
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, X-ray characterization, and DFT calculations of gold–nucleobase complexes: on the importance of regium bonds and anion–π interactions†
Jordi
Buils‡
 ,
Angel
Terrón
,
Miquel
Barceló-Oliver
,
Juan Jesús
Fiol
,
Angel
García-Raso
,
Rosa M.
Gomila
and
Antonio
Frontera
*
,
Angel
Terrón
,
Miquel
Barceló-Oliver
,
Juan Jesús
Fiol
,
Angel
García-Raso
,
Rosa M.
Gomila
and
Antonio
Frontera
*
Department of Chemistry, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain. E-mail: toni.frontera@uib.es
First published on 28th May 2025
Abstract
In this manuscript, we report the synthesis and X-ray characterization of two new Au(III)–cytosine systems: AuCl3(CytC6) (1) and (HCytC6)2[AuCl4]Cl (2), where CytC6 is N1-hexylcytosine. Compound 1 is an inner sphere complex where the AuCl3 unit is coordinated to N3, while compound 2 is an outer sphere complex (salt) where two N1-hexylcytosinium cations are charge compensated by one chloride anion and one tetrachloroaurate anion. Inner sphere complexes of Au(III) with cytosine and nucleobases, in general, are scarcely found in the CSD. In fact, compound 1 is only the third example of a cytosine derivative coordinated to Au(III). Such complexes remain elusive for other nucleobases. The formation of regium bonds in the solid state of compound 1 has been analysed using DFT calculations and characterized with several computational tools, including molecular electrostatic potential (MEP), energy decomposition analysis (EDA), quantum theory of atoms in molecules (QTAIM), and noncovalent interaction plot (NCIplot).
1 Introduction
The interaction between gold and nucleobases has been studied for various purposes, one of which is the presence of special noncovalent interactions like regium bonds and aurophilic interactions.1 Purines preferentially bond through N7, while pyrimidines bond through N3. An example is the recently published gold N1-hexylcytosine coordination complex by our research team.2The number of well-characterized gold–nucleobase complexes or their derivatives, confirmed by X-ray diffraction, is quite low. One published structure shows gold binding to a cytosine–guanine base pair within an RNA structure, but the resolution is too low to confirm the oxidation state of gold or the exact coordination positions.3 Apart from this, there are few other coordination complexes, with only one involving methyl-cytosine.4 Other notable structures include two organometallic uracil–gold complexes5 with metal–carbon bonds and a guanine6 derivative gold complex.
Gold coordination chemistry mainly involves two oxidation states: gold(I) and gold(III). The redox chemistry of gold(III) can be challenging with biological ligands due to the redox properties affecting the stability and reactivity of gold compounds.7 It has been demonstrated that gold coordinated to guanine can lead to guanine degradation. Additionally, the degradation of GMP coordinated to gold complexes in aqueous solution has been described.8
A key question is whether aurophilic interactions could be comparable to argentophilic ones and whether they could dictate the formation of infinite 1D chains. To the best of our knowledge, there are very few X-ray crystal structures showing Au⋯Au aurophilic interactions in nucleobase derivatives. One example is an Au(I)–isonitrile derivative of guanosine, which forms tetramers or octamers via self-assembly in the presence or absence of potassium ions, respectively, and exhibits switchable emission based on the Au+⋯Au+ interaction9 present only in the octameric assembly. Another example by Blasco et al. involves [Au(adeninate-N9)(PR3)] (PR3 = PMe3 or 1,3,5-triaza-7-phosphaadamantane) complexes with Au+⋯Au+ interactions (Au⋯Au distances of 3.208 and 3.0942 Å). Depending on the phosphine co-ligand used, they observed the formation of ultrathin nanowires leading to a blue-luminescent hydrogel or single crystals.10
Regium bonds could play a role in the interaction between group 11 metal ions and nucleic acids. Recently, a search was conducted to investigate the prevalence of regium bonds in tetra-coordinated gold complexes. The results indicated that these bonds are common in outer sphere complexes in X-ray crystal structures and have likely been overlooked due to the underestimated van der Waals radius value for gold.11
Several reports have shown that nucleic acids and nucleobases are able to participate in anion–π interactions.12,13 An example of an anion–π interaction with aurate(III) is the X-ray crystal structure of inosinium tetrabromidoaurate(III), where the [AuBr4]− anion is located over the centre of the hypoxanthine ring.14
Recently an Au(I)-S6-tGua (tgua: 6-tioguanosine) presents aurophylic interactions that permits conductivity in 1D dimension after activation.15
In this study, we report the synthesis, X-ray characterization and DFT study of two more X-ray examples of Au(III)–cytosine systems: AuCl3(CytC6) (1) and (HCytC6)2 [AuCl4] Cl (2), where CytC6 is N1-hexylcytosine. Compound 1 is an inner sphere complex and compound 2 is an outer sphere compound (salt), see Scheme 1. The formation of regium bonds and anion–π interactions have been studied using DFT calculations and characterized by means of QTAIM, NCIPlot, NBO and MEP surface analysis.
 | ||
| Scheme 1 Compounds 1–2 studied in this work. | ||
2 Experimental
2.1 Materials and methods
Some of the reagents were obtained from commercial products from VWR CHEMICALS (NaAuCl4 and HAuCl4) without further purification. N1-hexylcytosine and N1-hexylcytosinium bromide were synthesized according to bibliography.16 Elemental analysis was carried out by “Servei de Microanàlisi del CSIC Barcelona”. Carbon, hydrogen, and nitrogen were determined using Carlo-Erba (1106–1108) and Microanalyzer Thermo Finnigan Flash 1112. IR spectra were obtained in a Bruker Tensor between 4000–400 cm−1 in KBr tablets.2.2 Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 outer sphere complex, confirmed by SC-XRD.
