
Time dependence of water-induced phase transition in nano- and microcrystalline Eu3+-doped MOF-76(Y): different luminescence responses with memory effect†
Zulma L.
Moreno Botello‡
 a,
Marcos
Illescas
b,
Beatriz C.
Barja
b,
Sebastian E.
Collins
c,
Griselda E.
Narda
a and
María C.
Bernini
*a
a,
Marcos
Illescas
b,
Beatriz C.
Barja
b,
Sebastian E.
Collins
c,
Griselda E.
Narda
a and
María C.
Bernini
*a
aFacultad de Química, Bioquímica y Farmacia, Universidad Nacional de San Luis, Instituto de Investigaciones en Tecnología Química (INTEQUI-CONICET), San Luis, Argentina. E-mail: mcbernin@email.unsl.edu.ar
bINQUIMAE-DQIAQF, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Pab. II (1428), Buenos Aires, Argentina
cInstituto de Desarrollo Tecnológico para la Industria Química (CONICET, UNL), Facultad de Ingeniería Química, Universidad Nacional del Litoral (UNL), Güemes 3450, Santiago del Estero 2829, 3000 Santa Fe, Argentina
First published on 8th May 2025
Abstract
MOF-76 compounds were solvothermally synthesized, producing micrometer-sized needle-like crystals, while bean-shaped nanoparticles were successfully obtained by modifying the synthesis conditions and using sodium acetate as a modulator. The water-induced transition of MOF-76(Y1−xEux) x = 0, 0.05 in nano and microcrystalline forms was studied and followed by PXRD, FTIR, SEM, and TEM. The phase transition involves changing the 3D MOF-76 (tetragonal) structure to a 1D Ln-BTC (monoclinic) structure upon water exposure. The influence of particle size, mechanic stress (ultrasound treatment), and thermal activation on the kinetics of the structural and morphological transformation was studied. Besides, the changes in photophysical properties resulting from the phase transition are also analyzed. Photophysical properties of MOF-76(Y0.95Eu0.05) were studied before and after water exposure, revealing efficient UV absorption and characteristic Eu3+ emissions for all samples. Although the emission spectra of the samples synthesized in nanometric and micrometric forms exhibit the same peaks and almost the same intensity, the corresponding spectra of the compounds upon water immersion are different, indicating a likely memory effect in transformed MOF-76 compounds regarding the initial particle size of each one. This research provides valuable insights into the material stability and phase behavior with a MOF-76 structure under different operating conditions. These results are critical for their practical applications in sensing and other technological fields.
1. Introduction
Metal–organic frameworks (MOFs) have emerged as interesting materials derived from 2D or 3D coordination networks that exhibit varied applications. After over two decades of research, along with a deep understanding of their building unit connectivity, the number of MOF structures has grown to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000+ in the CSD subset.1 In such a large number of crystal structures, the most outstanding fields of applications for these materials include gas adsorption/separation properties,2,3 catalytic4 and photocatalytic5 behaviours, chemical4 and biochemical6 sensing, and controlled drug release.7 All the properties present in these curious materials are derived from the combination of selected metal ions and organic linkers that bind in a self-assembled process, leading to a specific MOF architecture. If the chemical composition or the crystal structure changes, the physicochemical properties of MOFs (frequently) change8 as well since the composition-structure-properties are correlated. Some chemical compositions embedded in a particular crystal structure or “phase” are more appropriate for some applications than others. For example, selecting some lanthanide ions and aromatic organic linkers to develop luminescent MOFs can lead to the formation of successful emission materials.9 The application of luminescent MOFs based as supramolecular sensors9 is a research field that has attracted a lot of attention in the scientific community, and several lanthanide-MOF structures were synthesized and evaluated as novel platforms for chemical or physical (temperature) sensing.10,11
000+ in the CSD subset.1 In such a large number of crystal structures, the most outstanding fields of applications for these materials include gas adsorption/separation properties,2,3 catalytic4 and photocatalytic5 behaviours, chemical4 and biochemical6 sensing, and controlled drug release.7 All the properties present in these curious materials are derived from the combination of selected metal ions and organic linkers that bind in a self-assembled process, leading to a specific MOF architecture. If the chemical composition or the crystal structure changes, the physicochemical properties of MOFs (frequently) change8 as well since the composition-structure-properties are correlated. Some chemical compositions embedded in a particular crystal structure or “phase” are more appropriate for some applications than others. For example, selecting some lanthanide ions and aromatic organic linkers to develop luminescent MOFs can lead to the formation of successful emission materials.9 The application of luminescent MOFs based as supramolecular sensors9 is a research field that has attracted a lot of attention in the scientific community, and several lanthanide-MOF structures were synthesized and evaluated as novel platforms for chemical or physical (temperature) sensing.10,11
Within this large subset of luminescent MOFs, the so-called MOF-76 stands out, whose structure was first reported by O. M. Yaghi et al.12 as one example of a 3D MOFs containing rod-shaped secondary building units (SBUs). The authors used Tb3+ ion as a metal node and 1,3,5-benzenedicarboxylic acid (BTC) as a ligand. Later, several lanthanides (Ln3+ = Nd3+,13 Eu3+,14,15 Gd3+,16 Tb3+,13,15 and Ho3+,17) and rare earth (Y3+) ions have been employed as metal centres in the MOF-76 structure. The members based on Eu3+,15,18–24 and Tb3+,15,19,20,22,25–27 have received more attention due to their strong red and green emissions, respectively, allowing the exploration of the performance of these materials as luminescence sensors.14,15,18,21,22,26–34
Similar to other archetypical MOFs, synthesis conditions to obtain nanocrystalline solids with the MOF-76 structure have been explored.15,19,27,31 These efforts have been driven to improve or enlarge the resultant applications, such as building homogeneous thin films for luminescent sensing devices. Importantly, in addition to particle size,35,36 dimensionality,37–41 and composition,42 the phase of MOFs; that is, the particular ordered arrangement of building blocks, is also a key parameter to tune the performances of MOFs. Notably, different phases of MOFs can exhibit different linker connectivity and lanthanide symmetry sites, which could also modify the luminescent performances of MOFs.43,44 In this sense, it is important to remark that as in other cases of metal–ligand systems, structures other than MOF-76 can be obtained by combining the same Ln3+ ions and the BTC linker and changing the synthesis conditions. Thus, an isostructural family of 1D coordination polymer based on Ln3+ ions and BTC ligand has been reported34,45–50 and the member based on Eu3+ has also been widely studied as a luminescence sensor of picric acid,32 dipicolinic acid,22 and so on. Unfortunately, both materials i.e. the 3D MOF-76 and the polymer 1D-coordination are frequently indistinctly referred to as LnBTC, which is not only unclear but also inconvenient since the luminescence property strongly depends on the crystal structure.8
Knowledge of the structural stability of a specific phase is crucial for shortening the way between fundamental studies and applications. Although the research about phase transformation is essential,8 the huge number51 of reported MOF architectures do not correlate appropriately with phase stability studies. In this sense, even though the phase transition of the MOF-76 structures into the 1D coordination polymer driven by water has already been reported,34 i) the kinetics involved in this process depending on the particle size of synthesized MOF-76 and ii) the differences in the luminescence properties of the transformed phase concerning the particle size of their precursors have not been investigated yet. Regarding the cost of lanthanide ions as well as the existence of concentration quenching issues, if the whole emissive matrix is made completely by the emissive lanthanide ion itself; a common strategy in the development and studying of luminescent materials involves the use of a non-emissive host matrix (for example an Y3+-based MOF) and diluting the emitter Ln3+ ions (i.e., Eu3+ or Tb3+) as dopants.19,52,53 This allows for high-intensity luminescence emission by using small amounts of expensive lanthanide emitter ions. These premises have driven our interests in studying the water stability and phase transition of both the MOF-76(Y) material as well as the MOF-76(Y0.95Eu0.05) doped one. Furthermore, the last material allowed us to evaluate and determine how the water-induced phase transition impacts the luminescence properties. A comparison of the stability of different particle-sized solids exhibiting the MOF-76 structure is analysed in the present work when such samples are in contact with water, ethanol, or methanol. These solvents are selected since they are the most frequently used to dissolve analytes for their determination by luminescence sensing.14,32,34 Moreover, the incidence of incorporating ultrasound during the contact time with solvents and the effect of thermal activation of the synthesized solids (micro- and nanocrystalline versions) before the immersion in different solvents are also investigated. Throughout the whole experimental study, the influence of particle size, mechanic forces, and thermal activation on the kinetics of the structural transformation from (3D) MOF-76(Y) → (1D) LnBTC in water is demonstrated for the first time for this metal–organic system. Furthermore, the variations in the luminescence of MOF-76(Y:Eu) phases before and after the phase transition with nano and micro sized particles as starting materials are evaluated and discussed.
