3).
 | ||
| Fig. 3 On the left: molecular structure of 3 with atomic displacement plot shown at 50% probability; on the right: fragment of the 1D supramolecular chain, bridged by water molecules. | ||
The angle between the mean planes of the imidazole rings is 54.20° and the angles between the mean planes of the imidazole rings and the naphthalene core are 89.89° and 86.02°, respectively, indicating more similarity with 1 than with 2 (Fig. S1†).
In this case, the water molecule interacts with both imidazole rings of the ligand molecule via strong O–H⋯N hydrogen bonds, O23–H23B⋯N3 (O⋯N distance of 2.887(1) Å, O–H–N angle of 170°) and O23–H23A⋯N20 (O⋯N distance of 2.873(1) Å, O–H–N angle of 178°), forming 1D zig-zag chains, which are extended into a 3D assembly via C–H⋯O hydrogen bonds. This assembly is further stabilised through C–H⋯N hydrogen bonds and π–π interactions, which involve adjacent naphthalene cores with a centroid–centroid distance of ca. 3.8 Å. Interestingly, C–H⋯π interactions are absent in this hydrate.
4 crystallizes as a dihydrate in the P21/c space group of the monoclinic system. It shows the presence of one ligand molecule and two water molecules in the asymmetric unit (Fig. 4). The angle between the mean planes of the imidazole rings is 63.73° and the angles between the mean planes of the imidazole rings and the naphthalene core are 83.85° and 87.01°, respectively. The water molecules interact with each other in this case, forming 1D chiral chains along the c axis via O23–H23B⋯O24 (O⋯O distance of 2.754(3) Å, O–H–O angle of 174°), and O24–H24A⋯O23 (O⋯O distance of 2.791(3) Å O–H–O angle of 167°) hydrogen bonding. These chains further interact with the ligand molecules via O–H⋯N hydrogen bonds, with O–N distances of ca. 2.8 Å and O–H–N angles oscillating around 167°, yielding 2D layers in the a–c plane which are stabilised via π–π interactions between adjacent naphthalene cores with a centroid–centroid distance of ca. 3.7 Å. These layers are extended in the third dimension via weak C–H⋯O hydrogen bonds and further stabilised via C–H⋯N hydrogen bonds. Like in 3, C–H⋯π interactions are absent. Interestingly, it was noticed before during studies on hydrates of five and six-membered N-heterocyclic aromatic compounds containing hydrogen bond acceptors, that hydrates in which water formed 1D chains showed a preference to form π–π interactions.10 The relative contributions (percentages) of selected intermolecular contacts to the Hirshfeld surface were calculated to further analyse the distribution of the intermolecular interactions. As shown in Fig. 5, the C–H⋯N and C–H⋯π hydrogen bonds gradually disappear upon hydration in favour of hydrogen bonds involving the O atoms and π–π interactions (see also Table 2).
 | ||
| Fig. 4 On the left: molecular structure of 4 with atomic displacement plot shown at 50% probability; on the right: fragment of the 2D supramolecular layers, bridged by water molecules. | ||
 | ||
| Fig. 5 The relative contributions (percentages) of selected intermolecular contacts to the Hirshfeld surface. | ||
3.2. Study on bulk material and hydrate conversions
PXRD studies indicated that all samples, isolated from different solvents, are mixtures containing products of different degrees of hydration (Fig. S2†), the relative contributions of which are influenced by time and storage conditions. However, upon heating the samples for a couple of minutes at 100 °C, the bulk material transforms to the anhydrate, which can be hydrated again by immersion of a powdered sample in water (Fig. 6).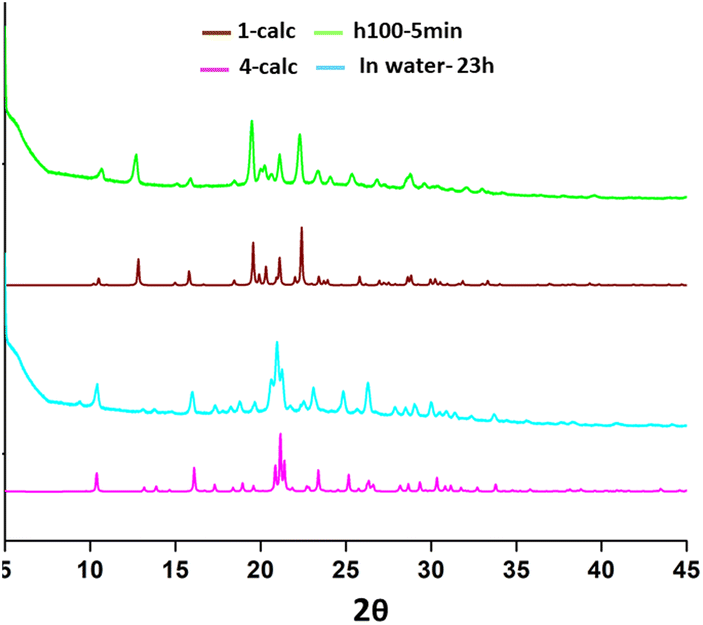 | ||
| Fig. 6 Exemplary experimental PXRD patterns of a sample obtained by crystallisation from THF: heated for 5 minutes at 100 °C, immersed for 23 h in water upon heating, and the simulated PXRD patterns for the anhydrate 1 and the dihydrate 4, respectively. | ||
TGA of the hydrated samples indicates that dehydration occurs in separate steps: the first up to approximately 45 °C, the next up to 70 °C, and the last up to around 141 °C (melting), showcasing the gradual process of dehydration (Fig. S3†).