:2 outer sphere complex, confirmed by SC-XRD.
C: 31.48% (31.33) H: 4.57% (4.73) N: 10.53% (10.96), elemental analysis calculated for asymmetric unit.
IR: 3433 (m) 3371 (m) 3212 (m) 2955 (m) 2920 (m) 2852 (m) 1922 (w) 1726 (s) 1682 (m) 1649 (s) 1490 (m) 1374 (m) 1200 (w) 788 (m) 616 (m) 502 (w).
2.3 Crystallography
Single crystals of two compounds were selected, covered with Parabar 10320 (formally known as Paratone N) and mounted on a Cryoloop on a D8 Venture diffractometer, with a Photon III 14 detector, using an Incoatec high brilliance IμS DIAMOND Cu equipped with an Incoatec Helios MX multilayer optics. The crystals were kept at 100.0 K during data collection Data reduction and cell refinements were performed using the Bruker APEX3 program.17 Scaling and absorption corrections were carried out using the SADABS program in all cases.18 Using Olex2,19 the structure was solved with the XT structure solution program20 using intrinsic phasing and refined with the XL refinement package21 using Least Squares minimization. All non-hydrogen atoms were refined with anisotropic thermal parameters by full-matrix least-squares calculations on F2. Hydrogen atoms were generally inserted at calculated positions and refined as riders.As usual in this kind of molecules,16 some of the aliphatic chains from CytC6 are disordered over two complimentary positions (50% for each one in 1 and 75%/25% in 2).
The structures were checked for higher symmetry with help of the program PLATON.22 The graphical material have been prepared with the help of Mercury software.23 Crystallographic details are summarized in Table 1.
| 1 | 2 | |
|---|---|---|
| Empirical formula | C10H17AuCl3N3O | C20H36AuCl5N6O2 |
| Formula weight | 498.58 | 766.76 |
| Temperature (K) | 100.0 | |
| Crystal system | Monoclinic | Triclinic |
| Space group | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) | 16.1471(11) | 6.8686(9) |
| b (Å) | 8.4748(5) | 10.7183(14) |
| c (Å) | 11.7690(7) | 20.600(3) |
| α (°) | 90 | 84.708(6) |
| β (°) | 105.399(3) | 86.889(6) |
| γ (°) | 90 | 72.364(5) |
| Volume (Å3) | 1552.69(17) | 1438.6(3) |
| Z | 4 | 2 |
| ρ calc (g cm−3) | 2.133 | 1.770 |
| μ (mm−1) | 22.504 | 14.120 |
| F(000) | 944.0 | 756.0 |
| Crystal size (mm3) | 0.19 × 0.15 × 0.08 | 0.28 × 0.08 × 0.05 |
| Radiation | CuKα (λ = 1.54178) | |
| 2Θ range for data collection (°) | 11.368 to 135.994 | 8.624 to 137.34 |
| Index ranges | −19 ≤ h ≤ 19 | −8 ≤ h ≤ 7 |
| −10 ≤ k ≤ 9 | −12 ≤ k ≤ 12 | |
| −14 ≤ l ≤ 13 | −24 ≤ l ≤ 24 | |
| Reflections collected | 10792 |
78925 |
| Independent reflections | 2775 [Rint = 0.0531, Rsigma = 0.0472] | 5201 [Rint = 0.0777, Rsigma = 0.0272] |
| Data/restraints/parameters | 2775/12/210 | 5201/7/309 |
| Goodness-of-fit on F2 | 1.063 | 1.177 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0493 | R 1 = 0.1094 |
| wR2 = 0.1094 | wR2 = 0.2871 | |
| Final R indexes [all data] | R 1 = 0.0641 | R 1 = 0.1113 |
| wR2 = 0.1203 | wR2 = 0.2880 | |
| Largest diff. peak/hole (e Å−3) | 2.97/−1.18 | 7.68/−3.92 |
| CCDC reference codes | 2440200 | 2440201 |
2.4 DFT calculations
Theoretical calculations were computed at the PBE0-D4/def2-TZVP level of theory,24–26 using the crystallographic coordinates within the Turbomole 7.7 program.27 The quantum theory of “atoms-in-molecules” (QTAIM)28 and noncovalent interaction plot (NCIplot)29 analyses were performed at the same level of theory using the Multiwfn program.30 The QTAIM/NCIplot analysis was represented using the VMD software.31 The energy decomposition analysis (EDA) was performed using the Turbomole 7.7 program27 using the Kitaura–Morokuma partition scheme.32 The NBO analysis33 was performed using the NBO 7.0 program.34 The MEP surface plots were generated using GaussView 6.0, employing a 0.001 a.u. electron density isovalue as an approximation of the van der Waals envelope. The corresponding cube files were produced with Multiwfn, based on wavefunctions obtained at the PBE0-D4/def2-TZVP level of theory.3 Results and discussion
3.1 Structural characterization of 1 and 2
A polymorph structure for AuCl3CytC6 was obtained after direct reaction of tetrachloridoaurate with N1-hexylcytosinium hexafluoroantimoniate. The asymmetric unit corresponds to the complex unit (see Table S1† for coordination distances and angles). Dihedral angle of cytosine planes and tetrachloridoaurate anion plane is 90°. Both amino hydrogen atoms stay coplanar with cytosine ring and form an angle of 120 with N(4). Angles around N(4) are all three of 120°, being a plane triangular geometry (Fig. 1). Crystal packing consists of four complex units where the hexyl chains establish hydrophobic interactions. | ||
| Fig. 1 Asymmetric unit of complex AuCl3(CytC6). The aliphatic chain of the cytosine is disordered and have been solved over two equivalent (50% each) positions. | ||
This polymorph differs with the previously described complex,2 in the unit cell parameters and the layout of the asymmetric unit which is the same (Fig. 2).