2. Experimental section
2.1 Reactants and solvents
Powder precursors: yttrium(III) oxide (99.99%) and trimesic acid H3BTC (95%) were from Sigma-Aldrich. Europium(III) oxide (99.9%) was from Alfa Aesar. Sodium acetate trihydrate (99.99%) was from Merck. Solvents: anhydrous N,N-dimethylformamide (DMF) (99.8%), ethanol (99.5%) and anhydrous (99.8%) were from Biopack. Nitric acid (65%) was from Cicarelli.2.2 Synthesis of microcrystalline MOF-76(Y) and MOF-76(Y0.95Eu0.05)
The microcrystalline solids were synthesized following this procedure: yttrium(III) oxide (to obtain 0.15 mmol of Y(NO3)3) and H3BTC (0.195 mmol) were combined in a 100 mL glass autoclave flask with a mixture of DMF (3 mL), ethanol (3 mL), distilled H2O (2.4 mL) and nitric acid (0.1 mL) and heated at 80 °C for 24 hours. The solid product named MOF-76(Y)_m was collected by vacuum filtration, washed several times with 40 mL of a 1:1 DMF/ethanol mixture, and dried under atmospheric conditions before characterization. The europium doped compound was named MOF-76(Y0.95Eu0.05)_m and was synthesized using a similar procedure, except that yttrium(III) oxide (to obtain 0.1425 mmol Y(NO3)3) and europium(III) oxide Eu(NO3)3 (to obtain 0.0075 mmol) were included into the reactant mixture, with the remaining reaction parameters kept unchanged.
2.3 Synthesis of nanocrystalline MOF-76(Y) and MOF-76(Y0.95Eu0.05)
The nanocrystalline version of these MOFs was obtained by modifying a previously reported synthesis route.31 In this procedure, 0.15 mmol of Y(NO3)3 (obtained by dissolving yttrium(III) oxide in nitric acid) and 0.39 mmol of sodium acetate were dissolved in distilled water (2.4 mL) to form solution 1, which was put in a 100 mL glass autoclave flask. H3BTC (0.195 mmol) was dissolved in DMF (3 mL), resulting in solution 2. Solution 2 was added to the solution 1 flask without stirring and a white precipitate immediately appeared. The resultant suspension was heated at 80 °C for 5 hours. The product was labelled MOF-76(Y)_n and was collected by centrifugation, washed several times with a 1:1 DMF/ethanol mixture, and dried in the air before characterization. The europium-doped compound named MOF-76(Y0.95Eu0.05)_n was synthesized by a similar procedure, except for adding 0.1425 mmol Y(NO3)3 and 0.0075 mmol Eu(NO3)3 into the reactant mixture, with the remaining reaction parameters kept unchanged. The formation of the sample structures was monitored using powder X-ray diffraction (PXRD). The crystal size and morphology were evaluated using scanning electron microscopy (SEM) and transmission electron microscopy (TEM).
2.4 Stability test of micro and nanocrystalline MOF-76 solids
Both microcrystalline- and nanocrystalline MOF-76(Y) compounds were left in contact with solvents (ethanol, methanol, and water) with and without ultrasound to examine the structural stability of MOF-76 in these conditions under different contact times.2.5 Preparation of transited MOF-76(Y0.95Eu0.05)_m-T and MOF-76(Y0.95Eu0.05)_n-T phases for luminescence study
One hundred and fifty mg of MOF-76(Y0.95Eu0.05)_m was sonicated in 5 mL of deionized water for 60 min at room temperature. After that, the supernatant solvents were extracted, and the solids were dried under atmospheric conditions. The solid was labelled MOF-76(Y0.95Eu0.05)_m-T and analysed by powder XRD analysis. The same procedure was carried out with the MOF-76(Y0.95Eu0.05)_n to obtain theMOF-76(Y0.95Eu0.05)_n-T material, but the sonication time was 15 min. In these notations, “T” means “transited phase,” regarding the water-induced structural transformation.2.6 Characterization techniques
Powder X-ray diffraction (PXRD) patterns of solids were collected with a Rigaku Ultima IV type II diffractometer using Cu-Kα (λ = 1.5418 Å) radiation in the 2θ range of 5–40°, in continuous mode, with increments of 0.02° and a scan speed of 3°min−1. Infrared spectra (FTIR) were measured using KBr pellets from 4000 to 225 cm−1 using a Nicolet Protégé 460 spectrometer with 64 scans and a spectral resolution of 4 cm−1. The morphology of the samples was inspected using a scanning electron LEO1450VP microscope (LABMEM-UNSL). High resolution (HRTEM) and high angle annular dark field scanning transmission (HAADF-STEM) images of compounds MOF-76(Y)_n and MOF-76(Y0.95Eu0.05)_n, as well as those of the transited samples obtained by water ultrasonic treatment, were registered using a JEOL-2100 Plus microscope (INTEC-UNL) operated at an acceleration voltage of 200 kV. This analysis allowed us to determine the average size of synthesized nanoparticles and characterize the morphological change upon water/ultrasonic treatment. The structural resolution of the equipment in the TEM mode was 0.19 nm with Scherzer defocus conditions, while the probe used in STEM was 0.5 nm. This analysis allowed us to determine the average size of synthesized nanoparticles and characterize the morphological change upon water/ultrasonic treatment. Energy dispersive analysis by X-rays (EDX) was applied to obtain Y3+/Eu3+ mapping in TEM images of MOF-76(Y0.95Eu0.05)_n nanoparticles.In both the SEM and TEM techniques, colloidal ethanolic and water suspensions obtained by the ultrasound treatment described above were dropped onto the sample holder and allowed to evaporate. Luminescence excitation and emission spectra were measured on a PTI Quanta Master QM-4 spectrofluorometer. The excitation and emission spectra were measured in a front-face arrangement under the same conditions of thickness and slit widths.