This observation was further confirmed by collecting the adsorption/desorption isotherms of H2O. The adsorption branch follows an almost horizontal course up to a relative pressure of about 0.8 p/p0, which could indicate that the tested material is rather hydrophobic in nature. When the relative pressure of water vapor exceeds the value of 0.85, the structure reorganizes, allowing capillary condensation and reaching near-maximum adsorption. The observed type V adsorption isotherm results from the formation of only a limited number of adsorption layers within the adsorbent pores due to their hydrophobicity. It is evident that the adsorbent–adsorbate interactions are significantly weaker than the adsorbate–adsorbate interactions, leading to condensation in the adsorbent pores at high relative pressures, resulting in significant hysteresis. The formation of bridged water molecules via hydrogen bonds in micropores is the main reason for the observed phenomenon. Interestingly, the desorption branch of the isotherm clearly reflects the gradual nature of the process, specifically the transitions between forms with different degrees of hydration (Fig. 7). The plateau observed at maximum relative pressure corresponds to the presence of ca. 1.7 adsorbate molecules in the adsorbent structure (the discrepancy between the number of water molecules in the crystal structure obtained from the SCXRD and the adsorption studies is most likely caused by the statistical distribution of crystallite grains). The next plateau appears at a relative pressure of about 0.7 p/p0 and corresponds to the presence of the monohydrate (about 1.1 molecules of H2O per ligand). At a relative pressure below 0.15 p/p0, partial desorption is observed, leading to the third plateau whereby approximately 0.5 molecules of H2O are associated with each molecule of the adsorbent (hemihydrate formation).
 | ||
| Fig. 7 Water adsorption/desorption isotherms as a function of relative pressure, with markings indicating the formation of 1–4. | ||
The revealed structural transitions taking place in the bulk let us have one more look at the crystal structures 1–4, whereby crystal packing similarities, despite changes in crystallographic parameters (Table 1), support the gradual mechanism of the dehydration process. The process is connected with a significant reorganization on molecular level by forming new intermolecular interactions upon water removal, achieved through molecular shifts and small conformational adjustments of the ligand (Fig. 8), the changes of which were followed in a single-crystal to single-crystal fashion.
 | ||
| Fig. 8 Packing diagrams of 1–4 shown along the b (1–3) and c (4) axis, small apertures present in 1 indicated in yellow, water molecules shown in space filling representation. | ||
There are small apertures in the anhydrous form (that could be mapped with a probe of 0.9 Å per contact surface), which could be sufficient to initiate water uptake, as a related phenomenon was observed in the case of an apohost of an organic macrocycle undergoing reversible uptake of THF in a single-crystal.45 However, immersion of anhydrous bulk material in water leads to the formation of the dihydrate form via recrystallization, as shown in Fig. S4.†
3.3. Crystal lattice energy calculations and stabilisation energy of hydrates
Hydrates exhibit different thermodynamic stability and solubility compared to their pure anhydrous forms. Typically, the introduction of water into the crystal lattice leads to a decrease in lattice energy per unit cell, as water molecules can form additional intermolecular hydrogen bonds, increasing the number of interactions. This can also be observed for the hydrates presented here, for which the lattice energy decreases upon hydration (Table 3). Furthermore, the stabilisation energy of the hydrates (Estab) was determined following the procedure reported by Braun et al.46 whereby Estab was calculated in order to predict the occurrence of particular hydrates. The stabilisation energy relates the different hydrates stoichiometries to the anhydrate and ice. By evaluating this energy, it is possible to determine which hydrates are more thermodynamically stable compared to the anhydrous form. The lattice energy of ice was calculated at the same theory level as in mentioned above report, varying in our case from −60 to −64 kJ mol−1 for the ordered ice polymorphs.47 All determined Estab (Table 3) are lower than the lattice energies calculated for the ice polymorphs, suggesting that the hydrates obtained are thermodynamically stable, which is in agreement with the experimental results.| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| E latt/per unit cell (kJ mol−1) | −198.7 | −263.3 | −266.9 | −365.8 |