 | ||
| Fig. 2 Comparison (front and top views) between the two AuCl3CytC6 polymorph structures, where the molecules in part (a) correspond to two different views of compound 1 and the molecules in part (b) are the previously reported polymorph complex.2 The different disposition of the units and the distinctive interaction patterns can be observed. Aliphatic chains have been omitted for clarity. | ||
Packing of complex 1 contains a AuCl3CytC6 complex unit nearly perpendicular to an adjacent complex unit. These are connected by hydrogen bonds. In particular a hydrogen bond between H(4a) and Cl(1) and another one between H(4b) and a Cl(1) (see Fig. 3) with similar distances (see Table 2).
 | ||
| Fig. 3 View of hydrogen bond formed between the NH2 group and the chloride ligands in 1. Distances in Å (aliphatic chains have been omitted for clarity). See Table 1 for N⋯Cl distances with esd values. | ||
| D–H⋯A | d (D–H) (Å) | d (H⋯A) (Å) | d (D⋯A) (Å) | < (DHA) (°) |
|---|---|---|---|---|
| N(4)–H(4a)⋯Cl(1) | 0.88 | 2.57 | 3.41(1) | 158.9 |
| N(4)–H(4b)⋯Cl(2) | 0.88 | 2.85 | 3.37(1) | 120.0 |
| N(4)–H(4b)⋯Cl(1) | 0.88 | 2.67 | 3.47(1) | 150.7 |
| C(6)–H(6)⋯O(2) | 0.95 | 2.46 | 3.26(2) | 141.9 |
An interesting contact is observed between the Au and a proximal chloride, that can be understood as a regium bond interaction between a π-hole on Au, and a π-lump on the chlorido ligand (see Fig. 4). The formation of such dimer is most likely enhanced by two NH⋯Cl contacts, as analysed below (theoretical section). Such contacts were also described in its polymorph,2 where molecules self-assemble via N–H⋯Cl and Au⋯Cl contacts to form centrosymmetric dimers.
 | ||
| Fig. 4 View of regium bond formation in compound 1. Distances in Å (aliphatic chains have been omitted for clarity). The RgB distance is 3.574(3) Å. | ||
Complex 2 is an outer-sphere complex (see Table S2† for coordination distances and angles), comprising two cytosinium cations and two anions (Cl and AuCl4) in the asymmetric unit (Fig. 5). The two protonated cytosine molecules form an angle of 78° with each other, while the tetrachloridoaurate anion is oriented at a 5° angle relative to one of the cytosine rings and at an 82° angle to the other. The chloride anion is positioned at the intersection of the two cytosine planes, contributing to the overall structural arrangement (Fig. 5).
 | ||
| Fig. 5 View of asymmetric unit of X-ray structure for complex 2. The aliphatic chain of one cytosine is disordered and have been solved over two positions (C7A–C12A, 75% occupancy; C7C–C12C, 25% occupancy). The lower occupancy chain has been omitted for clarity. | ||
Complex units form a 1D infinite supramolecular polymer along the “a” axis, where cytosine rings and tetrachloridoaurate anions stack alternately on each other (see Fig. 6). The second cytosine ring is positioned perpendicular to the planes of both the first cytosine ring and the tetrachloridoaurate anion. Meanwhile, the hexyl tails align in such a way that the entire system resembles a lipid bilayer structure, contributing to the overall stability and organization of the assembly.