3. Results and discussion
3.1 Structure and morphology of MOF-76(Y)_m, MOF-76(Y0.95Eu0.05)_m, MOF-76(Y)_n and MOF-76(Y0.95Eu0.05)_n
Micro-sized MOF-76(Y)_m crystals and nano-sized MOF-76(Y)_n with the formula [YBTC(H2O)](DMF) were obtained via solvothermal synthesis as described in experimental section 2.2. The characterization by powder X-ray diffraction (PXRD) allowed us to confirm the formation of the three-dimensional (3D) MOF-76 structure (S.G. P43), which was in good agreement with the corresponding MOF-76 (Ho) simulated pattern (COD 1426949) (Fig. 1(a)–(c)). Besides, MOF-76(Y0.95Eu0.05)_m, MOF-76(Y)_n and MOF-76(Y0.95Eu0.05)_n was also successfully synthesized (Fig. 1(a) and (b)). FTIR spectra of the four obtained materials are displayed in Fig. S1 (see ESI†).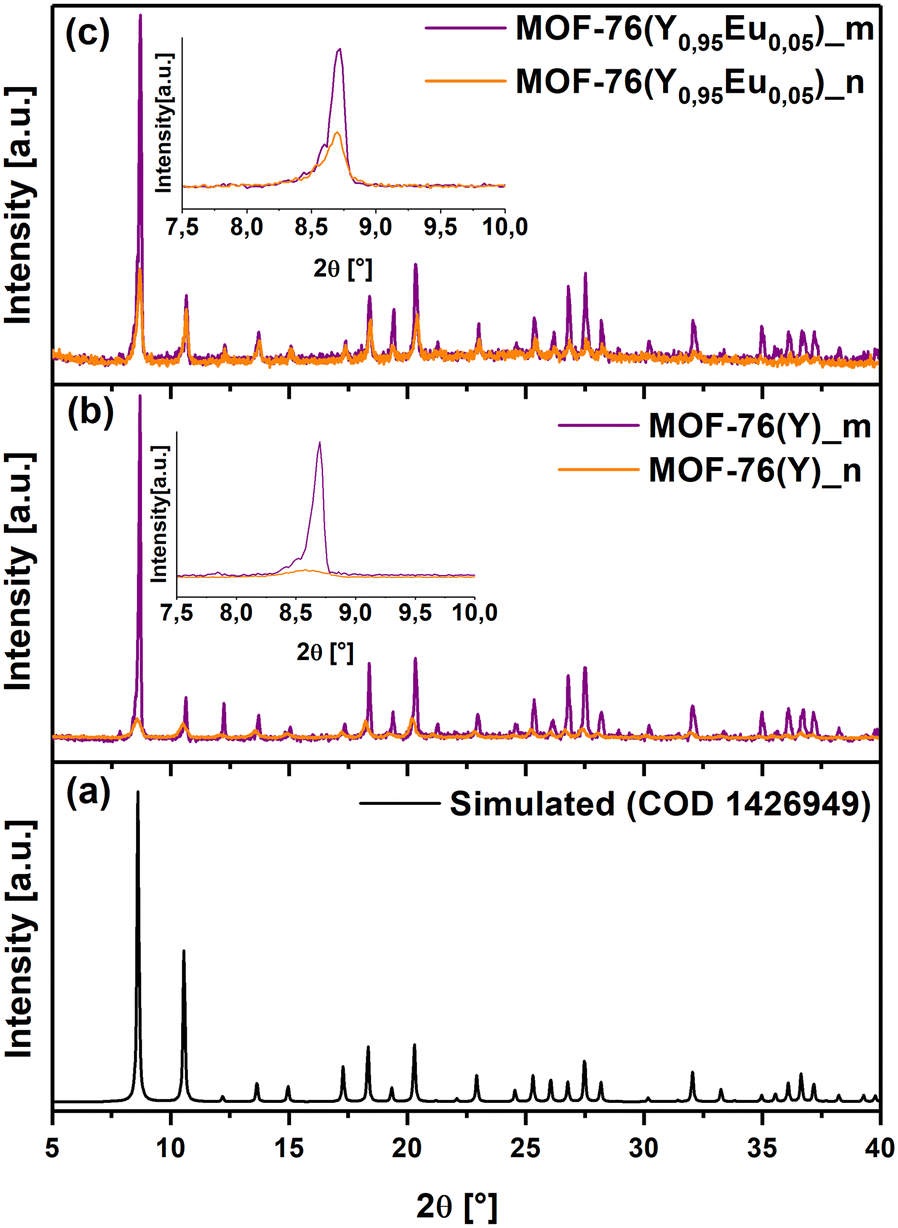 | ||
| Fig. 1 Diffraction patterns of (a) Simulated MOF-76(Ho), (b) MOF-76(Y)_m and MOF-76(Y)_n, and (c) MOF-76(Y0.95Eu0.05)_m and MOF-76(Y0.95Eu0.05)_n. | ||
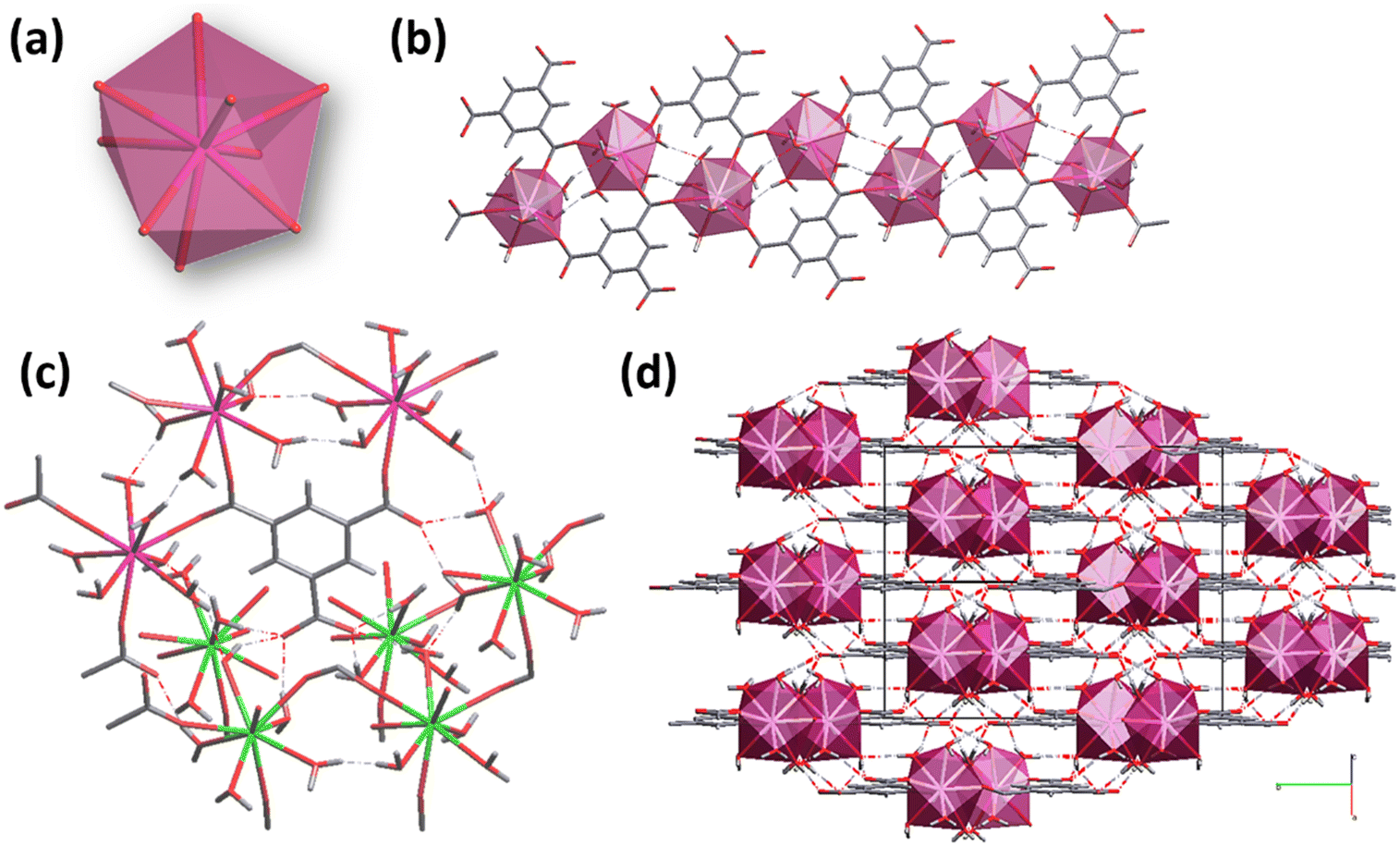
According to the structural features of the single crystal XRD data (COD 1426949), each asymmetric unit contains one rare earth cation (Ln3+), one BTC3− ligand, one coordinated water molecule, and one DMF molecule located in the cavities.54 The Ln3+ cations are hepta-coordinated with six oxygen atoms derived from six carboxylate groups of BTC3− linkers and one oxygen from a terminal water molecule, giving a distorted pentagonal bipyramid coordination geometry (Fig. 2(a)).17,32
 | ||
| Fig. 2 (a) Coordination environment of Ln3+ ions in the MOF-76 structure. (b) Helical chains propagating along the c-axis. (c) View of the structure along the b-axis shows the six-connected nodes on BTC linkers in detail. (d) MOF-76 3D-structure showing cavities propagating along the c axis. DMF molecules were omitted for clarity. | ||
The building blocks form a 3D framework in which Ln3+ ions are connected via carboxylate groups and arranged in one-dimensional left-handed helical chains propagating along the c-axis (Fig. 2(b)).17,54 Considering the connectivity of the Ln3+ and BTC3−, the framework can be described as six-connected nodes (Fig. 2(c)) and the whole network contains 1D channels running parallel to the [001] direction (Fig. 2(d)).17
Moreover, the observed pattern in the MOF-76(Y0.95Eu0.05)_m diffractogram confirmed that the compound is isostructural after the introduction of 5% molar of europium in the same reaction conditions as those employed for MOF-76(Y)_m (Fig. 1(b) and (c)). Additionally, the PXRD patterns of MOF-76(Y)_n and MOF-76(Y0.95Eu0.05)_n compounds (Fig. 1(b) and (c)) show the broadening of the peaks and a remarkable decrease in intensity compared with the diffractograms of microcrystalline solids, indicating a noticeable particle size diminution by changing the synthesis conditions.
Scanning electron microscopy (SEM) images showed the growth of needle-shaped microcrystals of MOF-76(Y)_m that are several hundred nm in length when the synthesis conditions are described in experimental section 2.2 (Fig. 3(a)). This characterization determined that the crystal morphology is maintained with the europium doped MOF-76(Y0.95Eu0.05)_m compound (Fig. 3(d)).
 | ||
| Fig. 3 SEM images of (a) MOF-76(Y)_m needle-shape microcrystals. (b) Panoramic view of MOF-76(Y)_n modulated by adding sodium acetate. (c) MOF-76(Y)_n showing the nanoparticles in detail. (d) MOF-76(Y0.95Eu0.05)_m needle-shape microcrystals. (e) MOF-76(Y0.95Eu0.05)_n modulated. (f) MOF-76(Y0.95Eu0.05)_n nanoparticle. | ||
The SEM analysis of the MOF-76(Y)_n solid at the lowest magnification verified that the grains were formed by a cumulus of small nanoparticles (Fig. 3(b)), and higher magnification determined that these aggregates showed a homogeneous distribution of bean-shaped nanoparticles in the MOF-76(Y)_n compound15,19,32 (Fig. 3(c)). A similar morphology could be observed for the doped MOF-76(Y0.95Eu0.05)_n material (Fig. 3(e) and (f)). As expected, this radical change in morphology and size is due to the function of sodium acetate as a crystal growth modulator.19,32,33 Nonetheless, Brunckova et al.32 previously obtained quite uniform rod-like nanocrystals with lengths of 110–850 nm and widths of 20–400 nm of bimetallic MOF-76 (Eu, Gd), MOF-76 (Eu, Gd) and MOF-76 (Eu, Tb) compounds using sodium acetate modulated synthesis at 60 °C for 48 h. Thus, each synthesis variable has a primary role in modulating crystal growth in MOF materials. Regarding the synthesis conditions employed here, using a mixture of only DMF/water (without ethanol)19 also seems to influence the crystal size.
The water present in the synthesis acts as both reactant and solvent, promoting the nucleation and crystal growth processes. Additionally, the smaller the number of hydroxyl groups in the solvent mixture, the greater the possibility of crystals sharing them and forming spheres instead of rods.32 Finally, the decrease in reaction time from 24 to 5 hours also impacts crystal size and morphology, reducing the opportunity for continued growth for the crystal nucleus.