| E stab (kJ mol−1) | −67.0 | −68.2 | −83.6 |
Conclusions
The crystalline material based on bipodal heterocyclic ligand with rigid naphthalene core appears to undergo a series of transformations induced by water uptake/removal. The process is connected with the molecular reorganisation, whereby weak C–H⋯N and C–H⋯π hydrogen bonds present in the anhydrate are replaced with strong O–H⋯N and O–H⋯O hydrogen bonds upon water incorporation into the crystalline material and dihydrate formation (channel hydrate; formed in water via recrystallization of the anhydrous form). Upon desorption, this channel hydrate gradually transforms to the anhydrate via mono- and hemihydrate phases, which is clearly reflected in the desorption branch of the H2O isotherm and it is possible to follow these transformations in a single-crystal. The crystal lattice energies show significant differences between the most stable dihydrate and the metastable phases of mono- and hemihydrate, which could be the reason that a one-step adsorption isotherm is observed, whereas the stabilisation energy confirms the feasibility of isolating all three hydrates experimentally. Computational studies, allowing to model such transitions, would provide valuable insights, but they are not yet feasible.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
SC, RML and LD would like to thank the National Science Centre – Poland for grant no. 2014/14/E/ST5/00611. We would also like to thank the programme Excellence Initiative – Research University for funding the research group of Crystal Engineering and Advanced Solid-State Characterisation, as well as the Polish high-performance computing infrastructure PLGrid (HPC Centers: ACK Cyfronet AGH, WCSS) for providing computer facilities and support within computational grant no. PLG/2024/017644.References
- C. H. Görbitz and H. P. Hersleth, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 526–534 CrossRef PubMed.
- G. R. Desiraju, J. Chem. Soc., Chem. Commun., 1991, 426–428 RSC.
- L. Infantes, L. Fábián and W. D. S. Motherwell, CrystEngComm, 2007, 9, 65–71 RSC.
- K. R. Morris, Polymorphism in Pharmaceutical Solids, ed. H. G. Brittain, Marcel Dekker, Inc., New York, 1999, pp. 125–181 Search PubMed.
- K. R. Morris and N. Rodriguez-Hornedo, Encyclopedia of Pharmaceutical Technology, ed. J. Swarbrick and J. C. Boylan, Dekker, New York, 1993, vol. 7, pp. 393–441 Search PubMed.
- U. J. Griesser, Polymorphism in the Pharmaceutical Industry, ed. R. Hilfiker, Wiley-VCH, Weinheim, 2006, pp. 211–257 Search PubMed.
- J. E. Werner and J. A. Swift, CrystEngComm, 2020, 22, 7290–7297 RSC.
- E. Tieger, V. Kiss, G. Pokol, Z. Finta, M. Dušek, J. Rohlíček, E. Skořepová and P. Brázda, CrystEngComm, 2016, 18, 3819–3831 RSC.
- A. Bajpai, H. S. Scott, T. Pham, K. J. Chen, B. Space, M. Lusi, M. L. Perry and M. J. Zaworotko, IUCrJ, 2016, 3, 430–439 CrossRef CAS PubMed.
- R. Sanii, E. Patyk-Kaźmierczak, C. Hua, S. Darwish, T. Pham, K. A. Forrest, B. Space and M. J. Zaworotko, Cryst. Growth Des., 2021, 21, 4927–4939 CrossRef CAS PubMed.
- S. K. Rai, A. Gunnam, G. J. O. Beran, J. A. Kaduk and A. K. Nangia, Cryst. Growth Des., 2023, 23, 1179–1188 CrossRef CAS.
- P. Klitou, I. Rosbottom and E. Simone, Cryst. Growth Des., 2019, 19, 4774–4783 CrossRef CAS.
- A. T. Hulme and S. L. Price, J. Chem. Theory Comput., 2007, 3, 1597–1608 CrossRef CAS PubMed.
- R. Mizoguchi and H. Uekusa, Cryst. Growth Des., 2018, 18, 6142–6149 CrossRef CAS.
- A. A. Malik and A. A. Dar, J. Mol. Struct., 2024, 1310, 138254 CrossRef CAS.
- E. Jurczak, A. H. Mazurek, Ł. Szeleszczuk, D. M. Pisklak and M. Zielińska-Pisklak, Pharmaceutics, 2020, 12, 1–25 CrossRef PubMed.
- P. Chakravarty and K. Nagapudi, TrAC, Trends Anal. Chem., 2021, 140, 116276 CrossRef CAS.