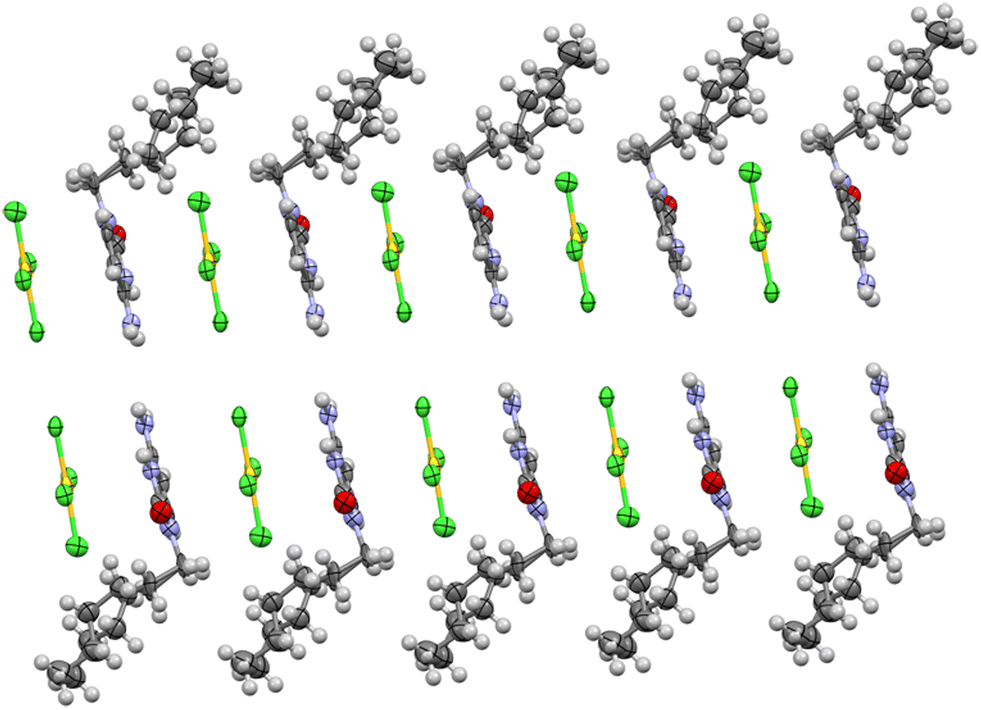 | ||
| Fig. 6 View of crystal growing through the “b” axis, with anion–π interaction between cytosine rings and AuCl4 displayed in the “a” direction. | ||
Several hydrogen bonds are present in this structure. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group from the cytosine ring serves as an acceptor for two hydrogen bonds: one donated by the N3 atom of a perpendicular cytosine ring and another by the C5 atom of a coplanar cytosine. Additional hydrogen bonds are observed where the free chloride from the asymmetric unit participates in up to four hydrogen bonds, interacting with three amino groups and the N3 atom of a different cytosine. Furthermore, the tetrachloridoaurate anion also engages in a hydrogen bond, with one of its chloride atoms acting as the acceptor. This bond involves the C8 hydrogen as the donor (see Fig. 7 and Table 3).
O group from the cytosine ring serves as an acceptor for two hydrogen bonds: one donated by the N3 atom of a perpendicular cytosine ring and another by the C5 atom of a coplanar cytosine. Additional hydrogen bonds are observed where the free chloride from the asymmetric unit participates in up to four hydrogen bonds, interacting with three amino groups and the N3 atom of a different cytosine. Furthermore, the tetrachloridoaurate anion also engages in a hydrogen bond, with one of its chloride atoms acting as the acceptor. This bond involves the C8 hydrogen as the donor (see Fig. 7 and Table 3).
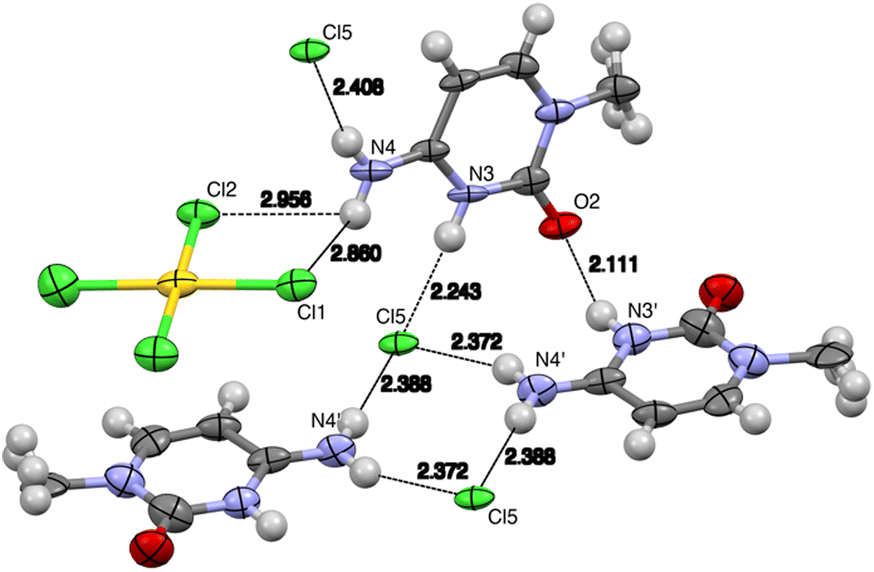 | ||
| Fig. 7 View of the hydrogen bonds in the solid-state X-ray structure of complex 2. Distances in Å (aliphatic chains have been omitted for clarity). See Table 2 for the N⋯Cl distances with esd values. | ||
| D–H⋯A | d (D–H) | d (H⋯A) | d (D⋯A) |
|
|---|---|---|---|---|
| N(3′)–H(3′)⋯O(2) | 0.88 | 2.10 | 2.93(1) | 155.6 |
| C(5)–H(5)⋯O(2) | 0.95 | 2.43 | 3.17(2) | 134.8 |
| N(4)–H(4b)⋯Cl(5) | 0.88 | 2.41 | 3.27(1) | 166.2 |
| N(4′)–H(4′a)⋯Cl(5) | 0.88 | 2.38 | 3.21(1) | 157.6 |
| N(4′)–H(4′b)⋯Cl(5) | 0.88 | 2.39 | 3.27(1) | 177.4 |
| N(3)–H(3)⋯Cl(5) | 0.88 | 2.24 | 3.12(1) | 176.5 |
| C(8)–H(8)⋯Cl(4) | 0.99 | 3.08 | 3.43(2) | 102.2 |

3.2 DFT study
The MEP surfaces of compound 1 and the (HCytC6)Cl fragment of compound 2 are shown in Fig. 8. For compound 1, the MEP minimum is located at the chloride ligands of Au(III) (−42.3 kcal mol−1), followed by the O-atom of the cytosine ring (−38.4 kcal mol−1). The MEP maximum is found in the region influenced by one of the H-atoms from the amino group and the adjacent H-atom of the cytosine's CC double bond (67.8 kcal mol−1). Additionally, a positive MEP is observed over the Au atom (37.7 kcal mol−1), indicating the presence of a π-hole, with the positive value enhanced by the adjacent N–H bond directed towards the π-hole. The MEP is also notably positive over the center of the cytosine ring.