3.2 Stability test of micro and nanocrystalline MOF-76 solids in different solvents
Based on the previously stated, supramolecular sensing applications of luminescent MOFs require knowing exactly what structure or MOF phase is being used as a sensor and that such a phase is stable under the experimental conditions of the analyte determination. Concerning the MOF-76 structure, there is ambiguous information on its structural stability in water. For example, J. D. Xiao et al.28 evidenced the structural transformation of MOF-76(Tb) into 1D coordination polymer when they were investigating the synthesis of nano MOF-76(Tb) by applying ultrasound; thus, the phase transition was detected indirectly during the evaluation of the synthesis reactions products. In this study, they realized that MOF-76 or 1D coordination polymer were obtained as products by applying “short” or “long” ultrasound times, respectively during the synthesis reaction. T. W. Duan et al.30 reported that by immersing 5 mg of a microcrystalline sample of MOF-76(Y99Eu0.1) in water for 24 h, they obtained the corresponding 1D coordination polymer. On the contrary, Y. Yang et al.54 have used MOF-76(Tb0.96Eu0.04) to detect analytes in water medium, claiming that this phase with MOF-76 structure is stable. However, the experimental conditions for these stability tests are not described. Furthermore, Y. Yang et al. provide evidence that such structural stability is derived from experiments that involve immersing the MOF-76 phase in Eu3+ aqueous solutions of different concentrations and measuring the detected Eu3+/Tb3+ concentration ratio by ICP. Such a methodology could apport evidence about a solid that lixiviates; however it does not consider the rearrangement that the lanthanide ions can suffer in the solid state during a phase transition. With this background in mind, a series of experiments were designed to consider the influence of several variables, such as solvent, particle size, the assistance of ultrasound, contact time, and activation treatment to perform a deep study on the structural stability of MOF-76(Y). | (1) |
| AT = AMOF-76 + ALnBTC | (2) |
 | ||
| Fig. 4 MOF-76(Y)_n in water: (a.1) sonicated, (b.1) non-sonicated. MOF-76(Y)_m in water: (a.2) sonicated, (b.2) non-sonicated. All samples were not thermally activated for this experiment. The asterisk symbol over the peaks indicates the reflection of the 1D LnBTC-compound. | ||
Considering the % converted from MOF-76(Y)_n to the 1D coordination polymer at 5 minutes of ultrasonic treatment in water, it is possible to estimate that the structural transformation rate in these experimental conditions is 12.4 % min−1 (Fig. 5(a)). According to this estimation, it can be deduced that about 8 minutes were necessary to complete the phase transition in the applied experimental setup. As a result, no peaks can be observed in the tetragonal MOF-76 structure in the PXRD with a longer ultrasonic treatment (15 min), and all reflections match well with the crystallographic information of the CCDC 290771 LaBTC phase. On the other hand, the phase change also occurs if MOF-76(Y)_n is immersed in water without ultrasonic treatment, verifying that it is completed in a longer time (60 min), suggesting a slower structural transformation rate of about 1.7 % min−1 (Fig. 4(b.1) and 5(a)). Additionally, the tetragonal structure remains when nano-MOF-76 is immersed in ethanol or methanol without sonication treatment (Fig. S3(a) and (b)†).
 | ||
| Fig. 5 LnBTC percentage in each test for: (a) MOF-76(Y)_n and (b) MOF-76(Y)_m. | ||
Microcrystalline MOF-76(Y)_m compound treated with ultrasound in ethanol and methanol was stable, which agreed with that observed for MOF-76(Y)_n. The resultant PXRD patterns are shown in Supplementary Information (Fig. S2(c) and (d)†). On the contrary, when MOF-76(Y)_m is immersed in water, ∼52.70% of the initial material transited to the LnBTC phase after 15 min of contact time, giving this step a transition rate of 3.51 % min−1. Even though no PXRD was measured between 15 and 60 minutes of immersion in water, about ∼30 minutes could be enough to complete the structural transformation according to the estimated transition rate. The 100% phase transition was verified by measuring the PXRD after 60 min of contact time in water (Fig. 4(a.2) and 5(b)). These results indicate that the microcrystalline MOF-76(Y) material needs more time to complete the phase transition than the nanometric-sized version, showing greater stability in these experimental conditions. In the same way, the phase transition occurred at a slower rate when MOF-76(Y)_m was immersed in water without the assistance of ultrasound action, and after 60 min of contact time, a mixture of compounds with ∼52.52% of LnBTC could be observed (Fig. 4(b.2) and (5(b)). Additionally, microcrystalline MOF-76 exhibits stability when exposed to ethanol or methanol (Fig. S3(c) and (d)†). Likewise, the difference in the phase transition time between nano and microcrystalline MOF-76(Y) could be because their surface-to-volume ratio dramatically increases as the size of materials decreases to the nanometer scale, which may change the phase transition conditions.57 The nano-size particles usually show better exposure to active sites and shorter diffusion distances, facilitating the interaction with molecules by coordination, resulting in a structural rearrangement.58,59 In the same way, ultrasound treatment can generate high temperatures at a localized level and violent pressure changes.60 These conditions can promote the phase transition. Therefore, all previous tests reveal that the phase transition from tetragonal MOF-76 to monoclinic LnBTC occurs much faster than previously reported (24 hours),30 regardless of crystallite size. The resultant product of the structural transition is a compound that exhibits a general formula LnBTC[H2O]646 and are white solids insoluble in water when Ln = Y3+ and Eu3+ (as 5 mmol% dopant). Unlike MOF-76, Ln3+ ions are nine-fold coordinated in the LnBTC structure; the coordination sphere consists of three oxygen atoms from three carboxylate groups of the BTC ligands and six water ligands, forming a tricapped trigonal prismatic geometry (Fig. 6(a)).46,47Fig. 6(b) shows the structure displaying parallel 1D ribbon-like molecular motifs along the a axis.47 As shown in Fig. 6(c), the three carboxylate groups of the BTC ligand exhibit different connectivity in this structure; one of them is unidentate, the second one is bidentate, and the third one is a free carboxylate group that stands without forming coordination bonds.47 Finally, the combination of non-covalent interactions such as hydrogen-bonding and π–π stacking determine the formation of the 1D coordination polymer structure (Fig. 6(d)).47,48
 | ||
| Fig. 6 (a) Coordination environment of Ln3+ ions in the LnBTC structure. (b) 1D ribbon-like structure along the a axis. (c) View of the carboxylate groups of coordination modes. (d) LnBTC framework. | ||
 | ||
| Fig. 7 TEM images of (a) MOF-76(Y)_n sonicated for 5 min in EtOH. (b) MOF-76(Y0.95Eu0.05)_n sonicated for 5 min in EtOH. (c) MOF-76(Y)_n sonicated for 15 min in H2O. (d) MOF-76(Y0.95Eu0.05)_n sonicated for 15 min in H2O. | ||
 | ||
| Fig. 8 STEM-HAADF image, point EDS spectrum, and calculated elemental composition of (a) one nanoparticle in MOF-76(Y0.95Eu0.05)_n compound, and (b) MOF-76(Y0.95Eu0.05)_n sonicated for 15 min in H2O. *Copper signal comes from the TEM grid. | ||
A sample of MOF-76(Y0.95Eu0.05)_n recovered after sonication in H2O for 15 min was also analyzed by FTIR spectroscopy to evaluate the incidence of the structural transformation on the compound's vibrational behavior (see Fig. 9). For example, the bands located between 3700–3250 cm−1 related to the asymmetric and symmetric O–H stretching vibration of water molecules change from a wide band centered at ca. 3410 cm−1 for MOF-76(Y)_n to a stronger and sharper band containing three components at 3490, 3410 and 3340 cm−1 46,47 for the sample recovered after the ultrasound treatment in water. Such change is consistent with the presence of the transited structure consisting of six coordination water molecules exhibiting a complex H-bond network. At the same time, there is only one coordination water molecule in the initial compound. Additionally, the MOF-76 structure crystallizes with DMF molecules occluded in the pore framework; the stretching of the carbonyl group of DMF is evident in the spectrum of the initial sample at ca. 1670 cm−1,13 while it is absent in the spectrum of the transited sample since the 1D coordination polymer does not contain this kind of solvent in its crystal structure.
 | ||
| Fig. 9 FTIR spectra of the synthesized MOF-76(Y0.95Eu0.05)_n (orange) compound and after sonication for 15 min in H2O (MOF-76(Y0.95Eu0.05)_n-T, black), MOF-76(Y0.95Eu0.05)_m (purple) material and after sonication treatment for 60 min in H2O (MOF-76(Y0.95Eu0.05)_m-T, pink), (a) from 4000–2900 cm−1 and (b) from 2000–1250 cm−1. | ||
Moreover, the changes in the 1700–1350 cm−1 range can be associated with the variations in the coordination modes/interactions that suffer the OCO groups of the BTC linker; from bidentate for the three OCO groups in the MOF-76 structure to one bidentate and one monodentate coordination geometry, leaving only the remaining OCO groups involved in H-bond interactions.16,17,54,56,61 These changes in the selected spectral zones represent the most marked differences that involve functional groups strongly affected by the phase transition, and they are equivalent in the transited phase MOF-76(Y0.95Eu0.05)_m-T after 60 min of sonication in H2O (Fig. 9).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)),54 and at 1253 and 1061 cm−1 corresponding to the (C–N)63,64 stretching modes. In the same way, additional bands can be identified after the activation. First, at 1263 and 1215 cm−1 (C–H in-plane bending vibrations), 1084 and 1043 cm−1 (in plane C–C stretching vibrations), and 877 cm−1 (C–H in-plane) corresponding to the H3BTC linker.17,65 All the above changes can be interpreted based on the successful DMF solvent removal.
O)),54 and at 1253 and 1061 cm−1 corresponding to the (C–N)63,64 stretching modes. In the same way, additional bands can be identified after the activation. First, at 1263 and 1215 cm−1 (C–H in-plane bending vibrations), 1084 and 1043 cm−1 (in plane C–C stretching vibrations), and 877 cm−1 (C–H in-plane) corresponding to the H3BTC linker.17,65 All the above changes can be interpreted based on the successful DMF solvent removal.