- A. M. Healy, Z. A. Worku, D. Kumar and A. M. Madi, Adv. Drug Delivery Rev., 2017, 117, 25–46 CrossRef CAS PubMed.
- R. K. Khankari and D. J. W. Grant, Thermochim. Acta, 1995, 248, 61–79 CrossRef CAS.
- Clathrate Hydrates: Molecular Science and Characterization, ed. J. A. Ripmeester and S. Alavi, John Wiley & Sons, 2022 Search PubMed.
- A. Kumar, D. G. Madden, M. Lusi, K. J. Chen, E. A. Daniels, T. Curtin, J. J. Perry and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- Y. Ming, J. Purewal, J. Yang, C. Xu, R. Soltis, J. Warner, M. Veenstra, M. Gaab, U. Müller and D. J. Siegel, Langmuir, 2015, 31, 4988–4995 CrossRef CAS PubMed.
- S. Boothroyd, A. Kerridge, A. Broo, D. Buttar and J. Anwar, Cryst. Growth Des., 2018, 18, 1903–1908 CrossRef CAS.
- D. E. Braun and U. J. Griesser, Cryst. Growth Des., 2016, 16, 6405–6418 CrossRef CAS PubMed.
- R. Bobrovs, A. Kons, A. Berziņš, T. Rekis and A. Actiņš, Cryst. Growth Des., 2018, 18, 2100–2111 CrossRef CAS.
- R. Okura, H. Uchiyama, K. Kadota and Y. Tozuka, CrystEngComm, 2021, 23, 4816–4824 RSC.
- D. E. Braun, J. A. McMahon, R. M. Bhardwaj, J. Nyman, M. A. Neumann, J. Van De Streek and S. M. Reutzel-Edens, Cryst. Growth Des., 2019, 19, 2947–2962 CrossRef CAS.
- D. E. Braun, L. H. Koztecki, J. A. Mcmahon, S. L. Price and S. M. Reutzel-Edens, Mol. Pharmaceutics, 2015, 12, 3069–3088 CrossRef CAS PubMed.
- H. D. Clarke, K. K. Arora, H. Bass, P. Kavuru, T. T. Ong, T. Pujari, L. Wojtas and M. J. Zaworotko, Cryst. Growth Des., 2010, 10, 2152–2167 CrossRef CAS.
- O. Kaszubowski, K. Ślepokura, K. N. Truong and J. Wojciechowski, Cryst. Growth Des., 2024, 24, 8516–8527 CAS.
- S. Kuang, H. S. Shah and B. Zhao, Pharm. Res., 2024, 41, 1533–1541 CrossRef CAS PubMed.
- W. Kraus and G. Nolze, J. Appl. Crystallogr., 1996, 29, 301–303 CrossRef CAS.
- N. Doebelin and R. Kleeberg, J. Appl. Crystallogr., 2015, 48, 1573–1580 CrossRef CAS PubMed.
- Match! - Phase Analysis using Powder Diffraction, Crystal Impact - Dr. H. Putz & Dr. K. Brandenburg GbR, Kreuzherrenstr. 102, 53227 Bonn, Germany, https://www.crystalimpact.de/match.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- S. Feng and T. Li, J. Chem. Theory Comput., 2006, 2, 149–156 CrossRef CAS PubMed.
- C. Caron, X. N. T. Duong, R. Guillot, S. Bombard and A. Granzhan, Chem. – Eur. J., 2019, 25, 1949–1962 CrossRef CAS PubMed.
- Rigaku Oxford Diffraction, CrysAlisPro Sofware System, version 1.171.38.41, Rigaku Corporation, Oxford, UK, 2015 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- http://www.povray.org/ .
- S. Chaudhary, D. Kędziera, Z. Rafiński and L. Dobrzańska, RSC Adv., 2023, 13, 30625–30632 RSC.
- J. Thomas, G. Reekmans, P. Adriaensens, L. Van Meervelt, M. Smet, W. Maes, W. Dehaen and L. Dobrzańska, Angew. Chem., Int. Ed., 2013, 52, 10237–10240 CrossRef CAS PubMed.
- D. E. Braun, P. G. Karamertzanis and S. L. Price, Chem. Commun., 2011, 47, 5443–5445 RSC.
- F. Della Pia, A. Zen, D. Alfè and A. Michaelides, J. Chem. Phys., 2022, 157, 134701 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Overlays of the ligands: 1/2, 1/3 and 1/4, PXRD patterns obtained for the products isolated from different solvents, thermogram of the bulk hydrated sample. CCDC reference numbers: 2400655–2400658. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00130g |
| This journal is © The Royal Society of Chemistry 2025 |