 | ||
| Fig. 8 MEP surfaces of compounds 1 (a) and 2 (b). MEP values at selected points in kcal mol−1. Isovalue 0.001 a.u. | ||
In compound 2, the MEP minimum is located at the chloride anion (−82.2 kcal mol−1), while the maximum is found at one of the NH groups of the amino (80.9 kcal mol−1). A large positive MEP is also seen at the protonated N-atom of cytosinium (60.2 kcal mol−1) and at the center of the ring (54.6 kcal mol−1), explaining its ability to form anion–π interactions (see Fig. 5 and 6). The MEP is negative at the O-atom of cytosinium (−20.7 kcal mol−1).
The π-hole regium bonding (RgB) interactions depicted in Fig. 4 have been analyzed using a combination of QTAIM and NCIplot methods, which are highly effective in revealing interactions in real space. The analysis for the dimer of compound 1 is presented in Fig. 9a, confirming the presence of the RgB. This interaction is characterized by a bond critical point (BCP), represented by a small red sphere, and a bond path linking the Cl atom to the Au. Additionally, a green-colored reduced density gradient (RDG) isosurface appears at the location of the BCP, further validating the existence of the RgB and confirming its attractive nature. The QTAIM analysis also shows that the amino group is connected to two chloride ligands via BCPs, bond paths, and green RDG isosurfaces. The overall dimerization energy is significant (−12.2 kcal mol−1), resulting from the combined contributions of hydrogen bonds and the Au⋯Cl RgB π-hole interaction. To isolate the contribution of the RgB, we modeled a system in which the amino group was replaced by a hydrogen atom. This reduced the dimerization energy to −6.8 kcal mol−1, indicating the contribution of the RgB, with the difference attributed to hydrogen bonding (−5.4 kcal mol−1).
 | ||
| Fig. 9 (a) Combined QTAIM (BCPs as red spheres and bond paths as orange lines) and NCIPlot (RDG = 0.5, ρ cut-off = 0.04, color scale ±0.035 a.u.) of the RgB dimer of 1. Only intermolecular interactions are represented. The dimerization energies are indicated. (b) Donor and acceptors NBO corresponding to the LP(Cl) → σ*(Au–Cl). The second order perturbation energy is indicated. | ||
To further explore the RgB from an orbital perspective, we conducted an NBO analysis, which effectively identifies electron transfer in donor–acceptor interactions. The NBO analysis revealed electron donation from the lone pair on the Cl atom to the three Au–Cl antibonding orbitals, consistent with typical π-hole regium bonding interactions.35,36 One of these donor–acceptor interactions is illustrated in Fig. 9b, with the total stabilization energy estimated at 1.23 kcal mol−1.