To investigate if the thermal activation process has any influence on the structural transformation from the 3D MOF-76 structure to the 1D LnBTC coordination polymer, additional experiments with activated MOF-76(Y)_n sample were performed. The material was immersed in water and sonicated for different purposes. The FTIR spectra of the activated compound after 5, 15, and 60 min show no changes (Fig. S7†), which is consistent with the results of the PXRD analysis of this sample in the same period (Fig. 10(a.1)). According to the PXRD analysis for a longer contact time in ultrasonic treatment, the phase transition starts to occur, and the 1D coordination polymer structure can only be detected at 180 min.
 | ||
| Fig. 10 MOF-76(Y)_n in water: (a.1) sonicated, (b.1) non-sonicated. MOF-76(Y)_m in water: (a.2) sonicated, (b.2) non-sonicated. All the samples were thermally activated for this experiment. The asterisk symbol over the peaks indicates the reflections of the LnBTC-compound. | ||
Likewise, ∼54.86% of MOF-76(Y)_n compound was still present after 960 min in water without sonication (Fig. 10(b.1)). On the contrary, no obvious improvement was observed in terms of water resistance after activation of MOF-76(Y)_m (Fig. 10(a.2) and 9(b.2)) using the same conditions, and the transformation to LnBTC occurred with a similar transformation rate to that of the non-activated soaked material (Fig. 5(b)). A careful observation of the activated samples of MOF-76(Y)_n and MOF-76(Y0.95Eu0.05)_n materials by SEM (Fig. 11) allowed us to realize that nanoparticles tend to sinter with each other when high temperatures are applied, which reduces their surface energy. Therefore, the structural stability of the activated phase MOF-76(Y)_n is greatly increased compared with the non-activated ones.66,67
 | ||
| Fig. 11 SEM images of materials after activation: (a) MOF-76(Y)_n as-synthesized form, (b) MOF-76(Y0.95Eu0.05)_n as-synthesized form, (c) MOF-76(Y)_n after thermal activation, and (d) MOF-76(Y0.95Eu0.05)_n after thermal activation. | ||
3.3 Photoluminescence of MOF-76(Y0.95Eu0.05)_m and MOF-76(Y0.95Eu0.05)_n
A series of luminescence measurements were carried out to investigate how the particle size of the Eu3+ doped versions of MOF-76(Y) affect the corresponding luminescence properties before and after the phase transition in water.The luminescence tests were performed at room temperature, and the excitation and emission spectra were recorded for MOF-76(Y0.95Eu0.05)_m and MOF-76(Y0.95Eu0.05)_n in the solid state. The excitation and emission photoluminescence spectra of materials are displayed in Fig. 12. As can be seen as a first evaluation, the spectra of the materials are almost identical in the quantity and positions of the bands, both in the excitation and emission spectra. This confirms that the synthesized materials with different particle sizes and isostructural to the MOF-76 phase exhibit the same luminescence behavior. Only slightly lower intensity emission is observed for MOF-76(Y0.95Eu0.05)_n.
 | ||
| Fig. 12 Excitation (λEmi = 611 nm) (a) and emission spectra (λExc = 290 nm) (b) of microcrystalline MOF-76(Y0.95Eu0.05)_m compound and nanocrystalline MOF-76(Y0.95Eu0.05)_n material. | ||
The excitation spectrum of MOF-76(Y0.95Eu0.05)_m and MOF-76(Y0.95Eu0.05)_n are dominated by a wide band between 250 and 325 nm, suggesting that the material can efficiently transfer the excitation energy to sensitize the emission of the lanthanide ions.19 The band is composed of two maxima: the band at ca 290 nm corresponds to the π–π* electronic transitions of the linker (BTC),30 and the other one is assigned to the Eu3+–O2− charge-transfer (CT) band.29 The bands located at 393, 465 and 360 nm can be assigned to the 7F0,1 → 5L6, 7F0 → 5D2 and 7F0 → 5D4 transitions of Eu3+, respectively.
The emission spectra show the expected five characteristic narrow bands corresponding to the 5D0 → 7FJ (J = 0, 1, 2, 3, 4) transitions of Eu3+ ions.22,29 The intra 4f–f energy levels of Eu3+ are strongly shielded by the 5s2 and 5p6 shells and their position is not affected by the crystal field. However, the nature and transition probabilities among these states, together with the splitting of these bands, strongly depend on the symmetry of the site where the lanthanide ions are located. The forbidden electric-dipole 5D0 → 7F0 transition is a single band due to the non-degeneracy of 7F0 and 5D0 levels (2J + 1 = 1 for J = 0) and its intensity is typically extremely weak or even undetectable. When allowed, this band is always a single peak for a given symmetry site in a crystal. If the Eu3+ ions locate in different non-equivalent sites, a 5D0 → 7F0 band should be observed for each site. Therefore, its profile and FWHM (full width at half maximum) can give information on the environment of the Eu3+ ions.68 The 5D0 → 7F1 band corresponds to a magnetic dipole transition being allowed when the local symmetry of the Eu3+ site presents a center of inversion68 and its intensity is almost independent of the local environment around Eu3+ ions as expected.29 On the contrary, 5D0 → 7F2 is known as a hypersensitive band arising from an electric dipole transition and is always present when a center of inversion is absent68 (as in the case of the P43 S.G corresponding to the MOF-76 crystal structure). This band is responsible for the brilliant-red emission of the Eu3+ doped materials.29 The 5D0 → 7F3 transition exhibits a mixed magnetic and electric dipole character29 and does not offer much structural information. 5D0 → 7F4, as in the case of J = 2, is an electric dipole transition only allowed by a low symmetry.68 The intensity ratio I5D0 → 7F2 /I5D0 → 7F1 (5D0 → 7F2 to 5D0 → 7F1) is widely employed to compare the strength of hypersensitive transitions in different Eu3+ compounds,24 and it can roughly reveal the relative amounts of symmetric and asymmetric Eu3+ ions sites.68 Therefore, the ratio is strongly dependent on the local symmetry of the Eu3+ ions and it is often used to detect the local environments probe29 because the higher the ratio, the farther away ion is from a centrosymmetric geometry luminescent center.69 In the case of MOF-76(Y0.95Eu0.05)_m, the I5D0 → 7F2 /I5D0 → 7F1 ratio is 3.0 ± 0.1.
As was enunciated above, the excitation and emission spectra of MOF-76(Y0.95Eu0.05)_n are very similar to their microcrystalline counterparts except for a somehow lower intensity of some Eu3+ emission bands (Fig. 12). Such a variation in intensity could be the result of a large surface-to-volume ratio and the presence of a larger number of defects in the nanocrystalline compound. It is a known fact that radiative mechanisms compete with nonradiative ones, and the defects can lead to nonradiative recombination and luminescence quenching of surface centers.29,70 Moreover, the calculated I5D0 → 7F2/I5D0 → 7F1 ratio value is 3.5 ± 0.1 for MOF-76(Y0.95Eu0.05)_n, which is higher than the value for the micrometric material, indicating that Eu3+ ions in the nanocrystalline material have coordination environments of lower symmetry compared with MOF-76(Y0.95Eu0.05)_m.46,71 The chromaticity diagram (CIE) of both materials is shown in Fig. 13.
 | ||
| Fig. 13 Chromaticity (CIE) diagram comparing the color coordinates of synthesized MOF-76(Y0.95Eu0.05)_n and MOF-76(Y0.95Eu0.05)_m compounds, and the corresponding ones to the MOF-76(Y0.95Eu0.05)_m-T and MOF-76(Y0.95Eu0.05)_n-T phases. The purity color is summarized below the graph. The CIE image was made using the Sci-Sim website.70 | ||
As can be noticed, the x, y CIE coordinates of compounds are relatively close to the primary color set by the National Television System Committee (NTSC 1987) (0.63, 0.34).72,73 The purity color for both is relatively high and similar (82.13% for the micro-sized material and 80.08% for the nanosized material), suggesting that the crystallite size subtly affects the light emission properties of these materials. Additionally, the CIE coordinate findings strongly agreed with values reported in prior studies of similar compositions.19,20
A comparison between the luminescence properties of MOF-76(Y0.95Eu0.05)_m and MOF-76(Y0.95Eu0.05)_m-T was performed, as well as between MOF-76(Y0.95Eu0.05)_n and MOF-76(Y0.95Eu0.05)_n-T (see Fig. 12 and 14–16). In these notations, “T” means “transited phase”, regarding the described structural transformation to LnBTC-structure induced by water. In Fig. 14, large differences can be observed between the luminescence spectra of MOF-76(Y0.95Eu0.05)_m and MOF-76(Y0.95Eu0.05)_m-T. First, the excitation spectrum shows that the band in the UV region (250–325 nm) of MOF-76(Y0.95Eu0.05)_m-T sample is much less intense but broader than MOF-76(Y0.95Eu0.05)_m-T. The maximum of the π–π* band located at 290 nm in the non-transited sample is red-shifted from 290 nm to 300 nm. These changes induce a less efficient energy transfer process from the ligands to the lanthanide ions and can be attributed to a change in the local coordination of the ions. Specifically, the coordination number of the emitting Eu3+ atom changes from seven to nine as a result of the structural phase change, where six out of nine oxygen atoms come from three coordinated water molecules in the MOF-76(Y0.95Eu0.05)_m-T sample and three correspond to three carboxylate groups of the BTC ligands (Fig. 6(a)). The –OH oscillators of these three water molecules greatly diminish the probability of an efficient energy transfer from the matrix ligands to Eu3+ ions, favoring non-radiative processes which are noticed by the lower intensity of the excitation and emission spectra. This change in the local environment of the lanthanide ions directly impacts the profiles of the emission spectra, as observed by previous authors.46 In this case, the x, y CIE coordinates for the LnBTC material (MOF-76(Y0.95Eu0.05)_m-T) shift away (0.5, 0.482) from the primary color set (Fig. 12). The reduced color purity could be attributed to additional or secondary transitions in the emission spectrum of the material, or to crystal field effects generating emissions in nearby bands caused by the structural phase change (Fig. 13).
 | ||
| Fig. 14 (a) The excitation (λEmi = 611 nm) and (b) emission spectra (λExc = 290 nm) of microcrystalline MOF-76(Y0.95Eu0.05)_m and MOF-76(Y0.95Eu0.05)_m-T. | ||
 | ||
| Fig. 15 (a) Excitation (λEmi = 611 nm) and (b) emission spectra (λExc = 290 nm) of nanocrystalline MOF-76(Y0.95Eu0.05)_n compound and MOF-76(Y0.95Eu0.05)_n-T material. | ||
 | ||
| Fig. 16 (a) Excitation (λEmi = 611 nm) and (b) emission spectra (λExc = 290 nm) of MOF-76(Y0.95Eu0.05)_n-T and MOF-76(Y0.95Eu0.05)_m-T materials. | ||
Unlike the previous case, the structural transformation from nano-sized MOF-76(Y0.95Eu0.05)_n to the 1D compound MOF-76(Y0.95Eu0.05)_n-T significantly enhances the emission efficiency compared with the initial material (see Fig. 15). Comparing these two samples, the excitation spectrum of the transited material exhibits a lower intensity than the MOF-76(Y0.95Eu0.05)_n compound (Fig. 14). The UV band for MOF-76(Y0.95Eu0.05)_n-T is much broader (250–350 nm) than the MOF-76(Y0.95Eu0.05)_n and the π–π* band is red shifted from 290 nm to 320 nm. Conversely, the emission spectrum of MOF-76(Y0.95Eu0.05)_n-T displays a higher intensity for all transitions 5D0 → 7F0,1,2,3, except for 5D0 → 7F4.
The number of water molecules around the BTC ligands in the transited samples stabilizes the carboxylate anion, lowering the energy of the excited triplet state while broadening the spectrum due to the different intermolecular interactions (mainly H-bonding). The observed red shift that is more marked for the transited sample whose precursor is MOF-76(Y0.95Eu0.05)_n shortens the energy gap between the 5D0 level of Eu3+ ions and the excited triplet state of the BTC, favoring the energy transfer process and enhancing the luminescence of MOF-76(Y0.95Eu0.05)_n-T. This can partly explain the higher luminescence of the transited compound compared with MOF-76(Y0.95Eu0.05)_n, which in turn prevails upon the OH well-known luminescence quenching of the OH oscillators of the water molecules around the lanthanide ion. In addition, the 5D0 → 7F2 transition of MOF-76(Y0.95Eu0.05)_n-T splits due to a much lower symmetric crystal field experienced by Eu3+ ions, breaking the degeneracy of the energy levels of the 7F2 state.74,75 Furthermore, the analysis of the CIE chromaticity coordinates and purity color for MOF-76(Y0.95Eu0.05)_n and MOF-76(Y0.95Eu0.05)_n-T shows very close values, contrary to that observed for MOF-76(Y0.95Eu0.05)_m and MOF-76(Y0.95Eu0.05)_m-T (Fig. 13). In Fig. 16, the excitation and emission spectra of the transformed phases obtained from nano-sized (MOF-76(Y0.95Eu0.05)_n-T) and micro-sized MOF-76 (MOF-76(Y0.95Eu0.05)_m-T) materials are compared.
The comparison of both excitation and emission spectra of MOF-76(Y0.95Eu0.05)_n-T and MOF-76(Y0.95Eu0.05)_m-T materials marked differences not only in intensity but also in peak positions and splitting components. The differences in the spectra of both phases are outstanding, considering that they have the same 1D LnBTC-structure. The corresponding analysis of the separated spectrum was done above. Still, one of the most evident differences to remark on is the higher intensity emission lines of the spectrum of the compound coming from the nanocrystalline MOF-76(Y0.95Eu0.05) material. This finding is likely attributable to grain growth occurring from nanoparticles that allow for obtaining less defective and more homogeneous nanorod crystals. Such results suggest the existence of a memory effect that considers the particle size of the precursor material and directly affects the luminescence of the product resulting from the structural transformation.
To investigate the possible microstructural causes that justify the differences in the observed luminescence behavior, a comparative analysis of the PXRD patterns of both transited phases employed in the luminescence study was performed (Fig. 17). It is interesting to note that there are notable differences in the relative intensities of certain peaks between the two samples, although the PXRD diffractograms of both transited phases are in good agreement with the isostructural LaBTC compound (CCDC 290771).
 | ||
| Fig. 17 (a) XRD patterns of transited materials from nano and micro-sized compounds compared with (b) the simulated LaBTC (CCDC 290771) single crystal XRD data. Rietveld refinement of the transited materials (c) MOF-76(Y0.95Eu0.05)_m-T and (d) MOF-76(Y0.95Eu0.05)_n-T. | ||
Often, needle-shaped materials exhibit preferred orientations due to their elongated shape, inducing a preferred alignment of the crystals along a particular direction. The above causes certain reflections of crystallographic planes to be more intense or less represented in the diffractogram as in the case of the transited compounds. In the Rietveld FullProf refinement, the crystallographic texture could include the March-Dollase function, which specifies the direction and corrects the peak intensity. In this sense, the PXRD pattern of the material coming from the nanosized compound needs to be corrected for the preferred orientation in more related hkl planes than that coming from the microsized one. This is consistent with more anisotropic crystal nanorods grown by nanocrystal seed nuclei. In particular, specific regions of the diffractograms display distinct distributions of peak intensities without altering their positions, such as those indicated by areas a and b in Fig. 17. This phenomenon is often attributed to defects that can influence the electron density locally or the alignment of specific planes with the X-ray beam.76
Conclusions
In this work, we investigated the impact of particle size on the structural transformation of MOF-76(Y) and MOF-76(Y0.95Eu0.05) phases into a 1D coordination polymer upon exposure to water. Both micro- and nanocrystalline samples were synthesized and compared. We found that nanocrystalline MOF-76 undergoes a more rapid structural transformation than its microcrystalline counterpart. The application of ultrasound further accelerated this process, particularly for nanocrystalline materials, allowing for complete transformation within 8 minutes. However, pre-activation before ultrasonic treatment increased the structural stability of MOF-76, especially in the nanocrystalline form. This enhanced stability is likely due to the sintering of nanoparticles into larger, more interconnected structures. Morphological analysis revealed distinct transformations for both particle sizes. Nanocrystalline MOF-76 transformed into nanorods of YBTC, while microcrystalline MOF-76 yielded sea urchin-shaped aggregates of YBTC. The luminescent properties of MOF-76(Y0.95Eu0.05) were also influenced by particle size. The initial particle size appeared to have a lasting impact on the luminescence characteristics of the transformed material, suggesting a potential “memory effect.” Our findings highlight the importance of considering particle size and operational conditions when working with MOFs and nanoMOFs. By understanding these factors, we can optimize the design and synthesis of MOF-based materials for specific applications. This work contributes to the growing field of luminescent MOFs by providing valuable insights into their structural stability and optical properties.Data availability
Most of the data from the work entitled “Time Dependence of Water-Induced Phase Transition in Nano- and Microcrystalline Eu3+-Doped MOF-76(Y): Different Luminescence Responses with Memory Effect” that is being submitted for its publication in J. Mater. Chem. C are provided in Fig. 1–17 (see details below).Additionally, some data have been incorporated in the ESI.†
Details of data displayed in Fig. 1–17 of the manuscript.
Fig. 1. Powder X-ray diffraction patterns of the synthesized materials and simulated pattern for the isostructural model. Crystallographic data were taken from the MOF-76(Ho) phase, COD 1426949.
Fig. 2. Crystal Structure of the MOF-76 phase.
Fig. 3. SEM images of the synthesized materials with the MOF-76 structure.
Fig. 4. Powder X-ray diffraction patterns of the synthesized materials exposed to different operational conditions.
Fig. 5. Representation of the percentage of the LnBTC crystal structure at different times obtained by phase transition under different operation conditions.
Fig. 6. Crystal structure of the LnBTC phase.
Fig. 7. TEM images of MOF-76 sonicated in ethanol and water for different times.
Fig. 8. STEM-HAADF image, point EDS spectrum and calculated elemental composition.
Fig. 9. FTIR spectra of MOF-76 and LnBTC phases.
Fig. 10. Kinetic evolution of powder X-ray diffraction patterns of MOF-76 of different particle sizes put in contact with water in different experimental conditions.
Fig. 11. SEM images of activated nanosized MOF-76 materials.
Fig. 12. Excitation and emission spectra of MOF-76(Y0.95Eu0.05) materials with different particle sizes.
Fig. 13. Chromaticity (CIE) diagrams comparing different MOF-76(Y0.95Eu0.05) materials and the corresponding ones obtained by phase transition.
Fig. 14–16. Comparison of excitation and emission spectra of MOF-76(Y0.95Eu0.05) materials with different particle sizes, before and after phase transition.
Fig. 17. Comparison of powder X-ray diffraction studies of transited phases from MOF-76 materials exhibiting different particle sizes and Rietveld refinement analysis.