To further validate the RgB nature of the interaction and the role of Au(III) as an electron acceptor, we compared the electron density (ED) and electrostatic potential (ESP) values along the bond path connecting the Au and Cl atoms. This computational approach is particularly useful for examining the role of two interacting atoms in noncovalent interactions.37 Specifically, the minimum of the electron density (EDmin), corresponding to the location of the BCP, is shifted toward the electron acceptor, while the minimum of the electrostatic potential (ESPmin) is shifted toward the electron donor. In the case of the RgB dimer in compound 1, the plot is shown in Fig. 10. It reveals that the EDmin is closer to the Au atom, while the ESPmin is nearer to the Cl atom, confirming the electron-donating role of Cl. This observation is consistent with the results from the MEP surface and NBO analyses, further corroborating the RgB nature of the interaction.
 | ||
| Fig. 10 Plots of the electron density (ED) and electrostatic potential (ESP) along the path connecting the Au and Cl-atoms. | ||
For compound 2, we compared the two anion–π interaction modes observed in the solid state (see Fig. 5). In one mode, the [AuCl4]− anion is positioned parallel to the cytosinium ring, which is responsible for forming the 1D supramolecular polymer shown in Fig. 7. In the other mode, the [AuCl4]− anion is arranged perpendicular to the cytosinium ring, with two chloride ligands pointing toward the ring. Both binding modes were analyzed energetically and using QTAIM/NCIplot analysis. For the energetic calculations, we used the (HCytC6)Cl fragment as a neutral monomer to minimize the dominant coulombic attractions between counterions.
The results, shown in Fig. 11, reveal that in the parallel orientation, all atoms of the anion are connected by bond critical points (BCPs) and bond paths to the (HCytC6)+ moiety. Three atoms (Au, Cl, Cl) are linked to the π-system of (HCytC6)+ by BCPs and bond paths, with a large, extended RDG isosurface located between the anion and the cytosinium ring, characterizing the anion–π interaction. The QTAIM/NCIplot analysis also shows the formation of CH⋯Cl contacts involving the hexyl chain's CH groups. This intricate combination of contacts explains the large dimerization energy (−25.3 kcal mol−1), highlighting the importance of this assembly in the solid-state structure of 2 and supporting the formation of the 1D supramolecular structure through alternating anions and cytosinium moieties.
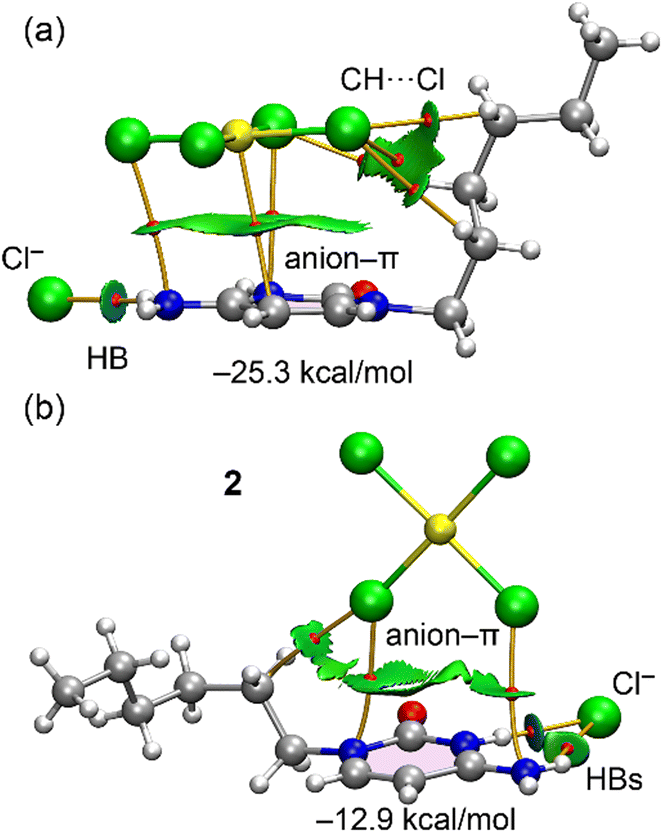 | ||
| Fig. 11 Combined QTAIM (BCPs as red spheres and bond paths as orange lines) and NCIPlot (RDG = 0.5, ρ cut-off = 0.04, color scale ±0.035 a.u.) for parallel (a) and T-shape (b) anion–π binding modes. Only intermolecular interactions are represented. | ||
In contrast, the T-shaped complex (Fig. 11b) exhibits a significantly lower interaction energy (−12.9 kcal mol−1) similar to the RgB dimer of compound 1. In this case, only two Cl atoms are linked to the (HCytC6)+ moiety, explaining the weaker nature of this binding mode compared to the parallel configuration. The chloride anion is linked to the cytosinium ring through two BCPs and bond paths corresponding to NH⋯Cl HBs. One of both is characterized by a blue, disk-shaped RDG isosurface, indicating the strong nature of this HB. The QTAIM parameters at the bond critical points (BCPs) shown in Fig. 9 and 11 are provided in the ESI† (Table S1). These values exhibit the characteristic features of weak interactions, including low electron density, a positive Laplacian, and positive total energy density, consistent with closed-shell noncovalent interactions.
Finally, an energy decomposition analysis (EDA) was conducted to assess the key contributions to the interaction energies in the RgB dimer of compound 1 and the anion–π binding modes of compound 2. The total interaction energies (Etot) were decomposed into exchange repulsion (Eex-rep), electrostatic (Eel), orbital (Eorb), correlation (Ecor), and dispersion (Edisp) components, as shown in Fig. 11.