Crystallographic analysis (powder-XRD patterns) of the synthesized samples was performed based on the crystallographic data reported for MOF-76(Ho). It has been deposited at the CCDC under accession number 1426949 and can be obtained from DOI: https://doi.org/10.1016/j.colsurfa.2015.10.048 (M. Almáši et al.17). For the XRD analysis of the phase transition products, crystallographic information for LaBTC (CCDC number 290771) was also considered. This file can be obtained from DOI: https://doi.org/10.5517/cc9rkq9 (Yi-Hang Wen et al.76).
For the crystallographic analysis, the CSD was used. “The Cambridge Structural Database, C. R. Groom et al.”77
CIE color coordinate analysis was performed using the CIE: https://sciapps.sci-sim.com/CIE1931.html, CIE 1931 web-based app. E. H. H. Hasabeldaim et al.78
The FullProf software “FullProf_Suite Windows (64 bits)”, used in this study for crystal structure refinement, is available at FullProf Suite: https://www.ill.eu/sites/fullprof/. Rodríguez-Carvajal, J. et al.79
https://www.ccdc.cam.ac.uk/solutions/software/mercury/. Macrae, C. F. et al.80
The author(s) are grateful to the FAIRE programme provided by the Cambridge Crystallographic Data Centre (CCDC) for the opportunity to access the Cambridge Structural Database (CSD) and associated software.
ImageJ, used for image analysis in this study, is available at the U.S. National Institutes of Health website: https://imagej.nih.gov/ij/. W. S. Rasband.81
OriginPro 2017 (OriginLab Corporation, Northampton, MA, USA), used for data analysis and graphing in this study, is commercially available from https://www.originlab.com/. OriginLab Corporation.82
Author contributions
Zulma L. Moreno Botello: investigation, methodology, data curation, visualization, writing original draft, writing, review & editing, and validation; Marcos Illescas: investigation and data curation; Beatriz C. Barja: investigation, writing original draft, and funding acquisition; Sebastian E. Collins: investigation, visualization, writing original draft, and funding acquisition; G. E. Narda: conceptualization, supervision, and writing original draft; María C. Bernini: conceptualization, supervision, writing original draft, visualization, writing, review & editing, funding acquisition, and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z. M. B. and M. I. acknowledge CONICET fellowships. M. C. B., G. E. N., S. E. C. and B. C. B. are members of CIC-CONICET. This work was supported by funds from CONICET (PIP-11220200102939CO), ANPCyT (PICT 2020-03713), and UNSL (PROICOs 02-0320 and 02-2320) projects.References
- P. R. Chandran and T. V. Arjunan, Int. J. ChemTech Res., 2015, 7, 974–4290 Search PubMed.
- A. Ahmed, S. Seth, J. Purewal, A. G. Wong-Foy, M. Veenstra, A. J. Matzger and D. J. Siegel, Nat. Commun., 2019, 10, 1568 CrossRef PubMed.
- C. Jiang, X. Wang, Y. Ouyang, K. Lu, W. Jiang, H. Xu, X. Wei, Z. Wang, F. Dai and D. Sun, Nanoscale Adv., 2022, 4, 2077–2089 RSC.
- M. S. Alhumaimess, J. Saudi Chem. Soc., 2020, 24, 461–473 CrossRef CAS.
- Q. Su, Q. Guo, H. Wang, M. Liu and C. Zuo, Carbon Resour. Convers., 2023, 100211 Search PubMed.
- L. Xiong, L. Yu, S. Li, L. Feng and Y. Xiao, Microchim. Acta, 2021, 188(7), 236 CrossRef CAS PubMed.
- M. Moharramnejad, A. Ehsani, M. Shahi, S. Gharanli, H. Saremi, R. E. Malekshah, Z. S. Basmenj, S. Salmani and M. Mohammadi, J. Drug Delivery Sci. Technol., 2023, 81(2), 104285 CrossRef CAS.
- C. Ma, L. Zheng, G. Wang, J. Guo, L. Li, Q. He, Y. Chen and H. Zhang, Aggregate, 2022, 3(1), e145 CrossRef CAS.
- J. Dong, D. Zhao, Y. Lu and W. Y. Sun, J. Mater. Chem. A, 2019, 7, 22744–22767 RSC.
- X. Wang, Y. Jiang, A. Tissot and C. Serre, Coord. Chem. Rev., 2023, 497, 215454 CrossRef CAS.
- Y. Zhao and D. Li, J. Mater. Chem. C, 2020, 8, 12739–12754 RSC.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- A. Garg, M. Almáši, R. Saini, D. R. Paul, A. Sharma, A. Jain and I. P. Jain, Environ. Sci. Pollut. Res., 2023, 30, 98548–98562 CrossRef CAS PubMed.
- B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693–1696 CrossRef CAS.
- X. Lian and B. Yan, RSC Adv., 2016, 6, 11570–11576 RSC.
- V. Zeleňák, M. Almáši, A. Zeleňáková, P. Hrubovčák, R. Tarasenko, S. Bourelly and P. Llewellyn, Sci. Rep., 2019, 9, 15572 CrossRef PubMed.
- M. Almáši, V. Zeleňák, J. Kuchár, S. Bourrelly and P. L. Llewellyn, Colloids Surf., A, 2016, 496, 114–124 CrossRef.
- G. E. Gomez, M. D. S. Afonso, H. A. Baldoni, F. Roncaroli and G. J. A. A. Soler-Illia, Sensors, 2019, 5, 1260 CrossRef PubMed.
- T. W. Duan and B. Yan, J. Mater. Chem. C, 2014, 2, 5098–5104 RSC.
- A. V. Mayeuski, D. Y. Poloneeva, E. A. Toshcheva, A. V. Bardakova, A. V. Shuruhina, A. V. Emeline and D. W. Bahnemann, J. Lumin., 2021, 235, 117970 CrossRef CAS.
- T. Kasper, M. Pavan and K. Müller-Buschbaum, J. Mater. Chem. A, 2024, 12, 769–780 RSC.
- Y. Zhang, B. Li, H. Ma, L. Zhang, H. Jiang, H. Song, L. Zhang and Y. Luo, J. Mater. Chem. C, 2016, 4, 7294–7301 RSC.
- J. F. S. do Nascimento, B. S. Barros, J. Kulesza, J. B. L. de Oliveira, A. K. Pereira Leite and R. S. de Oliveira, Mater. Chem. Phys., 2017, 190, 166–174 CrossRef.
- L. Pang, H. Zhou, Z. Qian, H. A. Munir, J. Li and H. Liu, ACS Appl. Nano Mater., 2023, 6, 20220–20230 CrossRef CAS.
- J. Li, X. Yuan, Y. nan Wu, X. Ma, F. Li, B. Zhang, Y. Wang, Z. Lei and Z. Zhang, Chem. Eng. J., 2018, 350, 637–644 CrossRef CAS.
- W. Yang, Z. Q. Bai, W. Q. Shi, L. Y. Yuan, T. Tian, Z. F. Chai, H. Wang and Z. M. Sun, Chem. Commun., 2013, 49, 10415–10417 RSC.
- K. Ren, X. F. Guo, Y. J. Tang, B. H. Huang and H. Wang, Analyst, 2020, 145, 7349–7356 RSC.
- J. D. Xiao, L. G. Qiu, F. Ke, Y. P. Yuan, G. S. Xu, Y. M. Wang and X. Jiang, J. Mater. Chem. A, 2013, 1, 8745–8752 RSC.
- Y. Li, M. Sun, Y. Yang, H. Meng, Q. Wang, C. Li and G. Li, J. Mater. Chem. C, 2021, 9, 8683–8693 RSC.
- T. W. Duan, B. Yan and H. Weng, Microporous Mesoporous Mater., 2015, 217, 196–202 CrossRef CAS.
- B. Xu, H. Guo, S. Wang, Y. Li, H. Zhang and C. Liu, CrystEngComm, 2012, 14, 2914–2919 RSC.
- H. Brunckova, E. Mudra, L. Rocha, E. Nassar, W. Nascimento, H. Kolev, M. Lisnichuk, A. Kovalcikova, Z. Molcanova, M. Strečkova and L. Medvecky, Inorganics, 2021, 9(10), 77 CrossRef CAS.
- H. Guo, Y. Zhu, S. Qiu, A. J. Lercher and H. Zhang, Adv. Mater., 2010, 22, 4190–4192 CrossRef CAS PubMed.
- X. Wang, Q. Cheng, Y. Yu and X. Zhang, Angew. Chem., 2018, 130, 7962–7966 CrossRef.
- K. Shen, L. Zhang, X. Chen, L. Liu, D. Zhang, Y. Han, J. Chen, J. Long, R. Luque, Y. Li and B. Chen, Thuwal 23955-6900, Saudi Arabia. 3 Imaging and Characterization Core Lab, KAUST, Thuwal 23955-6900.
- M. Zhang, G. Feng, Z. Song, Y. P. Zhou, H. Y. Chao, D. Yuan, T. T. Y. Tan, Z. Guo, Z. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241–7244 CrossRef CAS PubMed.
- M. Zhao, Y. Wang, Q. Ma, Y. Huang, X. Zhang, J. Ping, Z. Zhang, Q. Lu, Y. Yu, H. Xu, Y. Zhao and H. Zhang, Adv. Mater., 2015, 27, 7372–7378 CrossRef CAS PubMed.
- M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC.
- J. Guo, Y. Zhang, Y. Zhu, C. Long, M. Zhao, M. He, X. Zhang, J. Lv, B. Han and Z. Tang, Angew. Chem., 2018, 130, 6989–6993 CrossRef.
- C. Chang, W. Chen, Y. Chen, Y. Chen, Y. Chen, F. Ding, C. Fan, H. J. Fan, Z. Fan, C. Gong, Y. Gong, Q. He, X. Hong, S. Hu, W. Hu, W. Huang, Y. Huang, W. Ji, D. Li, L. J. Li, Q. Li, L. Lin, C. Ling, M. Liu, N. Liu, Z. Liu, K. P. Loh, J. Ma, F. Miao, H. Peng, M. Shao, L. Song, S. Su, S. Sun, C. Tan, Z. Tang, D. Wang, H. Wang, J. Wang, X. Wang, X. Wang, A. T. S. Wee, Z. Wei, Y. Wu, Z. S. Wu, J. Xiong, Q. Xiong, W. Xu, P. Yin, H. Zeng, Z. Zeng, T. Zhai, H. Zhang, H. Zhang, Q. Zhang, T. Zhang, X. Zhang, L. D. Zhao, M. Zhao, W. Zhao, Y. Zhao, K. G. Zhou, X. Zhou, Y. Zhou, H. Zhu, H. Zhang and Z. Liu, Wuli Huaxue Xuebao, 2021, 37(12), 2108017 Search PubMed.
- J. Dong, P. Shen, S. Ying, Z. J. Li, Y. Di Yuan, Y. Wang, X. Zheng, S. B. Peh, H. Yuan, G. Liu, Y. Cheng, Y. Pan, L. Shi, J. Zhang, D. Yuan, B. Liu, Z. Zhao, B. Z. Tang and D. Zhao, Chem. Mater., 2020, 32, 6706–6720 CrossRef CAS.
- X. Hu, Z. Wang, Y. Su, P. Chen, J. Chen, C. Zhang and C. Wang, Inorg. Chem., 2020, 59, 4181–4185 CrossRef CAS PubMed.
- P. Derias, J. Yu, R. P. Balamaran, J. Mashni and S. N. White, Chem. Commun., 2016, 52, 13031–13034 RSC.
- K. Sakamawari, M. Hagiwara and S. Fujihara, Opt. Mater., 2021, 116, 111111 CrossRef CAS.
- D. Y. Medina-Velazquez, B. Y. Alejandre-Zuniga, S. Loera-Serna, E. M. Ortiz, A. de J. Morales-Ramirez, E. Garfias-Garcia, A. Garcia-Murillo and C. Falcony, J. Nanopart. Res., 2016, 18, 10 CrossRef.
- K. Liu, H. You, G. Jia, Y. Zheng, Y. Song, M. Yang, Y. Huang and H. Zhang, Cryst. Growth Des., 2009, 9, 3519–3524 CrossRef CAS.
- F. Wang, K. Deng, G. Wu, H. Liao, H. Liao, L. Zhang, S. Lan, J. Zhang, X. Song and L. Wen, J. Inorg. Organomet. Polym. Mater., 2012, 22, 680–685 CrossRef CAS.
- J. F. Feng, X. Yang, S. Y. Gao, J. Shi and R. Cao, Langmuir, 2017, 33, 14238–14243 CrossRef CAS PubMed.
- K. Liu, Y. Zheng, G. Jia, M. Yang, Y. Huang and H. You, CrystEngComm, 2011, 13, 452–458 RSC.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef CAS.
- A. Garg, M. Almáši, J. Bednarčík, R. Sharma, V. S. Rao, P. Panchal, A. Jain and A. Sharma, Chemosphere, 2022, 305, 135467 CrossRef CAS PubMed.
- T.-W. Duan, B. Yan and H. Weng, Microporous Mesoporous Mater., 2015, 217, 196–202 CrossRef CAS.
- C. Serre, F. Millange, C. Thouvenot, N. Gardant, F. Pellé and G. Férey, J. Mater. Chem., 2004, 14, 1540–1543 RSC.
- Y. Yang, L. Zhao, M. Sun, P. Wei, G. Li and Y. Li, Dyes Pigm., 2020, 180, 108444 CrossRef CAS.
- K. Liu, H. You, Y. Zheng, G. Jia, L. Zhang, Y. Huang, M. Yang, Y. Song and H. Zhang, CrystEngComm, 2009, 11, 2622–2628 RSC.
- S. Ehrling, H. Miura, I. Senkovska and S. Kaskel, Trends Chem., 2021, 3, 291–304 CrossRef CAS.
- A. Garg, M. Almáši, D. Rattan Paul, E. Poonia, J. R. Luthra and A. Sharma, Front. Energy Res., 2021, 8, 604735 CrossRef.
- X. Cai, Z. Xie, D. Li, M. Kassymova, S. Q. Zang and H. L. Jiang, Coord. Chem. Rev., 2020, 417, 213366 CrossRef CAS.
- S. Kentish and M. Ashokkumar, in Ultrasound Technologies for Food and Bioprocessing, ed. H. Feng, G. V. Barbosa-Cánovas and J. Weiss, Springer, New York Dordrecht Heidelberg London, 2010, pp. 1–2 Search PubMed.
- D. Capková, T. Kazda, O. Čech, N. Király, T. Zelenka, P. Čudek, A. Sharma, V. Hornebecq, A. S. Fedorková and M. Almáši, J. Energy Storage, 2022, 51, 104419 CrossRef.
- J. E. Mondloch, O. Karagiaridi, O. K. Farha and J. T. Hupp, CrystEngComm, 2013, 15, 9258–9264 RSC.
- Q. Xie, Y. Li, Z. Lv, H. Zhou, X. Yang and J. Chen, Sci. Rep., 2017, 7, 1–15 CrossRef CAS PubMed.
- F. Israr, D. Chun, Y. Kim and D. Kyung, Ultrason. Sonochem., 2015, 31, 93–101 CrossRef PubMed.
- G. Mahalakshmi and V. Balachandran, Spectrochim. Acta, Part A, 2020, 124, 535–547 CrossRef PubMed.
- A. Williams, I. Altman, D. Burnett, E. G. Zorrilla, A. R. Garcia, C. Cagle, C. L. Croessmann and M. Pantoya, Appl. Surf. Sci., 2022, 579, 152185 CrossRef CAS.
- G. Okeke, R. B. Hammond and S. J. Antony, Chem. Eng. Sci., 2016, 146, 144–158 CrossRef CAS.
- X. Yi, J. Sun, X. F. Jiang, Y. Li, Q. H. Xu, Q. Zhang and S. Ye, RSC Adv., 2016, 6, 95925–95935 RSC.
- I. E. Kolesnikov, A. V. Povolotskiy, D. V. Mamonova, M. D. Mikhailov and A. V. Kurochkin, J. Rare Earths, 2018, 36, 474–481 CrossRef CAS.
- S. S. Kolesnik, V. G. Nosov, I. E. Kolesnikov, E. M. Khairullina, I. I. Tumkin, A. A. Vidyakina, A. A. Sysoeva, M. N. Ryazantsev, M. S. Panov, V. D. Khripun, N. A. Bogachev, M. Y. Skripkin and A. S. Mereshchenko, Nanomaterials, 2021, 11(9), 2448 CrossRef CAS PubMed.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- E. H. H. Hasabeldaim, H. C. Swart and R. E. Kroon, RSC Adv., 2023, 13, 5353–5366 RSC.
- A. Ali, Z. Ahmed, N. Rahisuddin and K. Iftikhar, Dalton Trans., 2023, 52, 14075–14087 RSC.
- P. Serna-Gallén, H. Beltrán-Mir and E. Cordoncillo, Ceram. Int., 2023, 49, 41078–41089 CrossRef.
- Z. Ahmed, R. dos S. Carvalho, A. M. dos Santos, F. Gambassi, E. Bandini, L. Marvelli, L. Maini, A. Barbieri and M. Cremona, Molecules, 2023, 28(11), 4371 CrossRef CAS PubMed.
- S. Chetry, M. F. Lukman, V. Bon, R. Warias, D. Fuhrmann, J. Möllmer, D. Belder, C. S. Gopinath, S. Kaskel, A. Pöppl and H. Krautscheid, Inorg. Chem., 2024, 63, 10843–10853 CrossRef CAS PubMed.
- Y.-H. Wen, J.-K. Cheng, Y.-L. Feng, J. Zhang, Z.-J. Li and Y.-G. Yao, CCDC 290771: Experimental Crystal Structure Determination, 2006.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179, DOI:10.1107/S2052520616003954.
- E. H. H. Hasabeldaim, H. C. Swart and R. E. Kroon, Luminescence and stability of Tb doped CaF2 nanoparticles, RSC Adv., 2023, 13, 5353–5366, 10.1039/D2RA07897J.
- J. Rodríguez-Carvajal, Recent advances in magnetic structure determination by neutron powder diffraction, Phys. B, 1993, 192(1–2), 55–69, DOI:10.1016/0921-4526(93)90108-I.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, Mercury 4.0: From visualization to analysis, design and prediction, J. Appl. Crystallogr., 2020, 53(1), 226–235, DOI:10.1107/S1600576719014092.
- W. S. Rasband, ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA, 1997–present Search PubMed.
- OriginLab Corporation, Origin (Version 2017), Northampton, MA, 2023 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ce00198f |
| ‡ Current position of Dr. Zulma L. Moreno Botello at Consiglio Nazionale delle Ricerche, Istituto di Tecnologie Avanzate per l'Energia “Nicola Giordano”, CNR-ITAE, via Salita Santa Lucia Sopra Contesse 5, 98126, Messina, Italy. |
| This journal is © The Royal Society of Chemistry 2025 |