For the RgB dimer, the dominant contribution comes from the electrostatic term, followed by the orbital component, with the dispersion and correlation terms being more modest. This predominance of the electrostatic term aligns with the MEP surface analysis and the presence of two H-bond interactions, where electrostatics plays the most significant role. More notably, the EDA analysis reveals clear differences between the two π-stacking modes in complex 2. In the parallel dimer, the electrostatic contribution is dominant, whereas in the T-shaped dimer, the orbital contribution surpasses the electrostatic. However, all interaction components are more pronounced in the parallel orientation, particularly the electrostatic and dispersion terms. The stronger electrostatic component is due to the proximity of all atoms of the anion to the cationic (HCytC6)+ moiety, while the increased dispersion results from the stacking of the anion over the π-system of cytosine, enhancing dispersion effects typical of π-stacking interactions (Fig. 12).
 | ||
| Fig. 12 The total, Etot (pink); exchange repulsion, Eex-rep (red); electrostatic, Eel (blue); orbital, Eorb (grey); correlation, Ecor (violet) and dispersion, Edisp (green) energies for the RgB dimer of compound 1 and the two π-stacking modes of compound 2. | ||
3.3 CSD search
To evaluate the potential of the AuX3(NR) synthon (X = Cl, Br) to engage in RgB interactions (see Fig. 13, top), we performed a search of the Cambridge Structural Database (CSD, version 2024.3.0, updated November 2024).38 Remarkably, we identified 83 structures (126 RgB contacts) for X = Cl and 7 (15 RgB contacts) for X = Br featuring Au⋯Y interactions (Y = O, S, N, F, Cl, Br, I) consistent with regium bonding. The complete list of hits, along with relevant geometric parameters, including Y⋯Au distances and Y⋯Au–X angles, is provided in Tables S2 and S3 (ESI†). | ||
| Fig. 13 Top: Histogram showing the distribution of CSD hits as a function of the Y⋯Au distance. Bottom: Scatterplot of the mean Y⋯Au–Cl angle (α) versus the Y⋯Au distance. | ||
For the more extensive X = Cl dataset, we analysed the distribution of Y⋯Au distances and the correlation between distance and directionality, represented in Fig. 13 as a histogram of Y⋯Au distances and a scatterplot of the mean Y⋯Au–Cl angle versus distance. As a geometric cutoff, we considered contacts within ∑RvdW + 0.5 Å, accounting for the fact that the van der Waals radius of gold in the CSD (based on Bondi)39 is underestimated.40 The histogram shows that the majority of hits fall between 3.35 and 3.65 Å, which is consistent with the Y⋯Au distance observed in compound 1.
The scatterplot shows that the mean angle (α1 + α2 + α3)/3 ranges from 80° to 96°, with the majority of data points clustered between 86° and 92°. The individual values of α1, α2, and α3 are summarized in Table S1,† revealing that in all cases, the angles span from 69° to 114°. This range confirms that the mean angle is a meaningful representation of the overall directionality and is not the result of compensating extremes. These observations support the interpretation of the interaction as a directional π-hole contact.
Most of the structures retrieved from the CSD search form self-assembled dimers stabilized by two symmetrically equivalent Au⋯Cl regium bonds, as exemplified by the structure shown in Fig. 14 (CSD code WIRGAH).41 In a few cases, such as WOQMEU,42 the regium bond facilitates the formation of a heterodimer. In this example, the electron-rich Br atom of the [AuBr]4− anion is positioned over the Au center of a neutral AuCl3Py molecule, establishing a directional regium bond. This interaction is further stabilized by a CH⋯Br hydrogen bond, highlighting the cooperative nature of noncovalent interactions in the assembly.
 | ||
| Fig. 14 Partial views of the CSD refcodes WIRGAH (a) and WOGMEU (b) forming homodimer and heterodimer, respectively, with indication of the RgBs. Distances in Å. | ||
Concluding remarks
In this study, the synthesis and structural analysis of two new Au(III)–cytosine complexes, AuCl3(CytC6) (1) and (HCytC6)2[AuCl4]Cl (2), have been reported. Through a combination of experimental techniques, including X-ray crystallography, and theoretical DFT calculations, we investigated the solid-state interactions that govern the stability and organization of these complexes. The analysis revealed that compound 1 exhibits significant regium bonding interactions, as evidenced by QTAIM/NCIplot and NBO results. These interactions, combined with hydrogen bonding, contribute substantially to the dimerization energy and the overall supramolecular structure. In addition, a CSD analysis of AuX3(NR)-type structures further confirms the prevalence and geometric consistency of regium bonding interactions, lending broader context and support to the findings from compound 1.In compound 2, the anion–π interactions, particularly between the [AuCl4]− anion and the cytosinium ring, play a dominant role in the formation of the 1D supramolecular assembly. Energy decomposition analysis (EDA) further demonstrated that in compound 2, the parallel π-stacking configuration exhibits a strong electrostatic contribution, whereas the T-shaped stacking is more dependent on orbital interactions.
Overall, these findings highlight the importance of both non-covalent interactions, including regium bonds and anion–π interactions, in the stabilization and architecture of Au(III)–cytosine complexes. The insights gained from this study pave the way for future exploration into the design of novel materials utilizing these unique interactions.
Data availability
All crystallographic data has been deposited at the CCDC under 2440200 and 2440201 deposition numbers and can be obtained from https://www.ccdc.cam.ac.uk/. All programs have been properly cited and are commercially available. The X-ray coordinates were used for the DFT study.Author contributions
The manuscript was prepared with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the MICIU/AEI of Spain (projects PID2020-115637GB-I00 and PID2023-148453NB-I00, FEDER funds). The authors also acknowledge the funds (project number 111/2024) received from the Universitat de les Illes Balears and the Pla anual d’impuls del turisme sostenible per al període 2023 (ITS2023-086 - Programa de Foment de la Recerca). We thank Prof. Dr. Bernhard Spingler (Department of Chemistry, University of Zürich) for his valuable advice on the structural resolution of compound 2.Notes and references
- M. Barceló-Oliver, A. Frontera, J. J. Fiol, A. García-Raso and A. Terrón, Met. Ions Life Sci., 2023, 25, 105 Search PubMed.
- A. Terrón, J. Buils, T. J. Mooibroek, M. Barceló-Oliver, A. García-Raso, J. J. Fiol and A. Frontera, Chem. Commun., 2020, 56, 3524–3527 RSC.
- E. A. Ennifar, P. Walter and P. Dumas, Nucleic Acids Res., 2003, 31, 2671–2682 CrossRef PubMed.
- M. S. Holowczak, M. D. Stancl and G. B. Wong, J. Am. Chem. Soc., 1985, 107, 5789–5790 CrossRef.
- F. Zamora, E. Zangrando, M. Furlan, L. Randaccio and B. Lippert, J. Organomet. Chem., 1998, 552, 127–134 CrossRef.
- A. Schimanski, E. Freisinger, A. Erxleben and B. Lippert, Inorg. Chim. Acta, 1998, 283, 223–232 CrossRef.
- M. D. Đurović, Ž. D. Bugarčić and R. van Eldik, Coord. Chem. Rev., 2017, 338, 186–206 CrossRef.
- S. Zhu, W. Gorski, D. R. Powell and J. A. Walmsley, Inorg. Chem., 2006, 45, 2688–2694 CrossRef PubMed.
- X. Meng, T. Moriuchi, M. Kawahata, K. Yamaguchi and T. Hirao, Chem. Commun., 2011, 47, 4682–4684 RSC.
- D. Blasco, J. M. López-de-Luzuriaga, M. Monge, M. E. Olmos, D. Pascual and M. Rodriguez-Castillo, Inorg. Chem., 2018, 57, 3805–3817 CrossRef.
- J. M. Salas, M. Quirós, M. P. Sánchez and A. L. Beauchamp, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1989, 45, 1874–1877 CrossRef.
- A. García-Raso, F. M. Albertí, J. J. Fiol, A. Tasada, M. Barceló-Oliver, E. Molins, D. Escudero, A. Frontera, D. Quiñonero and P. M. Deyà, Inorg. Chem., 2007, 46, 10724–10735 CrossRef PubMed.
- X. Lucas, A. Bauzá, A. Frontera and D. Quiñonero, Chem. Sci., 2016, 7, 1038–1050 RSC.
- A. Houlton, Met. Ions Life Sci., 1993, 25, 79–104 Search PubMed.
- L. L. G. Al-Mahamat, O. El Zubiri, D. G. Smith, B. R. Horrocks and A. Houlton, Nat. Chem., 2017, 8, 720 Search PubMed.
- M. Barceló-Oliver, B. A. Baquero, A. Bauzá, A. García-Raso, A. Terrón, I. Mata, E. Molins and A. Frontera, CrystEngComm, 2012, 14, 5777 RSC.
- Bruker, APEX III, Bruker AXS Inc., Madison, Wisconsin, USA, 2019 Search PubMed.
- G. K. Sheldrick, SADABS, Version 2008/1, Bruker AXS Inc., Germany, 2008 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef.
- J. Contreras-Garcia, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0., Theoretical Chemistry Institute, University of Wisconsin, Madison, 2018 Search PubMed.
- A. Daolio, A. Pizzi, G. Terraneo, M. Ursini, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 60, 14385–14389 CrossRef.
- A. Pizzi, M. Calabrese, A. Daolio, M. Ursini, A. Frontera and G. Resnati, CrystEngComm, 2022, 24, 3846–3851 RSC.
- L. Andreo, R. M. Gomila, E. Priola, A. Giordana, S. Pantaleone, E. Diana, G. Mahmoudi and A. Frontera, Cryst. Growth Des., 2022, 22, 6539–6544 CrossRef.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef.
- S. Alvarez, Dalton Trans., 2013, 42, 8617–8636 RSC.
- R. Mohammad-Nataj, A. Abedi and V. Amani, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2013, 43, 1375 CrossRef.
- K. Peters, E.-M. Peters, H. G. von Schnering, W. Honle, R. Schmidt and H. Binder, Z. Kristallogr., 2000, 215, 413 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2440200 and 2440201. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ce00363f |
| ‡ Present address: Institut Català d' Investigació Química ICIQ, Av. Països Catalans, 16, 43007 Tarragona, Spain. |
| This journal is © The Royal Society of Chemistry 2025 |